
Abstract
Leukocyte migration is a fundamental process in both innate and adaptive immune responses. This process is tightly regulated by chemokines and their cognate receptors. The bioavailability of chemokines is further modulated by atypical chemokine receptors (ACKRs), a subset of chemokine receptor–like molecules that lack coupling to canonical G protein–mediated signaling pathways. Among these, ACKR4 regulates dendritic cell migration through ligand scavenging and has been implicated in tumor progression in murine models. We previously established anti-mouse ACKR4 (mACKR4) mAbs, A4Mab-1, A4Mab-2, and A4Mab-3, by N-terminal peptide immunization. This study examined the binding epitopes of A4Mabs. Alanine (or glycine) scanning within the N-terminal region (amino acids 2–19) was performed using flow cytometry and Western blotting. Results showed that Tyr12 is required for recognition by A4Mab-1 in flow cytometry, whereas Tyr11, Tyr12, Glu14, Glu15, and Glu17 are required in Western blotting. For A4Mab-2, Tyr12, Glu15, and Asn16 are required in flow cytometry, whereas Tyr11, Tyr12, Tyr13, Glu15, and Asn16 are required in Western blotting. Additionally, Glu14, Asn16, and Glu17 are required for recognition by A4Mab-3 in flow cytometry. These findings contribute to the understanding of mACKR4 recognition by A4Mabs.
Introduction
Immune cell priming, effector responses, and memory formation are governed by chemokines and the spatially restricted expression of G protein–coupled receptors (GPCRs). 1 Chemokine receptors constitute a major class of seven-transmembrane receptors and are categorized as canonical GPCRs and atypical chemokine receptors (ACKR1–ACKR4).2,3 Ligand binding to canonical chemokine receptors activates heterotrimeric G proteins.1,4 In contrast, ACKRs, despite structural homology to GPCRs, do not couple to G protein signaling. Instead, they mediate β-arrestin–dependent internalization and degradation of chemokines, thereby functioning as scavenger receptors that regulate chemokine bioavailability.2,5 Cryogenic-electron microscopy analyses of agonist-bound ACKR3 revealed a distinct chemokine-binding mechanism and provided a structural basis for β-arrestin-biased receptor.6,7
ACKR4 is expressed in T lymphocytes 8 and in stromal compartments9,10 and binds CCL19, CCL20, CCL21, CCL22, and CCL25. 2 Through ligand sequestration, ACKR4 modulates CCR7-, CCR6-, CCR4-, and CCR9-dependent migratory responses. 2 This activity establishes chemokine gradients that direct dendritic cell trafficking from peripheral tissues to draining lymph nodes.9,11,12 ACKR4 is also expressed in a flow-dependent manner in afferent lymphatic collectors, where it removes CCL21 from the luminal surface and limits aberrant T-cell entry into inflamed dermal collectors. Consistent with this function, ACKR4 deficiency impairs T-cell migration to draining lymph nodes. 13
For functional analysis of ACKR4-expressing cells in mouse preclinical models, monoclonal antibodies (mAbs) against mouse ACKR4 (mACKR4) are required. Using immunization with the N-terminal mACKR4 peptide, we previously generated anti-mACKR4 mAbs, including A4Mab-1, A4Mab-2, and A4Mab-3. 14 This study determined their specific binding epitopes using an alanine scanning strategy.
Materials and Methods
Plasmid construction
The mACKR4 (Accession No. NM_145700.2) cDNA with N-terminal MAP tag was described previously. 14 Alanine (or glycine)-substituted mutants of mACKR4 were generated using QuikChange Lightning Site-Directed Mutagenesis Kits (Agilent Technologies Inc., Santa Clara, CA, USA). The PCR fragments containing the desired mutations were inserted into the pCAG-Ble vectors (FUJIFILM Wako Pure Chemical Corporation, Osaka, Japan). All mutants were confirmed by sequencing.
Cell line and plasmid transfection
The alanine- (or glycine)-substituted mutant plasmids were transfected into Chinese hamster ovary (CHO)-K1 cells (American Type Culture Collection, Manassas, VA, USA) using the Neon Transfection System (Thermo Fisher Scientific Inc., Waltham, MA, USA).
Antibodies
A4Mab-1 (rat IgG2b, kappa), A4Mab-2 (rat IgG2b, kappa), and A4Mab-3 (rat IgG2b, kappa) were established as described previously. 14
Flow cytometry
Cells were collected after brief exposure to 0.25% trypsin/1 mM ethylenediaminetetraacetic acid (Nacalai Tesque, Inc., Kyoto, Japan). After washing with 0.1% bovine serum albumin in phosphate-buffered saline, 2 × 105 cells were treated with 1 μg/mL of A4Mab-1, A4Mab-2, A4Mab-3, or an anti-MAP tag mAb (clone PMab-1 15 ) for 30 minutes at 4°C, followed by incubation with Alexa Fluor 488-conjugated anti-rat IgG (1:2000; Cell Signaling Technology, Inc., Danvers, MA, USA). Fluorescence data (from a total of 5000 cells per sample) were collected using the SA3800 Cell Analyzer (Sony Corp., Tokyo, Japan). Using FlowJo software (BD Biosciences, Franklin Lakes, NJ, USA), single cells were gated based on side scatter versus forward scatter, and the fluorescence intensity was plotted. For quantification, we measured the geometric mean of the fluorescence and calculated the normalized reactivity as the ratio of the geometric mean of A4Mabs to the geometric mean of PMab-1.
Western blotting
Cell lysates were boiled in sodium dodecyl sulfate sample buffer (Nacalai Tesque, Inc.). Proteins (10 µg/lane) were electrophoresed on 5%–20% polyacrylamide gels (FUJIFILM Wako Pure Chemical Corporation) and transferred onto polyvinylidene difluoride (PVDF) membranes (Merck KGaA, Darmstadt, Germany). After blocking with 4% nonfat milk (Nacalai Tesque, Inc.), PVDF membranes were incubated with 1 μg/mL of A4Mab-1, A4Mab-2, or PMab-1, followed by incubation with horseradish peroxidase-conjugated anti-rat IgG (1:10,000; Sigma-Aldrich Corp., St. Louis, MO). Finally, protein bands were detected with Pierce™ ECL Plus (Thermo Fisher Scientific, Inc.) or ImmunoStar LD (FUJIFILM Wako Pure Chemical Corporation) using a Sayaca-Imager (DRC Co. Ltd., Tokyo, Japan). For quantification, we used Image J, 16 and the band intensity of A4Mab-1 and A4Mab-2 was normalized by that of PMab-1.
Results
Determination of A4Mab-1, A4Mab-2, and A4Mab-3 epitopes by flow cytometry using alanine scanning
Alanine scanning was performed on the N-terminal region (aa 2–19) of mACKR4. Eighteen mutants with alanine (or glycine) substitutions in mACKR4 with an N-terminal MAP tag were created (Fig. 1). The MAP tag is essential for confirming the cell surface expression of each mutant. These mutant proteins were transiently expressed in CHO-K1 cells. Reactivity with A4Mab-1, A4Mab-2, and A4Mab-3 was assessed by flow cytometry. As shown in Figure 2A, A4Mab-1 did not react with the Y12A mutant. A4Mab-2 did not react with three mutants (Y12A, E15A, and N16A) (Fig. 2B). Moreover, A4Mab-3 did not react with three mutants (E14A, N16A, and E17A) (Fig. 2C). The cell surface expression of each mutant was confirmed with PMab-1, an anti-MAP tag mAb (Fig. 2D). The normalized reactivity (geometric mean of A4Mabs divided by the geometric mean of PMab-1) was calculated in Supplementary Fig. S1.
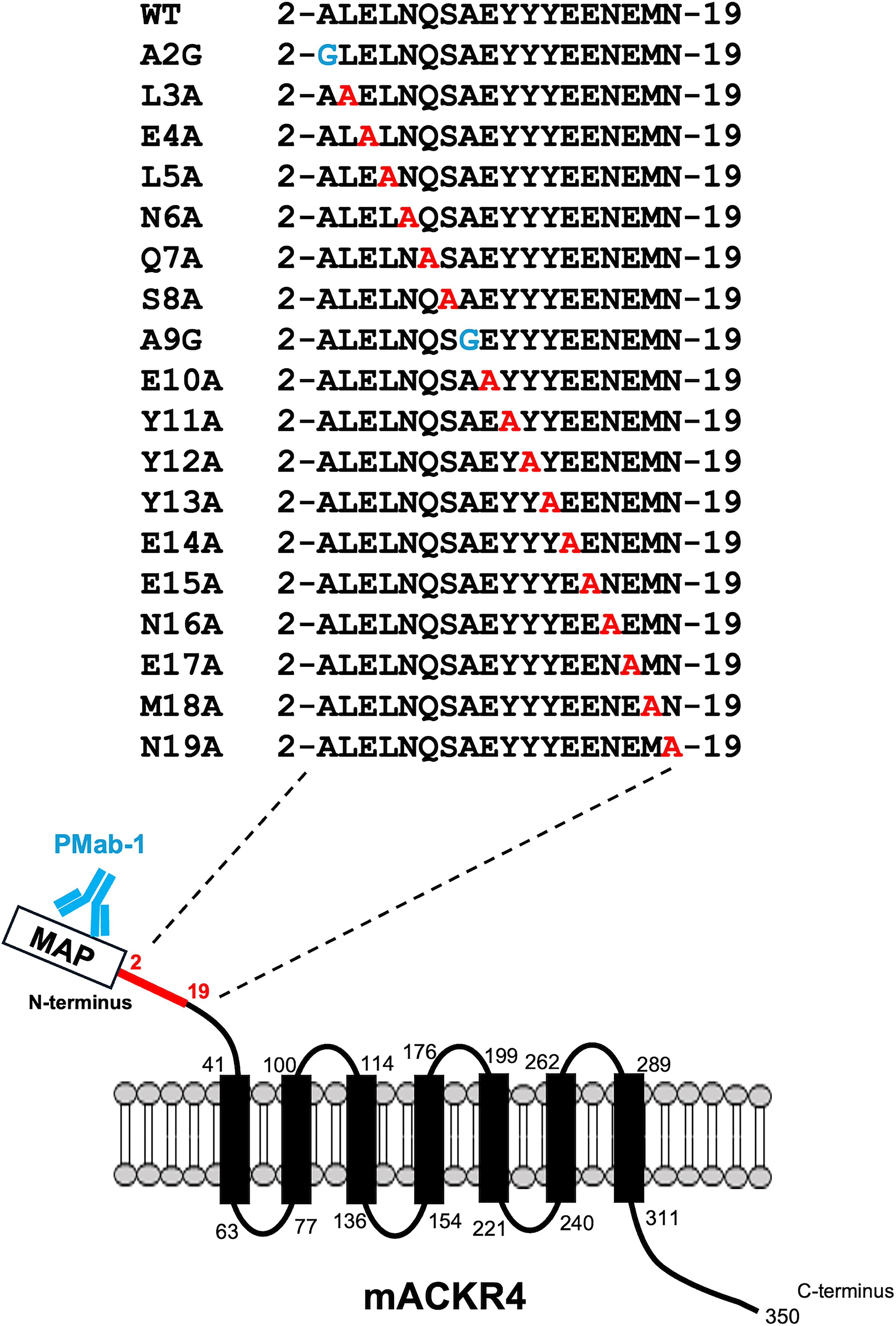
The illustration of alanine- (or glycine)-substituted mutants of mACKR4. The N-terminal amino acids of mACKR4 were substituted with alanine (or glycine). PMab-1 recognizes the N-terminal MAP tag and confirms the cell surface expression of each mutant.

Determination of A4Mab-1, A4Mab-2, and A4Mab-3 epitopes by flow cytometry using alanine scanning. CHO-K1 cells transiently expressing mACKR4 mutants and wild-type (WT) were treated with A4Mab-1 (1 µg/mL,
Determination of the A4Mab-1 and A4Mab-2 epitopes using Western blotting
We previously reported that A4Mab-1 and A4Mab-2 are suitable for Western blotting, but A4Mab-3 did not work in Western blotting. 14 Therefore, mutant lysates were prepared and analyzed by Western blotting using A4Mab-1 and A4Mab-2. As shown in Figure 3A, A4Mab-1 detected about 45 kDa major bands, which completely disappeared in the lysates of five mutants (Y11A, Y12A, E14A, E15A, and E17A). Furthermore, A4Mab-2 also detected 45 kDa major bands, which completely disappeared in the lysates of five mutants (Y11A, Y12A, Y13A, E15A, and N16A) (Fig. 3B). PMab-1 was used to confirm the expressions (Fig. 3C). The normalized band intensity (A4Mabs/PMab-1) was calculated in Supplementary Fig. S2.

Determination of A4Mab-1 and A4Mab-2 epitopes by Western blotting. Cell lysates from CHO-K1 cells transiently expressed mACKR4 mutants and wild-type (WT) were electrophoresed on 5%–20% polyacrylamide gels and transferred onto PVDF membranes. Membranes were then treated using A4Mab-1 (1 µg/mL,
Figure 4 summarizes the results and illustrates the epitope of A4Mab-1, A4Mab-2, and A4Mab-3 in the N-terminal region of mACKR4.

The schematic illustration of A4Mab-1, A4Mab-2, and A4Mab-3 epitopes.
Discussion
This study conducted the epitope mapping of anti-mACKR4 mAbs (A4Mab-1, A4Mab-2, and A4Mab-3) by alanine scanning. Results revealed that Tyr12 is required for recognition by A4Mab-1 in flow cytometry (Fig. 2A), while Tyr11, Tyr12, Glu14, Glu15, and Glu17 are required in Western blotting (Fig. 3A). Since Tyr12 is a commonly identified residue required for A4Mab-1 recognition, Tyr12 is thought to be a center of epitope. In A4Mab-2, Tyr12, Glu15, and Asn16 are required in flow cytometry (Fig. 2B), while Tyr11, Tyr12, Tyr13, Glu15, and Asn16 are required in Western blotting (Fig. 3B). Since Tyr12, Glu15, and Asn16 are commonly identified residues required for A4Mab-2 recognition, these are thought to be the center of epitope. Additionally, Glu14, Asn16, and Glu17 are required for recognition by A4Mab-3 in flow cytometry (Fig. 2C). The recognition mode by A4Mab-1 and A4Mab-2 may be different between the native structure in flow cytometry (Fig. 2) and the denatured structure in Western blotting (Fig. 3). Chemokine receptors and ACKRs possess a conserved disulfide bond between the N-terminus and the third extracellular loop. 17 Disruption of the disulfide bond may change the accessibility of A4Mabs in Western blotting. These results would contribute to the understanding of mAb–epitope interaction.
ACKR4 has been established as a critical regulator of dendritic cell trafficking through modulation of CCR7-dependent pathways via regulation of the abundance of CCR7 ligands, CCL19 and CCL21. 18 The N-terminal region of CCR7 contains sulfated tyrosine residues that are essential for high-affinity binding to these ligands.19,20 Although ACKR4 also binds to these ligands, the requirement for tyrosine sulfation in the ligand binding has not been investigated. A4Mabs were established by immunization with the N-terminal 1–19 peptide without any modification, including sulfation. 14 The epitope of A4Mab-1 and A4Mab-2 includes a Tyr triplet (Fig. 4), suggesting that A4Mab-1 and A4Mab-2 recognize nonsulfated tyrosine residues.
Unlike canonical GPCRs, ACKR4 does not elicit classical G protein–mediated signaling; instead, it functions as a scavenging receptor that facilitates chemokine clearance.21,22 Mechanistically, ACKR4 is coupled to the endocytic machinery via β-arrestin. Upon ligand engagement, ACKRs undergo internalization into endosomal compartments, followed by lysosomal degradation of the bound chemokine. 5 Although the N-terminal region of ACKR4 has not been definitively characterized as a ligand-binding interface, it remains of interest to determine the relationship between A4Mab epitopes and the neutralizing or receptor internalization effects. These activities would be expected to attenuate mACKR4 function, potentially leading to an accumulation of its cognate chemokines.
Authors’ Contributions
M.H., M.Y., and T.N. performed the experiments. T.T., M.K.K., and Y.K. designed the experiments. H.S. and Y.K. wrote the article. All authors have read and agreed to the published version of the article.
Footnotes
Author Disclosure Statement
The authors have no conflicts of interest.
Funding Information
This research was supported in part by the
Supplemental Material
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.