
Abstract
Objective
This study investigates how the inflammatory response of ATDC5 murine chondrogenic cells influences the activity of C6 (rat) and GL261 (mouse) glial cell lines. Prior research suggested nitric oxide (NO) involvement in cartilage-immune crosstalk. The current study explores whether NO, produced by inflamed chondrocytes, mediates signaling between chondrocytes and glial cells.
Design
Pre-challenged ATDC5 cells with 250 ng/ml of lipopolysaccharide (LPS) were cocultured with GL261 or C6 glioma cells for 24 h with a transwell culture system. Cell viability was assessed using MTT assay. Gene and protein expression were evaluated by qRT-PCR and WB, respectively.
Results
Real-time reverse transcription-polymerase chain reaction (RT-qPCR) indicated statistically significant upregulation of LCN2, IL-6, TNF-α, IL-1β, and GFAP in glial cells following 24-h coculture with challenged ATDC5 cells. Suppression of LPS-induced NO production by aminoguanidine decreased LPS-mediated LCN2 and IL-6 expression in glioma cells. We identified also the involvement of the ERK1/2 and AKT signaling pathways in the glial neuroinflammatory response.
Conclusions
This study demonstrates, for the first time, that NO produced by inflamed murine chondrocytes mediated pro-inflammatory responses in glial cells via ERK1/2 and AKT signaling, highlighting a potential mechanism linking cartilage NO to neuroinflammation and chronic pain in osteoarthritis.
Introduction
Accumulating evidence illustrates the intricate interplay among systemic responses, the immune system, and the brain that requires precise regulation for optimal biological functions.1 -3 Aberrant regulation of this dynamic balance, due to various conditions including inflammation, may disrupt the metabolic homeostasis, leading to adverse metabolic and inflammatory consequences.4 -6 Rheumatic diseases, stemming from autoimmune aggression on connective tissues, can influence brain functions leading to secondary neurological symptoms, either directly due to the immune system’s assault or indirectly through the chronic inflammatory processes and deterioration of supportive structures. 7 Peripheral injuries activate the glial component in both peripheral and central cellular circuitry, releasing stressors that contribute to neuroinflammation and adverse effects such as neuropathic pain.8,9
Primary osteoarthritis (OA), the prevailing type of arthritis, stands as a prominent cause of long-term disability and chronic pain. Osteoarthritis is now recognized as a “whole joint” disease involving not only cartilage but also bone and other joint structures. Knee and hip OA are the most common forms of OA. A meta-analysis reported that 19% to 43% of adults aged 40 years or older with asymptomatic, uninjured knees displayed OA features in magnetic resonance imaging. 10 In addition, a recent meta-analysis estimated the prevalence of hip OA at 8.55%. 11 Disrupted cartilage homeostasis in OA results in persistent low-grade inflammation and molecular alterations in chondrocytes, triggering catabolism and immune responses.12 -15 The persistence of this inflammatory state, coupled with cartilage degeneration and changes in surrounding structures, resulted in joint pain, articular swelling, and functional limitations experienced by individuals with OA. 13
In the OA cartilage, inflammatory mediators derived from chondrocytes, such as pro-inflammatory cytokines, prostaglandins, and nitric oxide (NO), crucially drive OA pathogenesis, exacerbating disease progression, and chronic pain.16,17 The role of NO and its redox derivatives seem to vary between normal and pathophysiological conditions within joints; however, excessive production in OA contributes to chondrocyte senescence, cartilage breakdown, and joint damage. 18 NO has been identified as a primarily catabolic factor in OA. It triggers the expression of pro-inflammatory cytokines as well as matrix metalloproteinases (MMPs), interferes with the synthesis of proteoglycans and collagen, and induces chondrocyte apoptosis.17,18 The inducible isoform, NO synthase type II (NOS2), exhibits prolonged expression upon activation by various factors, such as IL-1β and lipopolysaccharide (LPS). NO, with its capacity to diffuse locally, influences cellular signaling within the local microenvironment.19,20
Pain associated with degenerative cartilage in OA involves peripheral sensitization mechanisms, where nerve terminals undergo sensitization due to the release of inflammatory mediators such as nerve growth factor (NGF), bradykinin, prostaglandin E2 (PGE2), and NO in OA cartilage.21,22 Particularly, NO plays a crucial role as a signaling molecule in the mechanism of long-term potentiation (LTP), contributing to hyperalgesia. 23 This in turn results in alterations of joint nociceptors and subsequent activation of nociceptive processing within the spinal cord (dorsal horn), brain stem, and other cortical regions. It has been recently reported that OA, induced by surgical destabilization of murine meniscus (DMM) in mice, leads to late-stage dorsal horn microgliosis, 24 a condition that is thought to contribute to synaptic transmission in the dorsal horn and thereby promotes chronic pain. NO dependent-LTP of the nociceptive pathways, behind the long-term central sensitization maintained by peripheral input, has an impact on the central amplification and perpetuation of chronic pain perception. 22 Alongside the neuronal processes and central and peripheral sensitization, glial cells play crucial role as non-neuronal cells participating in the modulation of pain processing.25,26 In the neuropathic pain state, the spinal glial cells become activated and can sensitize the nociceptive signal process by releasing inflammatory cytokines such as IL-1, IL-6, and TNF, 27 which disrupt gap junction communication and open hemichannels, leading to increased pain sensitivity. Similarly, chondrocytes respond to endotoxins, like LPS, by altering gap junction communication, which disrupts cellular interactions and contributes to cartilage degradation. Both processes involve changes in connexin expression and calcium signaling, impacting pain and inflammation.
The aim of the present study is to analyze how the inflammatory response, elicited by LPS, in ATDC5 murine chondrogenic cells might affect the response of C6 (rat) and GL261 (mouse) glial cell lines via NO. To this regard, we have previously demonstrated the role of NO produced by inflamed J744 murine macrophages cells in activating LCN2 expression in ATDC5 murine chondrocytes, suggesting a potential role of NO in immunological-cartilage crosstalk. 28 Based on these findings, we hypothesize that inflammation-induced NO overproduced by the ATDC5 murine chondrocytes 29 might similarly trigger a signaling response that upregulates pro-inflammatory cytokines and astrocytic markers in C6 rat and GL261 mouse glial cells. To model this interaction, an in vitro indirect coculturing system, commonly referred to as transwell system, was used.
Materials and Methods
Reagents
Dulbecco’s modified Eagle’s medium (DMEM)/Ham’s F-12 medium (DMEM; BE17-605E), DMEM (Ref,41965-039), DMEM low glucose (Ref,12-707F),
Cell Culture Conditions and Treatments
Murine chondrogenic cell line ATDC5 cells (RIKEN Cell Bank, Tsukuba, Japan) were cultured in a monolayer as previously described. 29 C6 rat glioblastoma (ATTC, USA) were cultured in low-glucose DMEM supplemented with 2.5% heat-inactivated FBS, 15% heat-inactivated horse serum (Hyclone, Logan, UT), 2 mm glutamine, 100 μg/ml streptomycin, and 100 units/ml penicillin. GL261 murine cells were cultured in high-glucose DMEM supplemented with 10% heat-inactivated FBS, 2 mm glutamine, 100 μg/ml streptomycin, and 100 units/ml penicillin. All cells were incubated in a humidified incubator with 95% air and 5% CO2 at 37°C. For the coculture experiments, ATDC5 cells were seeded (3 × 105 cells/well) in plastic cell inserts (Corning, NY, USA, Ref, 353102) and treated with LPS 250 ng/ml for 24 h. The culture media of the treated ATDC5 cells (S1) was then recovered to be tested for nitrite accumulation. The cell inserts containing the challenged chondrocytes were washed thoroughly 2 times with fresh serum-free medium before initiating the coculture.
Coculture Conditions
C6 and GL261 derived from rat and murine glioblastoma tumor cell line, respectively. Both cell lines separately cocultured with ATDC5 chondrocyte cell lines using cell culture insert with 1.0 µm pore size (Corning, USA, Ref#353102). The chosen pore size only enables cellular communication via free soluble signaling factors (cytokines, growth factors) diffusing through the membrane while keeping the 2 cell types physically separated (Fig. 1). ATDC5 were seeded in the cell insert, while glia cells were cultured at the bottom of the plate. Each experiment was run in triplicate. After cell synchronization (seeding and harvesting), chondrocyte cells challenged with different stimuli were cocultured with glial cells at 37°C with 5% CO2.
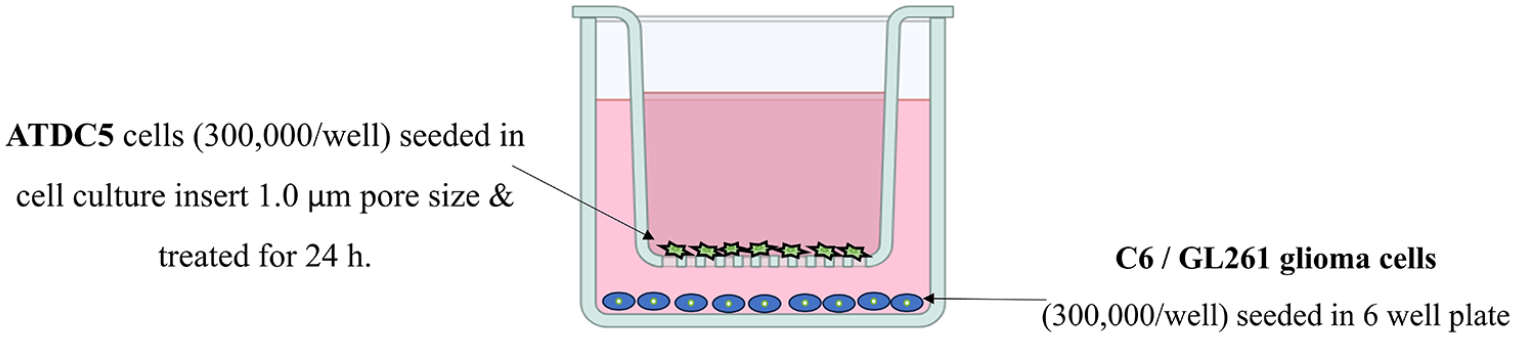
Co-culture layout represents a single well framework for a 6-well plate. ATDC5 were seeded in the cell insert, while the glial cells were cultured at the bottom in the 6-well plate. Cell inserts were of 1.0 µm pore size to avoid cell migration. Both medium well and the medium insert volumes were 1 ml. ATDC5: Murine chondrocyte cells. C6 glial cells. GL261 is a murine astrocyte-derived glioma cell.
Cell Viability (MTT)
The ATDC5 cell viability in various culture media was assessed using the methyl-thiazolyl-tetrazolium (MTT) assay. In 96-well plates, 8000 ATDC5 cells were seeded in each well and subsequently incubated in serum-free medium derived from either GL261 or C6 glioma cells. Viability was determined according to previously described protocols. 28
Nitrite Assay
Accumulation of nitrite in the culture media was quantified by Griess reaction as previously described. 28
RNA Extraction and Real-Time Reverse Transcription-Polymerase Chain Reaction
Total RNA was extracted and reverse-transcription was carried on as previously described.
30
A SYBR-green-based quantitative real-time reverse transcription-polymerase chain reaction (RT-qPCR) was conducted in Agilent AriaMX thermal cycler (Agilent Technologies, USA) following the standard protocol for RT2 SYBR Green qPCR Mastermix (Qiagen, Germany). Primers were designed in-house and synthesized by Invitrogen, UK (
Protein Extraction and Western Blot Analysis
Glioma cell total protein extraction and quantification were performed following the previously described protocols. 30 Sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) and blotting procedure were performed for protein separation as previously described. 30 Immunoblots were incubated with the appropriate antibodies against Phospho-p44/p42 mitogen-activated protein kinase (MAPK) (Erk1/2); MAPK 1/2 (Erk1/2) diluted 1:1000 (D13.14.4E; Cell Signaling, MA, USA), ERK1/2 (06-182), phospho-AKT diluted 1:1000 form Cell Signaling (Danvers, MA, USA). Blots underwent 4 washes with TBS-T (10 min each), followed by a 1-h incubation with anti-rabbit secondary antibody at a 1:5000 dilution (from GE Healthcare, IL, USA), and visualization using the Immobilon Western Detection kit (Millipore, MA, USA). Immunoreactive protein bands were detected using ChemiDoc MP Imaging System and analyzed with Image Lab 6.0.1 software. The protein expression levels of the kinases were normalized against GAPDH. Equal loading of sample in each membrane was verified by incubation with anti-GAPDH diluted 1:1000 (Sigma-Aldrich, USA). The bands in each gel figure originated from the same gel, although they may have been spliced.
Statistical Analysis
Statistical analyses were conducted using GraphPad Prism 9.3.1 software (GraphPad Software, CA, USA). The data are expressed as mean ± standard deviation (SD) from a minimum of 3 independent experiments. Group comparisons were assessed using 1-way analysis of variance (ANOVA), and statistical significance was estimated with Bonferroni’s multiple comparison test as the post hoc analysis. A P value < 0.05 was considered statistically significant.
Results
ATDC5 Cells Viability in Glioma Culture Media
As the experimental design included coculturing ATDC5 cells with C6 or GL261 glioma cells (
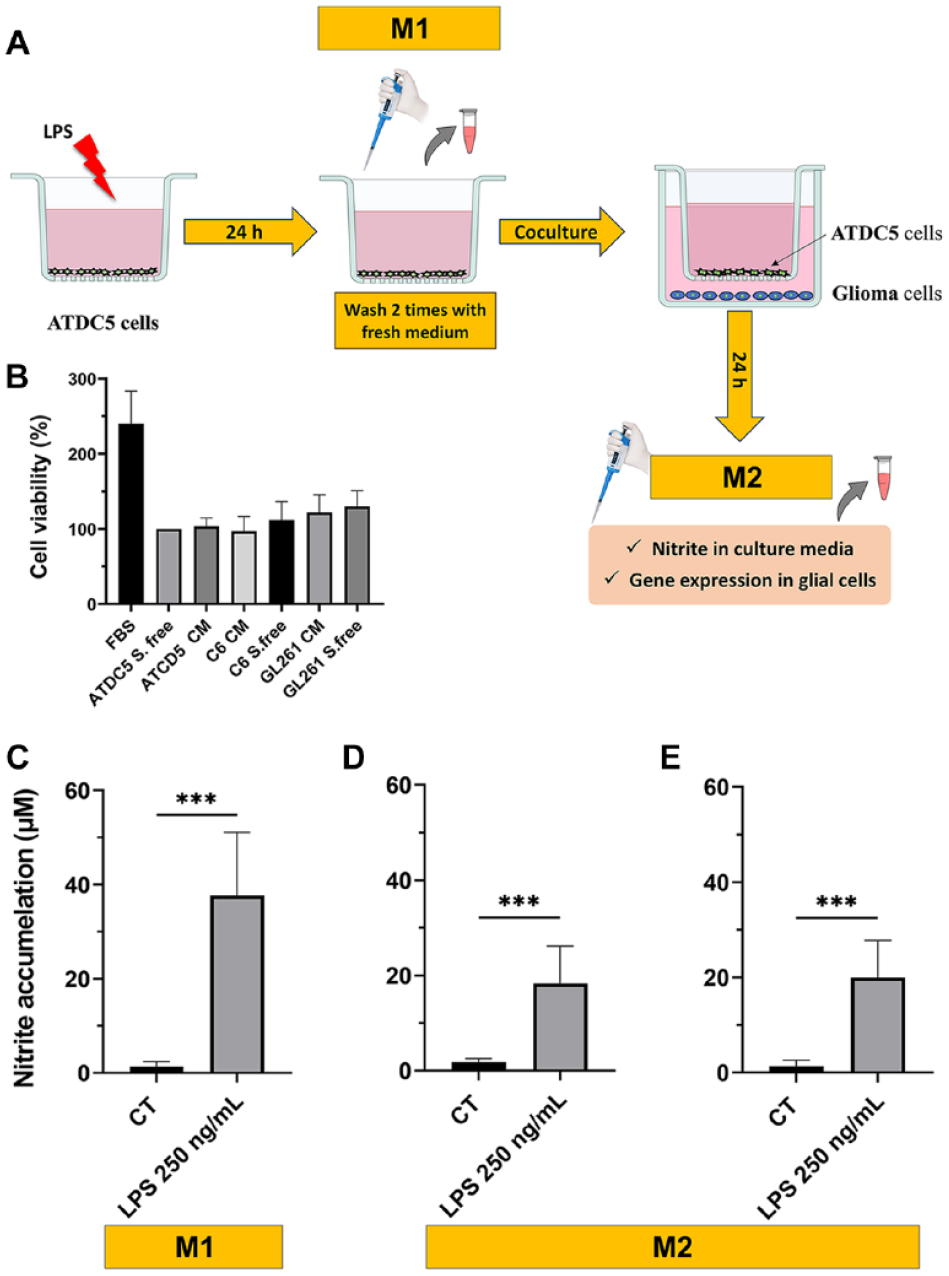
Schematic representation of the coculture experimental design (
The Challenged ATDC5 Cells Induce Inflammation in Glioma Cells
NO activation in ATDC5, as a result of a challenge with LPS (250 ng/ml), was assessed by measuring nitrite accumulation. Nitrites, stable metabolites of NO, are produced primarily by NO synthase type II in chondrocytes in response to pro-inflammatory stimuli. As shown in
Coculturing the glioma cells for 24 h with LPS-challenged ATDC5 upregulated the mRNA expression of pro-inflammatory cytokines such as lipocalin-2 (Lcn2), IL6, TNF-α, IL-1β, and also the astrocytic marker glial fibrillary acidic protein (GFAP) both in C6 rat and GL261 mouse glial cells (
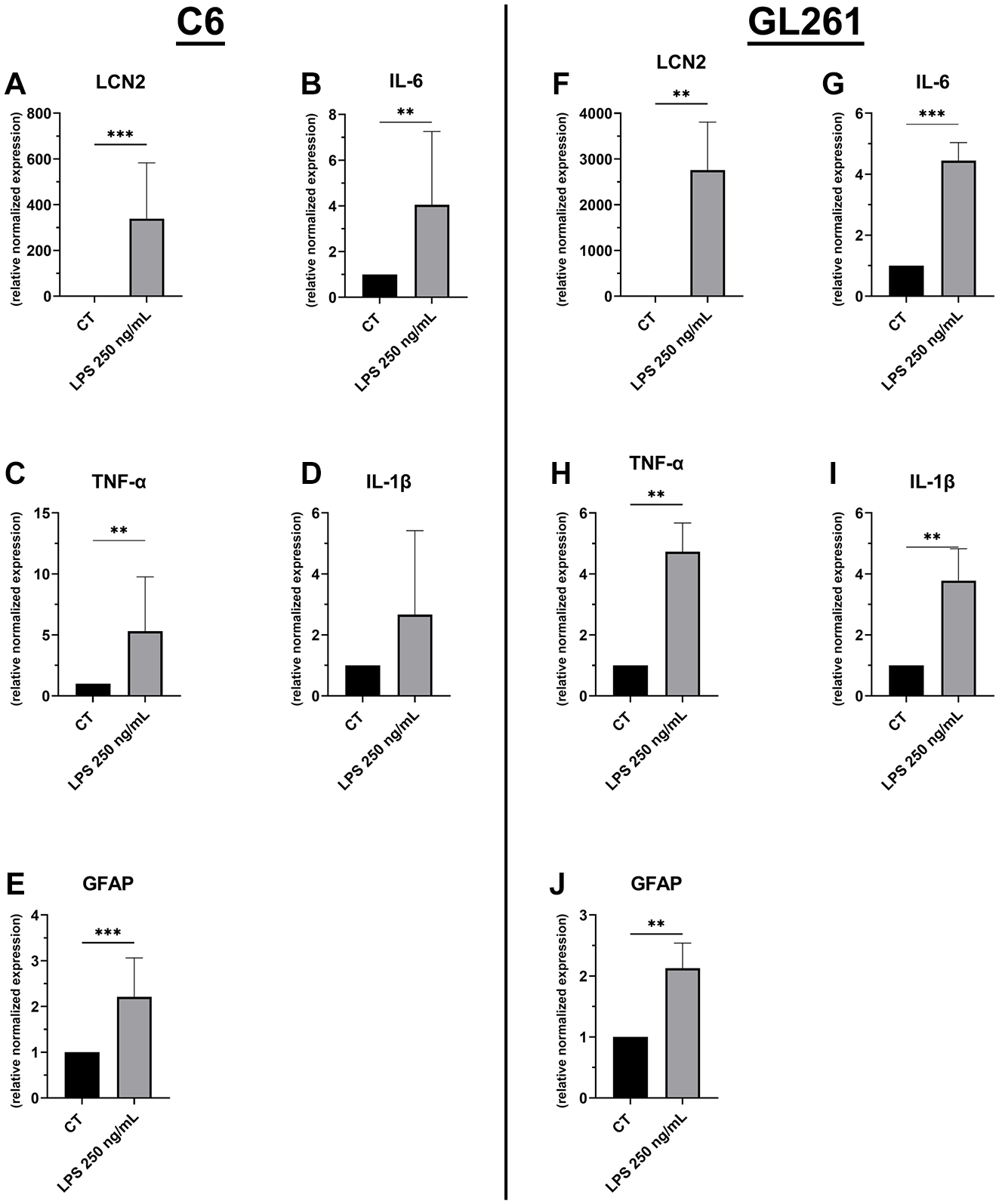
mRNA expression levels of pro-inflammatory cytokines in C6 rat glioma cells (
Inhibition of NO Synthase in ATDC5 Cells Reduces Neuroinflammation
To evaluate the involvement of NO produced by ATDC5 cells in upregulation of inflammatory factors in glial cells, ATDC5 cells were pre-treated for 1 h with the specific NOS2 inhibitor AMG before LPS stimulation. As shown in
Figure 4A
, inhibiting NOS2 reduced almost completely the production of NO from ATDC5 cells. The inhibition of NOS2 and the consequent statistically significant reduction of NO accumulation from the ATDC5 cells resulted in a statistically significant decreasing the mRNA expression of LCN2, IL6, and GFAP in C6 rat glioma cells (
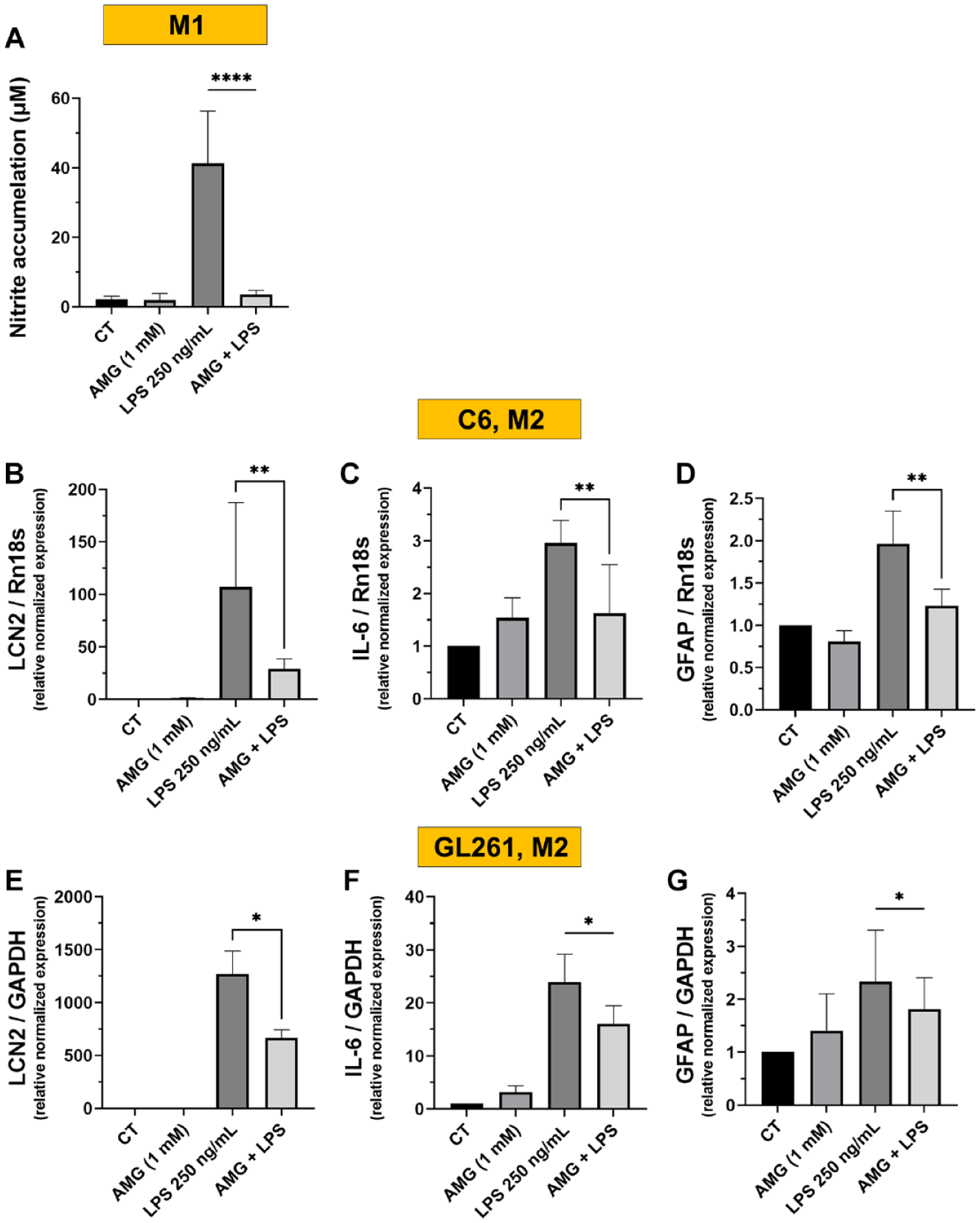
Nitric oxide (NO) accumulation at 24 h of incubating the ATDC5 cells with LPS 250 ng/ml with or without AMG pretreatment, at M1 phase (
Signaling Pathways Involved in the Peripheral-Central Inflammatory Crosstalking
To further investigate the paracrine signaling mechanism by which the inflammatory signals from challenged ATDC5 cells caused induction of pro-inflammatory effects in glial cells, we analyzed by western blot analysis, extracellular signal-regulated kinase 1/2 (ERK1/2 MAPK), and Akt intracellular signaling cascades via a time course experiment upon coculturing both pre-challenged chondrocytes (LPS 250 ng/ml for 24 h) with murine GL261 glioma cells for 2 h time course. In this study, an increase in the phosphorylation levels of ERK1/2 and AKT proteins was observed in glial cells following coculturing with LPS pre-challenged ATDC5 in a time-dependent manner (
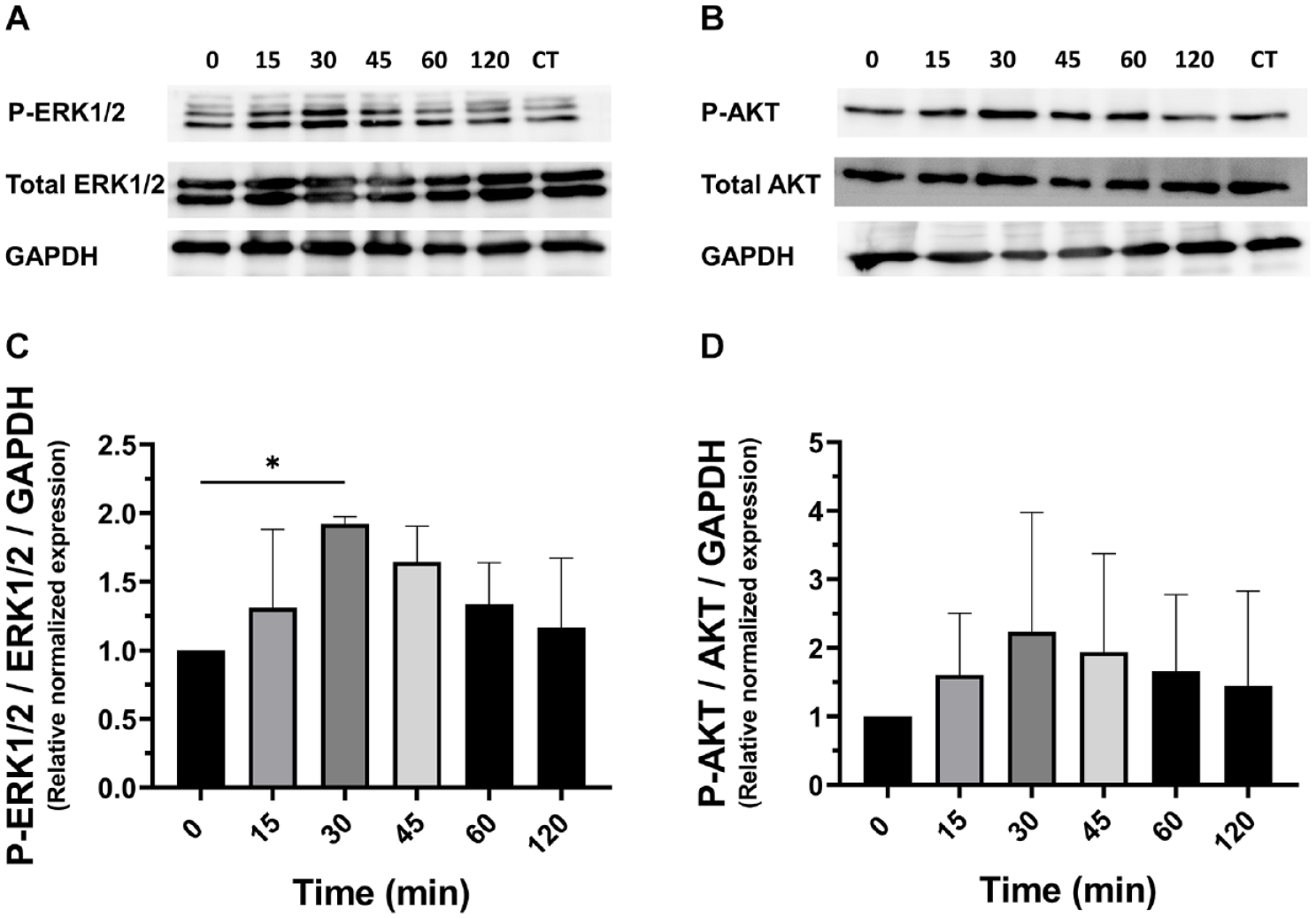
Protein expression of ERK1/2, p-ERK1/2 (
Discussion
Our study demonstrates that inflammatory stress in chondrocytes, induced by TLR4 activation with LPS, significantly influences glial cell behavior. We established a co-culture model revealing that NO produced by inflamed ATDC5 upregulates pro-inflammatory cytokines in C6 and GL261 glial cells, including IL-6, TNF-α, IL-1β, and LCN2. In addition, we identified that NO mediates inflammatory responses through the regulation of key signaling pathways, such as ERK1/2 and AKT, in glial cells. These findings suggest potential role of chondrocyte-derived NO in driving inflammation and chronic pain through glial cell activation.
Chronic pain is one of the main harmful symptoms associated with OA. Signals and mediators from OA joints are likely involved in the induction and perpetuation of pain. During the progression of OA, nociceptors can be sensitized by locally produced mediators. On the other hand, increased nociceptive inputs from the affected joints raise the activity and excitability in the spinal cord. Thus, OA pain may be caused by a remarkable excitability in pain pathways within both the peripheral and central nervous system.
31
In our study, we established a co-culture model to simulate the interplay between cartilage cells (chondrocytes) under inflammatory stress, elicited by the TLR4 activator LPS, and the subsequent abnormal response by glial cells. Our emphasis was on evaluating and understanding the communication dynamics. Specifically, we have focused our investigation on examining the role of NO released from chondrocytes in influencing the glial cells. Stimulation of TLR4 by LPS cascade in chondrocyte cells is a well-known stimulus that upregulates the expression of inducible nitric oxide synthase (NOS2),
29
the enzyme responsible for producing NO, promoting inflammatory cascade within joint environment and cartilage degeneration.18,28 We have demonstrated that NO accumulation driven by the activation of TLR4 in chondrocytes induced subsequent glial activation and upregulated several pro-inflammatory cytokines and mediators such as IL-6, TNF-α, IL1β, and LCN2. After the coculturing period, NO production persisted even after the removal of inflammatory stressors. The concentration of NO measured in the coculture medium appears to be exclusively released by chondrocyte cells, as none of glial cell lines produced nitrites (
One of the crucial inflammatory mediators upregulated in the glial cells during pathological inflammatory conditions is LCN2, a master modulator of inflammation both in the brain and periphery.36,37 It exerts catabolic activities in the arthritic joint. 38 In CNS, the basal level of LCN2 in the normal brain is from low to undetectable amounts. In certain conditions, elevated LCN2 levels in glial cells may have a neuroprotective role, promoting neuronal survival by modulating iron metabolism, oxidative stress, and apoptosis. 39 However, excessive and prolonged elevation might have neurotoxic effects that lead to astrocytes and microglial activation, thereby neuronal damage and dysfunction. 40 Moreover, increased levels of LCN2 within glial cells might influence the release and production of other cytokines. 39 Interestingly, NO produced by chondrocyte cells seems to have a regulatory role in the production of LCN2 and IL-6 in the glial cells. Notably, the inhibition of NOS2 by pretreatment of chondrocytes with AMG, the specific NOS2 inhibitor, caused a subsequent statistically significant decrease in LCN2, Il-6, and GFAP expression in both glial cell lines. These data indicate that NO released from stimulated chondrocyte cells can mediate the inflammatory response in glial cells. These results are consistent with our previous findings where NO mediated the macrophage-chondrocyte crosstalking and consequently modulated LCN2 expression in ATDC5 chondrocytes. 28
We went more in depth in the analysis of the molecular mechanisms potentially involved in the chondrocyte/glial crosstalk. Our results also suggested the potential involvement of ERK1/2 as well as AKT signaling pathways in the observed cellular response and crosstalk. Activation of the ERK1/2 is implicated in inducing the expression of NOS2 and COX2 as well as upregulating pro-inflammatory cytokines such as IL-6 and TNF-α. 41 ERK1/2 cascade is a potent trigger of neuroinflammation and neuropathic pain,41,42 and MAPK signaling activation in glial cells, astrocytes, microglia involving ERK is associated with chronic pain sensitization. 32 Microglial ERK is activated in the early phase of nerve injury, while ERK phosphorylation in the spinal astrocytes comes later. 32 In a neuropathic pain model, inhibiting the ERK activation by intrathecal administration of MAP kinase and ERK kinase inhibitor improved the painful reaction. 43
In addition, phosphatidylinositol 3-kinase/protein kinase B (PI3K/AKT) signaling pathway has been proved to have a pivotal role in the development and progression of CNS disease.44,45 Elevated ERK1/2 and AKT phosphorylation are also well known among the crucial pathways contributing to joint inflammation in OA.46,47 Therefore, the regulation of these pathways presents a dual opportunity: not only as a promising avenue for developing molecular targets in rheumatic disease treatment but also as an additional potential strategy for alleviating neurological comorbidities. The findings of this study highlight that the activation of these pathways in glial cells, triggered by inflammatory signals from chondrocytes, may establish a connection between peripheral inflammation and its consequences on activating other neural cells. Indeed, the impact of peripheral inflammatory insults, such as injuries or systemic and metabolic diseases, on glial activation and subsequent nervous system-related pathologies is supported by numerous studies.1,6,48,49 In response to inflammatory stimuli from the periphery, activated glial cells often exhibit increased expression of glial markers and pro-inflammatory cytokines contributing to a state of neuroinflammation within the CNS.8,50,51 Understanding the nuanced crosstalking between peripheral tissues and the nervous system is crucial for a thorough comprehension of both physiological and pathological processes.
Our findings established a valuable coculture model elucidating cell-to-cell communication and interaction between chondrocytes and glial cells, particularly under specific inflammatory conditions mediated by NO. We do not have precise indications about the potential mechanism/s involved in the chondrocytes-glial cells crosstalk. The fact that the NO, produced by chondrocytes, is able to increase pleiotropic cytokines such IL-6, TNF-α, IL-1β, further complicates this task. Thus, for the time being, we can speculate several possibilities but only for similarities with other cell types. In addition, we cannot rule out the role of other cell types potentially involved in the crosstalk mechanism/s. For instance, the inflammatory cytokines and mediators like NO can potentially alter glial hemichannels and gap junction connexins.52 -54 This can promote the propagation of calcium waves and enhance glutamate release in extracellular space of glial cells, 55 activating glial cells and creating a feedback loop that exacerbates neuroinflammation. 56 This crosstalk suggests a potential interplay and sheds light on the signaling molecules and mechanisms linking the peripheral inflammatory responses with activity of CNS cells. 57
It is important to acknowledge certain limitations of our study. First, we have to bear in mind that our results were obtained in an in vitro system, with established cell lines, which clearly simplifies the complex interactions that may occur in vivo. In addition, our results were obtained with cell lines, due to the scarcity of human primary cells, both from joint and glia. Thus, further studies with OA animal models are needed to validate our findings in a more pathophysiological context. However, our data are likely in line with published literature describing spinal microglial activation in a murine surgical model of knee OA.24,31 Another limitation is the use of the MTT assay, which primarily assesses mitochondrial activity linked to apoptosis. Since OA pathology also involves necrosis and chondroptosis, these cell death mechanisms are not fully assessed by this method. Nevertheless, we do think that our novel observations might contribute novel insights into the understanding of molecular mechanisms by which chronic pain is produced in OA.
Supplemental Material
sj-docx-1-car-10.1177_19476035241292323 – Supplemental material for Impact of Chondrocyte Inflammation on Glial Cell Activation: The Mediating Role of Nitric Oxide
Supplemental material, sj-docx-1-car-10.1177_19476035241292323 for Impact of Chondrocyte Inflammation on Glial Cell Activation: The Mediating Role of Nitric Oxide by Mariam Farrag, Alfonso Cordero-Barreal, Djedjiga Ait Eldjoudi, María Varela-García, Carlos Torrijos Pulpón, Francisca Lago, Amina Essawy, Ahmed Soffar, Jesus Pino, Yousof Farrag and Oreste Gualillo in CARTILAGE
Footnotes
Acknowledgment and Funding
O.G. and F.L. hold positions as Staff Personnel (I3SNS Stable Researcher) at Xunta de Galicia (Servizo Galego de Saude [SERGAS]), through a research staff contract (ISCIII)/SERGAS. O.G. is a member of the RICORS Program, RD21/0002/0025, through ISCIII and FEDER European Union and European Commission. F.L. is a member of Centro de Investigación Biomédica en Red de Enfermedades Cardiovasculares (CIBERCV). The research of O.G. and J.P. (PI23/00289, and PI20/00902) and F.L. (PI21/01145 and CB16/11/00226) is financially supported by ISCIII and FEDER European Union and European Commission. O.G. is a member of the COST action CA21110 (Building a European Network on Osteoarthritis [NETWOARK]), funded by the European Union and European Commission under the European Cooperation in science and technology program (COST). O.G. and J.P. received grants from GEER (Sociedad Española de Columna vertebral) becas 2020 and 2023. O.G. is the beneficiary of a grant funded by Xunta de Galicia, Consellería de Educación, Universidade e Formación Profesional and Consellería de Economía, Emprego e Industria (GAIN) (GPC IN607B2022/02. Y.F. is a “Sara Borrell” researcher funded by ISCIII and FEDER European Union and European Commission (CD21/00042). M.V.G. is beneficiary of a contract of the Rio Hortega program of the ISCIII (CM22/00070) funded by ISCIII and FEDER European Union and European Commission. C.T.P. is beneficiary of a predoctoral grant funded by Fundación IDIS and Caixa Rural.
Author Contributions
Drs. Oreste Gualillo, Yousof Farrag, and Ms. Mariam Farrag have full access to all the data in the study and take responsibility for the integrity of the data and the accuracy of the data analysis. Conception and design: Oreste Gualillo, Yousof Farrag, and Ms. Mariam Farrag. Acquisition, analysis, or interpretation of data: All authors. Drafting of the manuscript: Oreste Gualillo, Yousof Farrag, and Ms. Mariam Farrag. Critical revision of the manuscript for important intellectual content: All authors. Obtaining of funding: Oreste Gualillo, Jesus Pino. Final approval of the version to be published: All authors.
Declaration of Conflicting Interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.
Ethical Approval
This article does not contain any studies with human or animal participants.
Data Availability
The datasets generated during and/or analyzed during the current study are available from the corresponding author on reasonable request.
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.