
Abstract
The absence of induced pluripotent stem cell (iPSC) lines derived from Emirati patients with developmental disease hampers region-specific disease modeling and therapeutic research. Herein, we describe the creation of an iPSC line from peripheral blood mononuclear cells obtained from a 21-year-old Emirati female patient with ventricular septal defect (VSD) using Sendai virus-mediated delivery of reprogramming factors. The resulting line, UAEUi001-A, exhibited typical colony morphology, was mycoplasma negative, successfully generated embryoid bodies), and demonstrated strong alkaline phosphatase activity. These iPSCs were further characterized for pluripotency potential and their differentiation potential into the three germ layers under in vitro culture conditions through immunostaining using stage-specific markers. To the best our knowledge, this is the first reported generation of an iPSC line from an Emirati patient with VSD. Overall, this iPSC line may serve as a valuable model for establishing an Emirati-specific iPSC repository, supporting disease modeling and drug discovery relevant to the Emirati population.
Impact Statement
This study establishes the first induced pluripotent stem cell (iPSC) line derived from an Emirati patient with ventricular septal defect, addressing a critical gap in region-specific disease models. It provides a valuable platform for understanding developmental cardiac disorders in underrepresented populations and supports the development of precision medicine and targeted therapeutic strategies relevant to the Emirati population.
Keywords
Introduction
Congenital heart disease (CHD) is the most common birth defect worldwide and the number one killer of live-born infants. CHD accounts for 30% of fetal losses and affects about 1% of live births. CHD is classified into acyanotic and cyanotic types. The most common acyanotic CHDs include ventricular septal defect (VSD), atrial septal defect, and atrioventricular septal defect, whereas the most common cyanotic CHDs include tetralogy of Fallot and transposition of the great arteries. 1 VSD is not only the most common acyanotic congenital heart defect but also the most prevalent of all CHDs, accounting for 20–30% of cases. 2 Induced pluripotent stem cells (iPSCs) have revolutionized the study of CHD by enabling the exploration of genetic mechanisms using patient-specific and clinically relevant cardiac cell types, including cardiomyocytes of ventricular fate. 3 Utilizing iPSCs generated from patients with ventricular defects provide a versatile platform to investigate disease mechanisms through directed differentiation into multiple cardiac-relevant cell types, including cardiomyocytes, endothelial cells, smooth muscle cells, and fibroblasts. In addition, these cells can be used to generate more physiologically relevant models, such as cardiac organoids and engineered heart microtissues. 4 Given the limited understanding of the genetic and cellular underpinnings of VSD, patient-specific iPSC-derived models offer significant potential for identifying mechanisms driving disease progression and serve as a valuable platforms for therapeutic testing. 5
A 21-year-old Emirati patient with a VSD provided the cell source for generating an iPSC line using Sendai virus (SV)-mediated reprogramming. Differentiation of cardiac cells from this VSD-specific iPSC line would offer clinically relevant models for studying CHD and identifying potential therapeutic targets. Therefore, this iPSC line serves as a valuable model for establishing an Emirati-specific iPSC repository, thereby enabling population-relevant disease modeling and drug discovery tailored to the genetic and clinical landscape of the Emirati population.
Methods
Peripheral blood mononuclear cells extraction, culture, and reprograming to iPSCs
A 7 mL of peripheral blood sample was collected in ethylenediaminetetraacetic acid-containing tubes from a 21-year-old female patient previously diagnosed with VSD and transported immediately to the laboratory for processing. Peripheral blood mononuclear cells (PBMCs) were extracted from fresh blood samples using a Histopaque density gradient and cryopreserved in aliquots of 1–2 × 106 cells/tube. For reprograming of isolated PBMCs, 1 × 106 cells were seeded into one well of a P12 well plate in PBMC expansion medium (Roswell Park Memorial Institute Medium [RPMI 1640] supplemented with 10% fetal bovine serum, 1× penicillin/streptomycin [P/S], 1× L-glutamine, 100 ng/mL of Fms-like tyrosine kinase 3, thrombopoietin, granulocyte colony-stimulating factor, stem cell factor, and 10 ng/mL of interleukin-3) and cultured for 4 days using standard cell culture conditions. PBMCs were reprogramed using the CytoTune™-iPS 2.0 Sendai Reprogramming Kit (Invitrogen, Carlsbad, CA, USA) following manufacturer’s protocol. Briefly, on day 4 of PBMC culture in expansion media, cells were infected with SV constructs at a multiplicity of infection of 5:5:3 (KOS:c-Myc:Klf4) for 24 h, followed by removal of SVs. Cells were further cultured for 2 days in PBMC expansion medium. On day 7, infected cells were collected and replated onto two wells of Geltrex-coated (150–200 ug/cm2) P6-well tissue culture (TC) plates and cultured using human induced pluripotent stem cell hESCs; human embryonic stem cells (hiPSC) derivation medium (mTesR plus media supplemented with 1× CloneR and 1× P/S) with media change performed every other day until small iPSC colonies were visible around day 6 post replating. At this point, the medium was switched to hiPSC culture medium (mTesR plus media supplemented with 1× P/S) and cells were cultured until desired colony size was reached. Individual clones of iPSCs were manually picked and transferred onto Geltrex coated (1:50 dilution) P6-well TC plates and expanded using hiPSC culture media under standard cell culture conditions.
H9 hESCs and hiPSCs culture and embryoid body formation
All media and chemicals used for cell culture were purchased from GIBCO unless otherwise stated. Both H9 hESCs (WiCell Institute, Madison, WI, USA) and PBMC-derived hiPSC clones were routinely cultured using feeder-free conditions on Geltrex (diluted 1:50)-coated 6-well TC plates in mTeSR Plus media supplemented with 1× P/S. ROCK inhibitor (10 μM) was added on the day of splitting to avoid apoptosis and was withdrawn the following day of cell culture. Cells were routinely tested for mycoplasma contamination using the Mycoplasma PCR Detection Kit (Abcam, Cambridge, UK) following manufacturer’s protocol. For embryoid body (EB) formation, cells were seeded onto low-attachment cell surfaces (Corning, Corning, NY, USA) without Geltrex coating and maintained under suspension conditions for 48 h. The resulting EBs were photographed using a phase-contrast microscope.
Alkaline phosphatase live staining, fixed-cell staining, and immunostaining
Alkaline phosphatase (AP) analysis was performed using the Alkaline Phosphatase Live Stain (500×) Kit from Invitrogen using manufacturer’s recommendations. AP staining was also performed on fixed cells using the Stemgent Alkaline Phosphatase Staining Kit II following the protocol provided by manufacturer. For immunostaining, cells were seeded onto P4 well plates (Thermo Scientific, Waltham, MA, USA, 176740) and incubated for at least 48 h before fixation to allow for cells to grow as clones. When ready, cells were washed 3× with phosphate-buffered saline (PBS) to remove debris and dead cells, followed by fixation with 4% paraformaldehyde for 20 min at room temperature (RT). Permeabilization was done using 0.3% Triton X-100 for 20 min at RT, followed by blocking with 1% bovine Ssrum albumin (BSA) for at least 45 min at RT. Cells were then incubated with primary antibodies (Supplementary Table S1) diluted in 1% BSA overnight at 4°C. Next day, samples were washed 3× with PBS and incubated with appropriate Alexa Flour® conjugated secondary antibodies (Supplementary Table S1) diluted in 1% BSA for 1 h at RT. Finally, samples were washed with PBS 3×, counterstained with 4′,6-diamidino-2-phenylindole (DAPI), and imaged using fluorescence microscope (Zeiss, Axiovert).
Sendai transgene clearance
Total RNA was extracted from cells using the MasterPure RNA Isolation Kit (Epicenter) according to the manufacturer’s protocol. An aliquot equal to 1 µg of total RNA was subjected to cDNA synthesis using FIREScript RT cDNA Synthesis Kit (Solis BioDyne, Tartu, Estonia). Reverse transcription polymerase chain reaction (RT-PCR) was performed for 30 cycles using Hot Start Taq 2× Master Mix (NEB) with primers specific for the SV transgene and GAPDH as the internal control. Primer sequences as well as expected polymerase chain reaction (PCR) product sizes are listed in Supplementary Table S2. Nuclease-free water was used as the negative control for PCR, while SV-infected PBMCs harvested 72 h post infections served as the positive control. All samples were loaded on a 2% agarose gel and imaged.
Real-time PCR analysis
Total RNA extraction and cDNA synthesis from cells were performed as described above. The cDNA samples were diluted 10-fold, and reverse transcription quantitative polymerase chain reaction (RT-qPCR) was performed using SYBR Green Real time Master Mix (Solis Biodyne, Tartu, Estonia) in a total reaction volume of 10 µL. Primer sequences used for the analysis are listed in Supplementary Table S3. 2−ΔΔCt method was used to calculate relative gene expression data and normalized to that of 18S rRNA that served as internal control for the assay.
Chromosome stability assay
Genomic DNA was isolated from UAEUi001-A cells using a gDNA isolation kit (Promega, Madison, WI, USA) and subjected to chromosome stability assay using the hPSC Genetic Analysis Kit (STEMCELL Technologies, Vancouver, BC, Canada) following manufacturer’s protocol. Briefly, an aliquot equivalent to 300 ng of gDNA (from UAEUi001-A cells) and control DNA (provided with the kit) was mixed with 5-FAM-labeled probes, and samples were analyzed using the StepOne Real-Time PCR system. Cycle threshold (CT) values obtained after PCR were loaded to Genetic Analysis Tool (available online at STEMCELL Technologies website), and the copy number of each tested locus was calculated using the 2−ΔΔCt method by normalizing it to Chr 4p. Plots were exported directly from the website.
In vitro differentiation into three germ layers
For neuroectoderm differentiation, the dual-SMAD inhibition protocol was used following established protocols from our laboratory as previously described.6–8 In Brief, cells growing under monolayer culture conditions and at 90% confluency were treated with LDN193189 (200 nM) and SB431542 (10 μM) on day 1 in 100% KSR media followed by the addition of XAV939 (2 μM) for an additional 2 days. After that, percentage of KSR medium was gradually reduced to 100% N2 media over a period of 8 days while supplementing with LDN193189 (200 nM), SB431542 (10 μM), and XAV939 (2 μM). At day 12 of differentiation protocol, cells were fixed for immunostaining using PAX6 as a neuroectoderm marker. For mesodermal fate, monolayer cells at 70% confluency were treated with 12 μM of CHIR99021 in culture media (RPMI-1640 supplemented with 1× B27, 1× P/S, and 1× L-glutamine) for 24 h, followed by fixation for immunostaining using Brachyury as a mesodermal stage marker. Endodermal differentiation was achieved using an established protocol from our laboratory.9,10 Briefly, monolayer cells at 70% confluency were treated with 3 μM of CHIR99021 in definitive endoderm (DE) media (RPMI-1640 supplemented with 1× B27, 1× P/S, 1× L-glutamine, and 100 ng/mL of Activin) for day 1 of differentiation. Next day, CHIR99021 was removed from culture media, and cells were maintained in DE medium alone supplemented with a reduced concentration of Activin (50 ng/mL) for additional 24 h. On day 3, the medium was switched to DE alone after completely removing Activin before fixing cells on day 4 for immunostaining using FOXA2 as endodermal marker.
Institutional review board statement
The study was approved by the Abu Dhabi Health Research and Technology Ethics Committee, Department of Health (DOH) (approval numbers DOH/CVDC/2022/726 and DOH/ADHRTC/2024/2840) (Table 1).
Resource Table for the UAEUi001-A iPSC Line
Experiment
Reprogramming of PBMCs and iPSC characterization
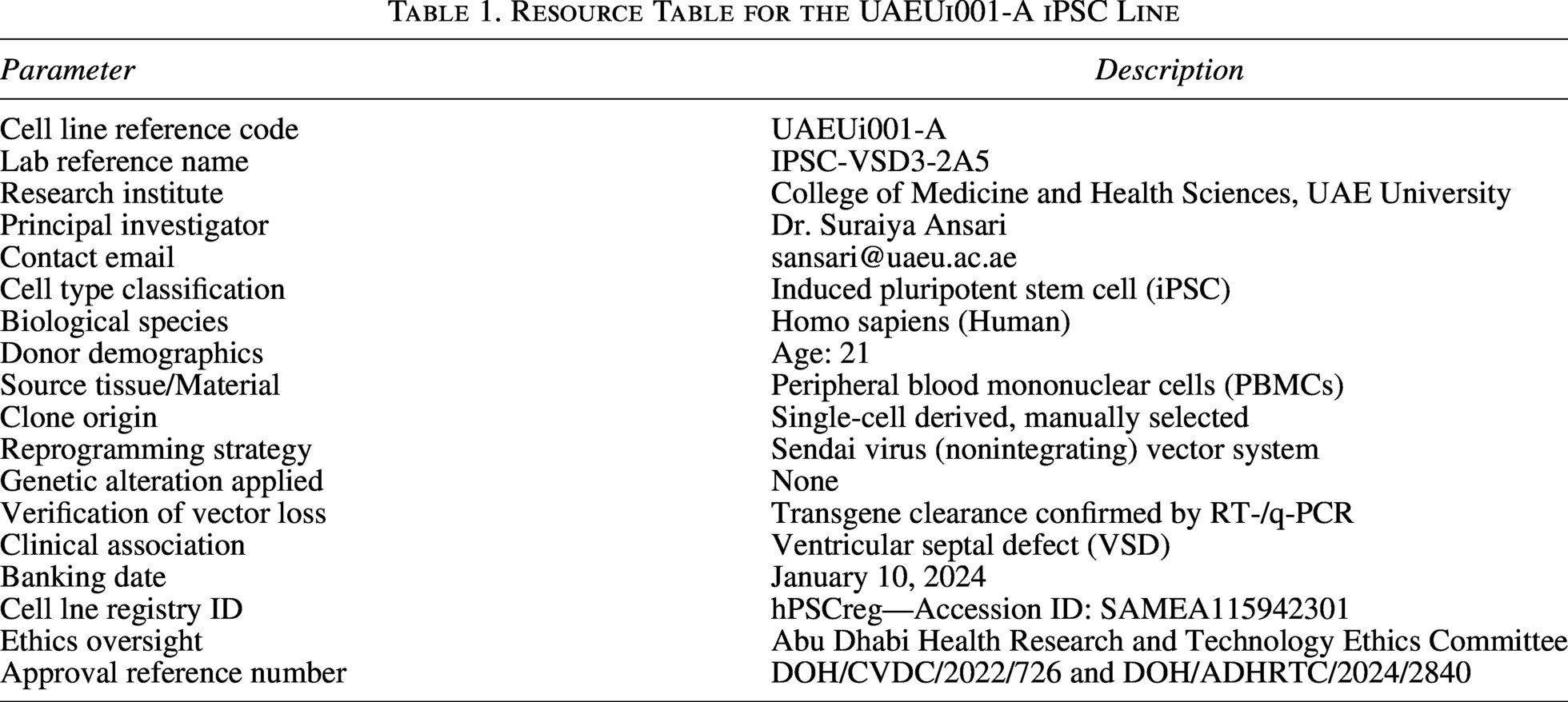
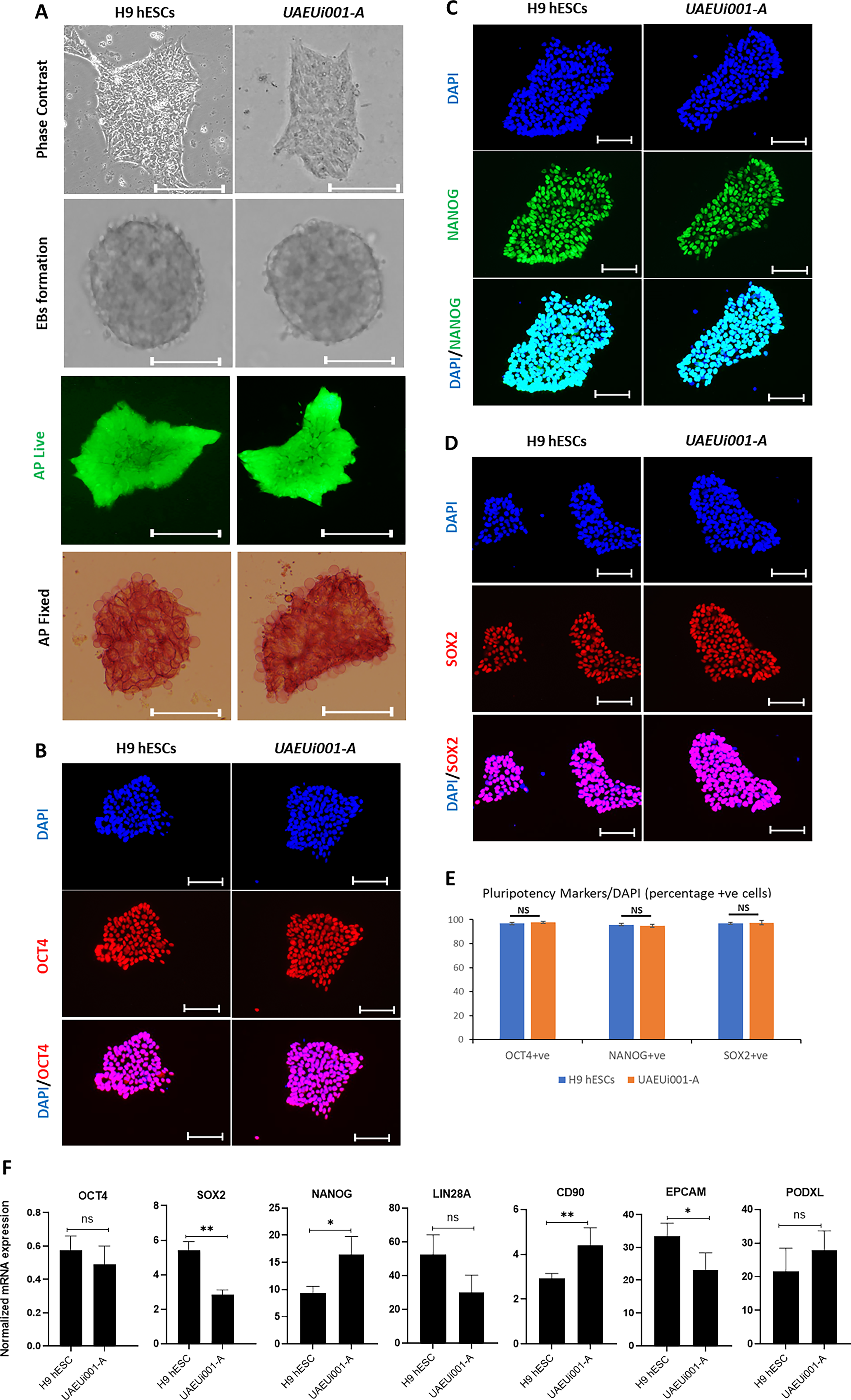
In this study, PBMCs obtained from a 21-year-old female VSD patient were reprogramed using CytoTune™2.0 Sendai reprogramming vectors, which ensure transgene-free expression of Oct3/4–SOX2, cMyc, and Klf4 with high efficiency to generate iPSC-like cells emerging as colonies. Of the 13 total colonies that emerged, five were manually picked and further cultured to observe morphologically for growth characteristics highly similar to that of hESCs. Among these, one clone (UAEUi001-A) showed remarkable colony morphology (at passage 14) similar to that of H9 hESCs (Fig. 1A), and hence selected for further processing and characterization. The iPSCs were expanded and cryopreserved, and different characteristics were compared with those of H9 hESCs to prove the potential of derived iPSCs. Both H9 hESCs as well as iPSCs were shown to be mycoplasma negative (Supplementary Fig. S1). PBMC-derived iPSCs successfully generated EBs under nonattachment cell culture conditions, and their differentiation potential was similar to that of H9 hESCs (Fig. 1A). The iPSC colonies showed abundant expression of AP, similar to that observed in H9 hESCs, as confirmed by both live-cell and fixed-cell staining analyses (Fig. 1A). Expression of the pluripotency markers OCT4, NANOG, and SOX2 was compared by using immunofluorescence analysis and was shown to be similar between iPSCs and H9 hESCs (Fig. 1B–D). Quantitative analysis of immunostaining results was expressed as the percentage of marker-positive cells, calculated by dividing the number of cells positive for specific markers by the total number of DAPI-stained nuclei. The quantification results were consistent with the initial observations, showing no significant differences between H9 and UAEUi001-A cells in the expression of pluripotency markers OCT4, NANOG, and SOX2, as determined by immunostaining (Fig. 1E). We also analyzed the mRNA expression of the core pluripotency markers, OCT4, SOX2, NANOG, and LIN28A, as well as the cell surface markers associated with pluripotent stem cells, CD90, EPCAM, and PODXL, by RT-qPCR in both H9 hESCs and UAEUi001-A iPSCs. All genes showed less than a twofold difference in expression levels between the two cell lines (normalized to 18S rRNA). Although the differences were statistically significant for SOX2, NANOG, CD90, and EPCAM, the direction of change varied: SOX2 and EPCAM were expressed at lower levels in UAEUi001-A cells, whereas NANOG and CD90 showed higher expression levels compared with H9 cells (Fig. 1F). Despite these differences, the overall expression profiles indicate that UAEUi001-A cells exhibit mRNA levels of pluripotency markers comparable to those of the H9 hESC line. Transgene clearance from iPSCs was confirmed by RT-PCR analysis using total RNA isolated from UAEUi001-A cells at passage 15. No PCR products were detected using primers specific for the Sendai virus transgenes (SeV, KOS, KLF4, and c-MYC), indicating the absence of residual transgene expression. The integrity of the cDNA was confirmed by amplification of GAPDH, which served as an internal control. No amplification was observed in the negative control reactions lacking cDNA, confirming the specificity of the assay. In contrast, abundant expression of all Sendai virus transgenes, as well as GAPDH, was observed in positive control samples showing that the PCR conditions were optimal for the detection of PCR products (Fig. 2A). Furthermore, a normal karyotype of UAEUi001-A cells was confirmed by performing a chromosome stability assay by using a qPCR-based loci analysis kit that utilizes probes for nine most commonly reported chromosomal abnormalities at chr 1q, chr4p, chr 8q, chr 10p, chr 12p, chr 17q, chr 18q, chr20q, and chr Xp. CT values obtained were analyzed by using an online tool provided by the manufacturer, and the report showed that all nine tested loci were normal compared with the control DNA provided with the kit (Fig. 2B).
iPSC characterization generated from whole PBMCs obtained from a 21-year-old female Emirati VSD patient.
The established iPSC line demonstrates the potential to differentiate into all three embryonic germ layers in vitro
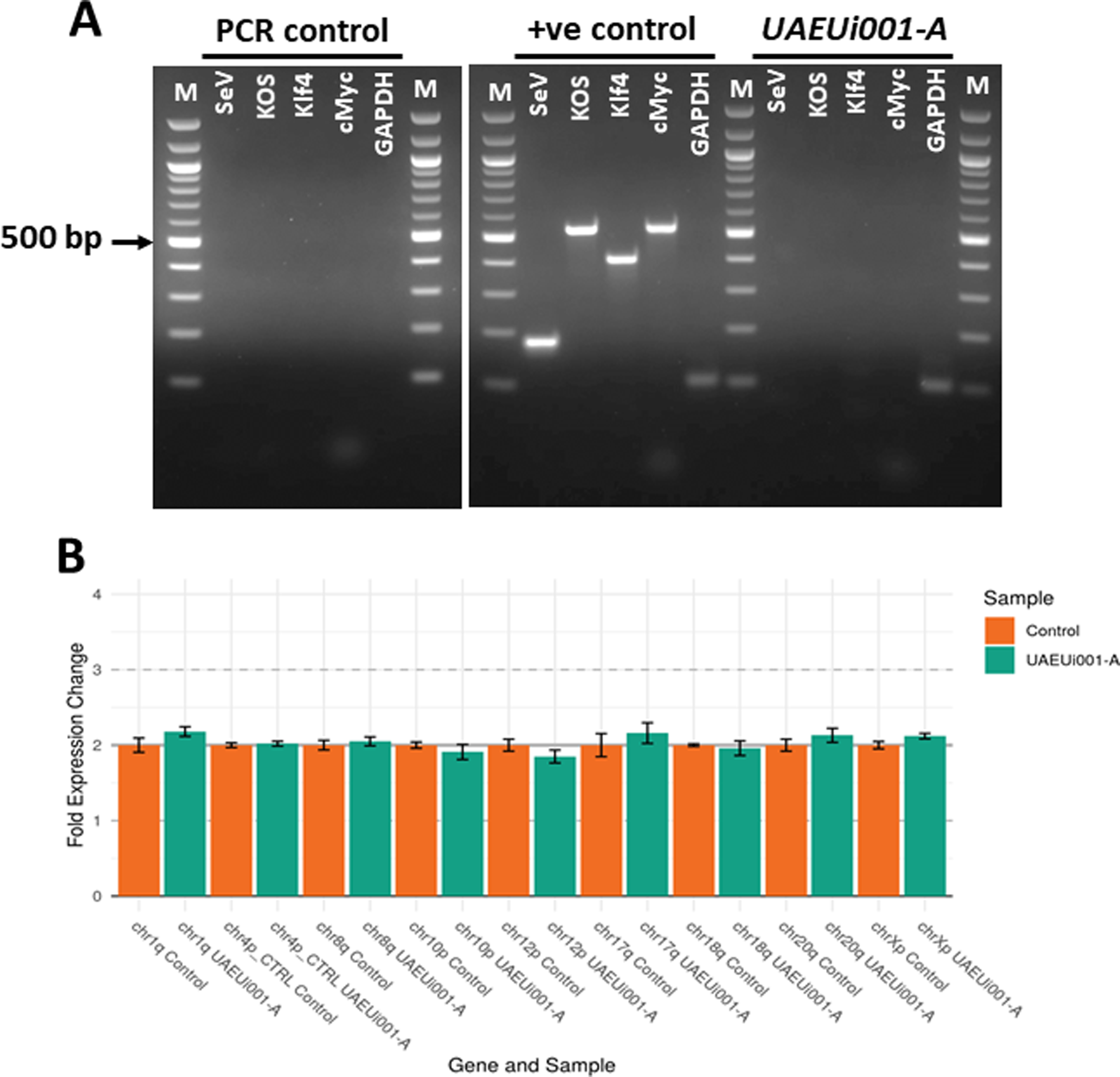
The differentiation potential of UAEUi001-A cells was studied toward all three germ layers under in vitro culture conditions using optimized protocols in our laboratory. Successful differentiation of the cells was demonstrated by immunostaining for PAX6, Brachyury, and FOXA2, marking ectoderm, mesoderm, and endoderm lineages, respectively (Fig. 3A–C), with differentiation potential comparable to that of H9 hESCs used as a control. Quantification of the immunostaining analysis was performed as described above and showed no significant differences between H9 and UAEUi001-A cells in terms of percentage of positive cells for each differentiation markers PAX6, Brachury, and FOXA2 (Fig. 3D). In summary, the presented iPSC line may be valuable for creating cell models of VSD, which would further facilitate disease modeling under in vitro culture conditions and advance drug discovery.
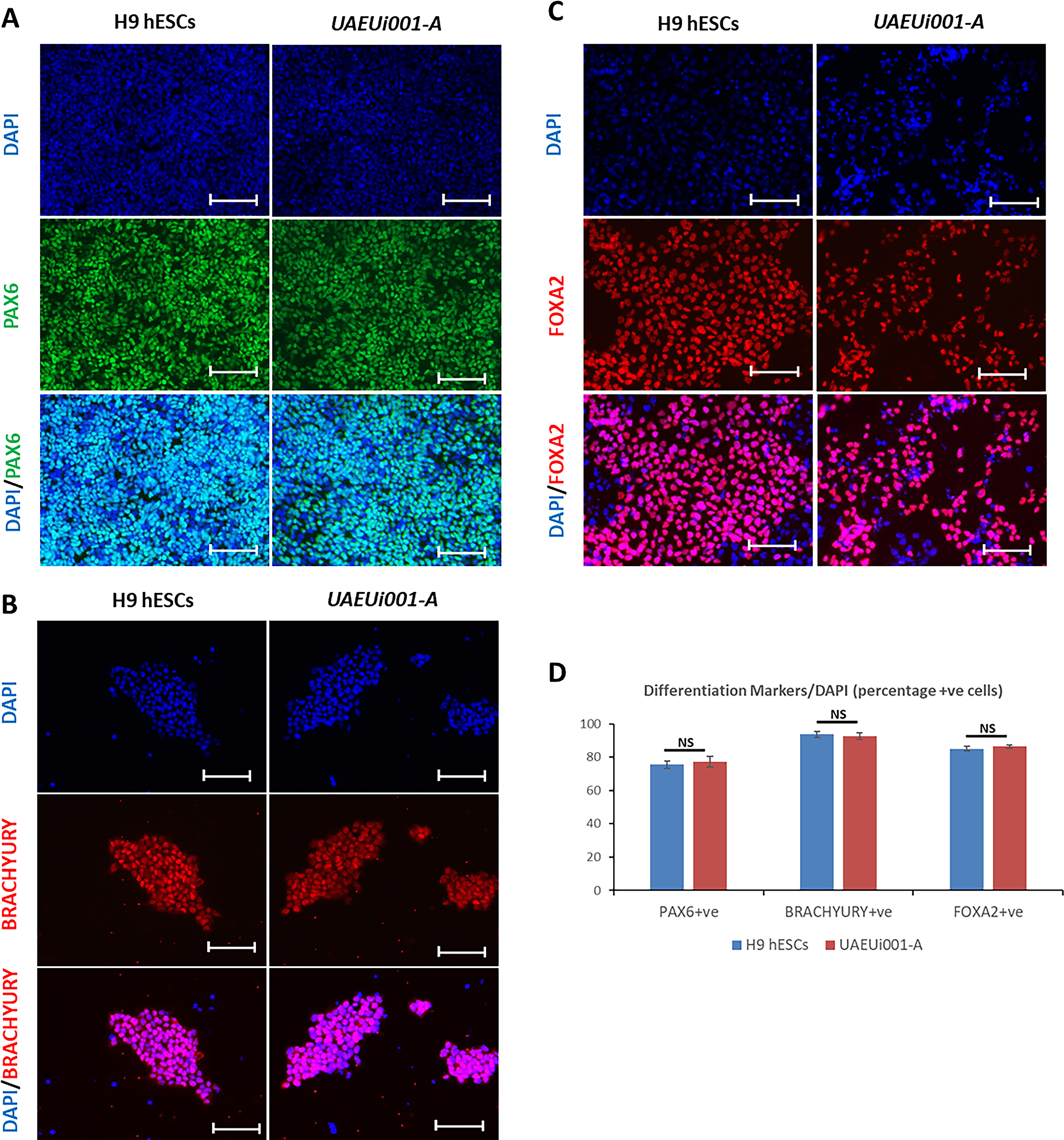
The iPSC line (UAEUi001-A) demonstrates the potential to differentiate into all three embryonic germ layers under in vitro conditions.
Discussion
We report here, for the first time, the generation of an iPSC line (UAEUi001-A) from peripheral blood mononuclear cells obtained from an Emirati patient with VSD. This line represents a foundational step toward establishing an Emirati-specific iPSC repository for patients with CHD (Table 1). As described above, the iPSC line was derived through single-cell clonal selection and underwent comprehensive characterization to confirm its pluripotency and evaluate any abnormalities that could compromise its functionality or safety. The line exhibited normal results in the targeted chromosome stability assay, supporting genomic stability and the absence of common reprogramming-associated chromosomal abnormalities, although additional cytogenetic analyses, such as conventional G-banded karyotyping, may provide a more comprehensive assessment of genomic integrity. Morphologically, the cells resembled H9 hESCs and showed robust alkaline phosphatase activity. Immunostaining further confirmed high expression of the core pluripotency markers NANOG, OCT4, and SOX2, with protein levels comparable to those observed in hESCs. Moreover, comparative RT-qPCR analysis of core pluripotency and cell surface markers revealed that UAEUi001-A iPSCs exhibited overall mRNA expression profiles comparable to those of H9 hESCs, with less than a twofold variation across all tested genes, supporting the pluripotent identity of the reprogrammed cell line. Functionally, the iPSC line demonstrated the capacity to differentiate into derivatives of all three germ layers, further affirming its pluripotent nature.
iPSC technology has emerged as a powerful platform for modeling CHDs, allowing patient-specific cells to be reprogrammed and differentiated into cardiomyocytes that retain the individual’s genetic background. Several studies have successfully used iPSC-derived cardiomyocytes to model a range of CHDs, including hypoplastic left heart syndrome, Tetralogy of Fallot, left ventricular noncompaction, and atrial and ventricular septal defects. 11 For instance, iPSC models have helped elucidate defects in sarcomere organization, calcium handling, and electrophysiological properties associated with genetic mutations linked to CHD.12,13 These in vitro systems have also enabled high-throughput drug screening and the identification of potential therapeutic compounds capable of reversing or mitigating disease phenotypes. 14 The creation of iPSC lines from genetically diverse populations remains limited, particularly in Middle Eastern cohorts, where CHD prevalence and severity may be influenced by unique genetic and consanguineous backgrounds. Therefore, the establishment of an Emirati iPSC repository, beginning with the current VSD-specific line, offers a significant opportunity to expand the understanding of CHD in this underrepresented population. Such efforts may lead to more precise, population-tailored therapeutic strategies and advance personalized medicine initiatives in cardiovascular research.
Authors’ Contributions
M.A.S. and T.G. did the experiments and generated data. E.M. collected blood samples and consents. M.E.M., B.S.E., E.H.A., and S.A.A. conceived the idea, designed the study, and received ethics approval. B.S.E., E.H.A., and S.A.A. acquired the funding and supervised the study. M.A.S. and S.A.A. wrote the original draft and all authors reviewed, edited, and approved the article.
Footnotes
Author Disclosure Statement
No competing financial interests exist.
Funding Information
This work was supported by research grants from the