
Abstract

Invited Talks
HO.03.02
HO.03.02 - Ultrasound as a Complement to EMG
1Mayo Clinic, United States
In recent years, high-frequency ultrasound has become increasingly utilized as a diagnostic tool in the workup of neuromuscular disease. With advances in technology, high resolution, portable units have become more affordable and increasingly prevalent. As a result, this modality is now a potential adjunctive tool in the evaluation of neuromuscular disease that is an ideal complement to electrodiagnosis. Prerequisites to successful implementation include an in-depth knowledge of anatomy, sound knowledge of ultrasound physics, and comfort with the machine itself in order to recognize and minimize artifacts and facilitate image optimization.
Ultrasound allows one to image in real time, providing diagnostic anatomical and functional information concurrent with electrodiagnosis, without exposing the patient to radiation. Advantages of ultrasound in the clinical setting, compared with MRI, include cost, accessibility, speed of the examination, ability to image the entire length of a nerve in a single study, and the capability to image both statically and dynamically. Ultrasound can provide equivalent or better spatial resolution than MRI, with individual fascicles visible in many peripheral nerves.
One of the disadvantages of needle EMG is its invasive and sometimes painful nature, with a small but finite risk of direct injury to nerves, blood vessels, and vital structures. Ultrasound provides high-resolution imaging of soft tissue, fascial planes, and neurovascular structures, and the use of real-time ultrasound guidance for needle placement during NCS and needle EMG can increase accuracy and decrease risk in certain settings.
Indications for ultrasound in the EMG lab not only include enhancing safety and accuracy of needle EMG and occasionally NCS; in the hands of a skilled operator ultrasound provides detailed anatomical and pathophysiological information about nerve and muscle disease and real-time information regarding muscle activation and movement patterns. Thus, ultrasound can be used in the diagnosis of entrapment neuropathies and generalized demyelinating neuropathies based on increased cross-sectional area of nerves, it can identify the underlying cause of nerve enlargement, such as tumors, hematomas, fascial bands, bony encroachment, tenosynovitis, and can identify muscle disease based on changes in echo intensity.
Ultrasound should be considered complementary to electrodiagnosis; the latter providing physiologic information, while ultrasound provides both structural and, in some cases, additional physiologic information. Just as the electrodiagnostic examination must be individualized for each patient and modified based on ongoing findings, the ultrasound examination is also dynamic; the sonographer can dynamically rule in or out various diagnoses based on the information acquired while performing the study. With ongoing developments in technology and a rapidly increasing body of supportive literature, the role of ultrasound in the diagnosis of neuromuscular disease will continue to rapidly evolve.
PL.01.03
PL.01.03 - Carrier Screening
1University of Western Australia, Australia
Carrier screening has a long and illustrious history of facilitating couples to avoid having children with severe genetic conditions if that is what the couple want to do. Carrier screening started with screening for sickle cell anaemia in one small town in Greece in 1966 because of one clinician’s drive to help the population he served. Carrier screening for Tay-Sachs disease started in the US in 1970 and then spread around the world. When the Duchenne gene complete cDNA probes became available in 1987, as soon as we started cascade carrier testing in known Duchenne families, we were able to finally, more than 100 years after Duchenne described the disease, highly accurately determine which females in the family were carriers and which were not. Telling a mother of a Duchenne boy that she was not a carrier, that her son had a de novo mutation, that there was nothing she could have done to avoid her son being affected could lift decades of guilt from her shoulders. Through meeting the families via the West Australian Muscular Dystrophy Association, I could see the personalities of these women change remarkably. As early as the late 1980s there was interest in trying to establish population-wide carrier screening to identify couples with a 1:4 chance of having a boy with Duchenne muscular dystrophy before they were identified as high chance by having an affected boy. In reality, however, the technology was not available to make this possible. It became possible with the advent of next generation sequencing (NGS). Through NGS, carrier testing for hundreds of genes was feasible. This led to consumer demand for such testing to be implemented. In Australia, in turn, this led to the Mackenzie’s Mission research project whose aims were to demonstrate that such expanded reproductive carrier screening could be taken to every corner of this giant country. Mackenzie’s Mission demonstrated this was possible, screening >9,000 couples for nearly 1,300 recessive disease genes, including for many severe neuromuscular diseases. Work is now ongoing into how to turn the results of Mackenzie’s Mission into a government funded national screening program similar to the cancer screening programs. In the meantime, since 1st November 2023, government funded, free, carrier screening for three diseases (cystic fibrosis, fragile X syndrome and spinal muscular atrophy) has been available to all Australians. As of May 2024, use of this three gene screening test is already running at a rate of 100,000 couples a year in a country that has around 300,000 births a year. This indicates a huge appetite for carrier screening in Australia. In the future, hopefully, expanded reproductive carrier screening will similarly be freely available to all Australians.
PL.01.04
PL.01.04 - Newborn Screening for SMA…and Other Diseases
1University Of Liege, Belgium
Newborn screening (NBS), historically performed using biochemical and metabolic techniques, has greatly improved the management of many serious and treatable conditions. In 2018, we established a pilot program for NBS of spinal muscular atrophy (SMA), which screened more than 250,000 children and identified 15 of them with SMA. We were able to evaluate the positive outcome of these children, who were treated rapidly. A health economic analysis of this screening also confirmed that it is more cost-effective to treat children rapidly at birth rather than waiting until symptoms appear. At the end of the pilot project, the health authorities added this new screening to the list of diseases routinely screened at birth.
As a follow-up to this project, we launched the Baby Detect study program in September 2022. Baby Detect aims to screen for 165 serious, treatable and early-onset genetic diseases. The gene list has been compiled by the pediatricians of Liege and evolves regularly. This prospective study aims to assess the potential and acceptability of newborn genetic testing to screen up to 8,000 newborns in 3 years for all early-onset serious diseases that could benefit from treatment or pre-symptomatic clinical testing. The primary outcome is the percentage of parents who accept the proposed screening compared to the number of mothers approached for consent. Information to parents was provided during pregnancy and after the baby's birth using flyers, videos, a website, and verbal information. On the first day of the child's life, almost all parents were visited by a member of the team to present the study. If they agreed, they would fill in an electronic consent form (paper consent was also possible). If they refused, we tried to find out the reasons for their refusal. As of 20 June 2024, 4,554 families had been contacted and 4,134 had agreed, giving an acceptance rate of 90.5%. The refusals do not seem to be related to a fear of genetic testing, but rather to a lack of knowledge and understanding of the project and the risks. The most common reasons for refusal were "my family doesn't have any diseases", "it's not mandatory, I don't do it".
This genomic NBS received strong support from the local IRB, local health professionals involved in pregnancy and neonatal care, with good acceptance from parents.
PL.01.05
PL.01.05 - Genomic Newborn Screening
1Murdoch Children's Research Institute, Australia
Newborn bloodspot screening (NBS) is a highly successful public health programme that uses biochemical and other assays to screen for severe but treatable childhood-onset conditions Genomic newborn screening (gNBS) brings the potential to increase the range of detectable disorders, including a range of neuromuscular conditions, but raises many practical and ethical issues. Although discussions about gNBS to date have been largely hypothetical, evidence is starting to emerge from the first gNBS pilot projects, including a number that currently underway in Australia. Selecting genes for testing is critical and needs to reflect that parents value the certainty of prediction over actionability. In 2024, the utility of NBS for most neuromuscular disorders is limited by the lack of effective and available treatments, but it is hoped that this will change over the next decade. It is also important that data is analyzed in a way that minimizes uncertainty and incidental findings. The expansion of traditional newborn screening to identify more life-threatening and treatable diseases needs to be balanced against the complexity of consenting parents of newborns for genomic testing as well as the risk that overall uptake of traditional NBS may decline. Overall, implementing gNBS will require a nuanced approach, including consideration of the views of diverse populations, the capabilities of health systems, and health economic implications. It will be essential to rigorously evaluate outcomes and ensure programs can evolve to maximize benefit.
PL.02.04
PL.02.04 - Clinical and Biological ALS Biomarkers: Role in Clinical Trials
1University Medical Center Utrecht, Netherlands
Determining biological and clinical efficacy is a primary objective of randomized clinical trials. Central to defining efficacy is the biomarker or outcome, which drives the success of a study. An outcome should be measured objectively to determine the intervention's benefit, while clinically oriented outcomes must also reflect the patient's well-being, functionality, or survival.
In clinical trials for amyotrophic lateral sclerosis (ALS), a wide array of clinical and biological outcomes have been employed, with new outcomes emerging annually. This diversity is largely driven by the unsuccessful drug development and numerous futile studies, highlighting the field's ongoing efforts to enhance clinical trial success rates.
This talk will review the state-of-the-art clinical and biological outcomes in ALS clinical trials. We will discuss the main challenges these outcomes face at various stages of clinical development and propose alternative strategies for future trials.
PL.03.03
PL.03.03 - Treatment Advances in Genetic Neuropathies
1UCL Queen Square Institute of Neurology, London, UK
Until recently the treatment of the hereditary neuropathies was largely managing symptoms and complications including pain management, physiotherapy, orthotics and foot surgery as needed.
The success of gene silencing therapy (ASO and SiRNA) for TTR amyloidosis has revolutionised the management of this condition with excellent outcomes especially if patients are diagnosed and treated early.
For Charcot Marie Tooth disease (CMT) and the related disorders Hereditary Motor Neuropathy (HMN) and Hereditary Sensory Neuropathy (HSN) there are no current treatments but there are ongoing trials in patients for pathogenetically based therapies and huge progress in the preclinical development of genetic therapies. Examples of ongoing trials include an aldose reductase inhibitor for SORD (sorbitol) related CMT and serine supplementation for SPTLC 1/2 related HSN1. Multiple SiRNA therapies aiming to reduce PMP22 expression in CMT1A are in late stage pre clinical development. For forms of CMT with loss of function including X-linked CMT due to GJB1 variants and many of the recessive forms of CMT including CMT4C (SH3TC2) and CMT4J (FIG4), gene replacement therapies are also at an advanced preclinical stage. Many other forms of CMT including CMT1B (MPZ) and CMT2A (MFN2) have also promising therapies in pre clinical development.
In this talk, I will provide an overview of the hereditary neuropathy therapy landscape with particular emphasis on ongoing trials and those therapies in advanced pre clinical development.
PL.03.04
PL.03.04 - Advances in Treatment of Diabetic Neuropathy
1University Of Michigan, United States
We will review both neuropathic pain treatments and potential disease modifying treatments for diabetic neuropathy. Neuropathic pain treatment guidelines consistently include multiple classes of effective oral medications. Evidence also supports use of some topical medications and non-pharmacologic interventions. Emerging evidence points to the downsides of opioids for the treatment of chronic pain. Current practice is far from ideal with frequent opioid use. Potential disease modifying treatments include glycemic control, exercise, dietary weight loss, surgical weight loss, and medication weight loss. Glycemic control has been shown to be more effective in type 1 diabetes compared to type 2 diabetes.
PL.04.02
PL.04.02 - Outcome Measures
1University Of Toronto, Canada
We will discuss the complexities involved in evaluating patients with MG, emphasizing the potential pitfalls that may arise during the assessment process. This will include addressing the fluctuating nature of MG symptoms, the interplay between different muscle groups, and the heterogeneous presentation of the disease. We will provide a comprehensive overview of the range of measures currently available for assessing MG patients, offering insights into their strengths and limitations.
Furthermore, we will explore the existing gaps in the literature concerning MG assessment. Additionally, we will address the challenges encountered in assessing outcomes in clinical trials and everyday clinical practice, emphasizing the need to strike a balance between efficiency and accuracy in measuring outcomes.
Attendees, including clinicians and researchers in the field, will gain a deeper understanding of the challenges faced in clinical trials and clinical practice, while being updated on the current standards of measuring in MG. This knowledge will not only enable clinicians to implement more accurate and comprehensive assessment strategies in their daily practice, thus improving patient care, but it will also provide researchers with valuable insights for study design.
PL.04.03
PL.04.03 - Biomarkers
1Uppsala University, Sweden
Myasthenia Gravis (MG) is an autoimmune disorder caused by autoantibodies, predominantly directed against the acetylcholine receptor (AChR) or muscle-specific tyrosine kinase (MuSK) at the muscle membrane. This results in disturbed neuromuscular signaling and fluctuating fatigable skeletal muscular weakness. MG is a very heterogeneous disease, with several subgroups into which patients can be divided based on antibody subtype, age at onset (juvenile, early-onset, late-onset, and very-late-onset), clinical phenotype (ocular or generalized), and association with thymus pathology. This heterogeneity and daily and day-to-day fluctuations in skeletal muscle weakness cause difficulties in predicting the clinical course. Despite antibody detection being important for MG diagnosis, antibody titers do not necessarily correlate with disease severity or response to treatment. Nevertheless, there are currently no available biomarkers in MG that can predict the disease course or treatment response.
This talk will explore the potential candidates for objective biomarkers in MG, including circulating microRNA (miRNA), proteins, metabolites, and blood cells. MiRNA emerged in recent years as possible biomarkers in several diseases, and studies performed in MG subgroups have highlighted specific circulating miRNA serum profiles that correlate with clinical response upon treatment and thymectomy. In particular, levels of miR-30e-5p and miR-150-5p have been associated with MG and change upon immunosuppression and thymectomy. Additionally, serum metabolomic profiles and the detection of clonal antibody-producing B cells show promise in predicting treatment response and disease relapse.
T-cell signatures could be promising cellular markers of disease severity, and plasma complement components, especially C3a, C5a, and the membrane attack complex C5b-9, offer insights into complement activation and MG subtype characterization. Moreover, inflammatory protein patterns, including cytokines and interleukins, distinguish MG patients from healthy controls, while serum neurofilament light and calprotectin exhibit stronger associations with MG than healthy controls.
Moving forward, the validation and multi-center evaluation of these promising biomarkers are essential to establish their sensitivity, specificity, and clinical utility. Ultimately, validated biomarkers hold the potential to revolutionize MG management by facilitating early intervention and personalized treatment strategies.
SS.01.02
SS.01.02 - Fundamental Pathobiology for the Clinician
1Harry Perkins Institute of Medical Research, Centre for Medical Research, University of Western Australia, Perth, Australia
The sarcomere, the fundamental unit of striated muscle contraction, comprises various structural components, including the Z-line, thin filament, thick filament, titin (the third filament), M-line, and sarcomere-associated proteins. Pathogenic variants in these components lead to diverse skeletal muscle diseases. Despite extensive research, the pathobiology of many gene defects remains unclear. A thorough understanding of the pathobiology of sarcomere-associated skeletal muscle disease variants is crucial for clinicians as it provides insights into the mode of inheritance, recurrence likelihood in families, prognosis, and potential treatments.
Sarcomeric protein diseases exhibit a wide range of onset, from fetal akinesia to old age, with some cases remaining asymptomatic into advanced age, indicating reduced penetrance. This spectrum of presentations results from different pathogenic variants causing distinct pathobiological mechanisms. Inheritance patterns can be dominant, recessive, de novo, or mosaic, with mosaicism potentially leading to asymmetric disease manifestations.
Pathobiologies of thin filament variants are quite diverse, for example, in different congenital myopathies, nemaline myopathies, and fetal akinesia deformation syndrome (FADS). They can present with hypertonia or hypotonia depending on the specific effect of the variant on calcium sensitivity. Notable variants occur in genes such as ACTA1 (skeletal muscle actin), TPM3 (slow muscle fibre tropomyosin), TNNI1 (slow skeletal muscle troponin I), NEB (nebulin), and CFL2 (cofilin 2). The pathobiology of these variants directly influences disease severity, exemplified by termination codon variants in ACTA1, resulting in severe nemaline myopathy.
Thick filament pathobiologies encompass myosin-related diseases, including myosin storage myopathy, Laing distal myopathy, scapuloperoneal myopathy, and distal arthrogryposis, highlighting the complexity of sarcomere-related pathobiologies. Notable variants occur in genes such as MYH3, MYH7, and MYH8. Intrafamilial variability in the severity, such as in MYH7-distal myopathy, suggests the presence of modifying factors, though these factors remain largely unidentified.
Titinopathies, caused by mutations in TTN, present a broad spectrum of clinical manifestations due to titin’s role as a molecular spring, contributing to the sarcomere’s elasticity and stability. These disorders underscore the intricate interplay of sarcomere components in muscle function and the necessity for a deeper understanding of their molecular mechanisms. A notable recent finding is the presence of SRPK3 deleterious variants in combination with heterozygous TTN variants, which results in a digenic inheritance and leads to progressive early-onset myopathy. This emphasises the need to explore digenic and modifying factors further to comprehend disease prognosis better.
In conclusion, the diverse pathobiologies of sarcomere-associated skeletal muscle diseases necessitate detailed research to unravel the molecular mechanisms underlying these conditions. Enhanced understanding will facilitate improved diagnosis, management, and treatment strategies for affected individuals.
SS.01.03
SS.01.03 - New Sarcomere Diseases
1University Of Western Australia, Australia
The sarcomere is the functional contractile unit that underlies striated muscle contraction; pathogenic variants in genes encoding key sarcomeric proteins are known to cause a range of different skeletal muscle diseases. These vary in severity and age of onset from severe paralysis in utero to milder conditions compatible with normal life expectancy. In recent years, several new insights have been gleaned into the genetics of skeletal muscle sarcomeric disorders, including the landmark association of digenic inheritance of variants in the X-linked gene SRPK3 with variants in titin. Deleterious variants in SRPK3 cause a progressive early-onset myopathy only when in combination with heterozygous pathogenic TTN variants (typically truncating variants). Other key recent discoveries include identification of dominant and recessive inheritance of TNNI1-related myopathy. TNNI1-encodes the slow skeletal muscle isoform of troponin I; the associated phenotypes range from severe-infantile weakness (recessive form) to an adult-onset hypercontractile disease with muscle cramping and myalgia (dominant form). Heterozygous missense variants in the gene encoding cardiac alpha-actin (ACTC1) have recently been identified to underlie rare cases of distal arthrogryposis, this phenotype likely results from ACTC1 expression in developing skeletal muscle. Bi-allelic loss-of-function variants in OBSCN, encoding the giant sarcomeric protein obscurin, have been identified to predispose individuals to recurrent and severe episodes of rhabdomyolysis. Given the size of OBSCN, it is tempting to speculate that it may underlie a substantial proportion of recurrent rhabdomyolysis. Expanding phenotypes are also recently described for variants in genes encoding alpha-actinin-2 (ACTN2) and embryonic myosin heavy chain (MYH3). It is likely that in time, diseases will be associated with defects in all genes encoding sarcomeric proteins.
SS.02.02
SS.02.02 - Gene Therapies
1University College London, United Kingdom
In the last decade there have been consistent advances in different approaches aimed at restoring muscle dystrophin production in patients with Duchenne muscular dystrophy. The earlier approaches focused on RNA therapies, with multiple splice-switching antisense oligonucleotides (ASOs), several of which having received conditional FDA approval, targeting specific “skippable” mutations. These efforts with first generation ASOs were generally well tolerated but resulted in low levels of restored dystrophin levels, requiring at least 3 years of weekly intravenous infusions to convincingly appreciate divergence from the natural history course of the disease. These early efforts are currently being followed by second generation ASOs, now in early clinical trials and demonstrating significantly higher levels of the surrogate dystrophin expression on muscle biopsy, while larger studies to demonstrate clinical efficacy are due to start this year. These new ASO have the advantage of requiring less common administrations, although we have less long term data on their tolerability.
More recently the efforts have taken advantages of different adeno-associated viral vectors (AAV), which can deliver to muscle and heart a much smaller version of the dystrophin protein, a minidystrophin. At the time of writing, of the 5 ongoing DMD AAV gene therapies, one has received in June 2024 full FDA approval for ambulant DMD boys and conditional approval for non-ambulant DMD individuals, while another product also in June 2024 unexpectedly failed to demonstrate efficacy following a large phase 3 clinical trial and is being discontinued.
While the progress of AAV in DMD is extremely encouraging, there are also issues related to the very high viral load administered systemically and the associated immune responses that require close surveillance from treating physicians. Other unknowns that the field will have to better understand over the next few years relate to the efficiency of transduction in muscle with a different degree of pathology, hence if the same level of efficacy and in turn of risk/ benefit will be observed across the entire spectrum of the condition, and the long-term durability of these episomal viral vectors.
Despite these unknown these parallel programs of translational research have brought a high level of competition and
SS.02.03
SS.02.03 - Exon-Skipping/PMO Therapies
1Murdoch University, Murdoch, Australia, 2Perron Institute for Neurological and Translational Science, Nedlands, Australia
The application of steric blocking antisense oligomers as potential exon skipping therapeutics now seems very obvious for many genes, especially large multi-exon genes encoding structural proteins with repeated domains. The unequivocal differences in disease severity between Duchenne Muscular Dystrophy (DMD) and the milder, in some cases asymptomatic, individuals with Becker Muscular Dystrophy, clearly indicate that many dystrophin exons are not necessary for near-normal function. Exceptions to the dystrophin reading frame rule that are unlikely to be amenable to exon skipping are the massive genomic deletions of more than 30 exons or mutations involving crucial exons encoding functionally important domains of the protein. The deletion/mutation hotspots in the dystrophin gene involve exons that should be amenable to targeted exon skipping with phosphorodiamidate morpholino oligomers (PMO), and hence an estimated 80% of DMD individuals could potentially benefit from this intervention.
Where from here? Improvements in the efficiency of delivery and uptake are essential and are currently being explored. Ongoing clinical trials of an exon 51 targeting morpholino oligomer coupled to a cell penetrating peptide are showing considerable promise in treating DMD with increased dystrophin expression induced after monthly, rather than weekly dosing.
What other conditions could be responsive to targeted exon skipping?
- Any gene compromised by mutations that lead to cryptic splicing and retention of pseudo-exons are potential targets, as suppression of the abnormal pre-mRNA processing should allow production of the normal protein.
- Specific gene down-regulation can be achieved by excising a crucial exon or disrupting the reading frame, so that the induced transcript would be non-functional and subjected to nonsense mediated decay.
- Other multi-exon genes encoding structural proteins may be potential candidates. While the collagens are compact, highly expressed genes that are generally composed of scores of small in-frame exons, additional challenges arise from fibril folding, particularly heterotrimer assembly of collagen fibrils.
- Marfan's syndrome arises from mutations in the fibrillin 1 gene that compromise normal dimer folding. FBN1 mutations are inherited in a dominant manner, and we have shown these gene lesions could be addressed by excising corresponding exons from the mutated and normal allele to allow correct protein folding.
Perhaps the most widely applicable applications for splice switching antisense oligomers will be in redirecting specific alternative splicing patterns. Most human genes express multiple isoforms through the use of alternative promotors and exon combinations, with some of the gene isoforms having directly opposing activities, e.g., pro-inflammatory vs anti-inflammatory). Redirecting expression of a disease-causing or associated isoform into a protective transcript should have enormous potential in addressing many conditions.
SS.02.04
SS.02.04 - Corticosteroids, Vamorolone and Other Oral Treatments
1University Of Pittsburgh, United States
The pathology of dystrophic muscle caused by dystrophin protein deficiency in Duchenne muscular dystrophy (DMD) and Becker muscular dystrophy (BMD) has a prominent inflammatory component. Treatment with corticosteroids, which are potent anti-inflammatory agents, was shown in both randomized, controlled trials and real-world evidence to result in a delay in loss of ambulation and upper extremity function for boys with DMD. However, corticosteroid treatment for DMD has resulted in side effects of poor linear growth, arrest/delay of puberty, weight gain, adrenal suppression, Cushingoid features, cataracts and behavior problems. The side effects of corticosteroids are only partly tolerated, as real-world evidence shows that most boys with DMD are dosed at lower doses of corticosteroids than are recommended. Real world data suggests there is a balance of clinically meaningful functional benefits on the backdrop of a markedly truncated lifespan and further reductions in quality of life due to corticosteroid side effects. The risk/benefit ratio of treatment with corticosteroids is even less favorable in the milder phenotypes that are classified as BMD despite the potential of benefit in this patient population. Vamorolone was developed as a first-in-class steroid that retains the anti-inflammatory benefit of corticosteroids with the goal of limiting typical corticosteroid-associated side effects. Phase 1, 2 and 3 studies were performed for vamorolone treatment in patients with DMD resulting in regulatory approval of vamorolone for DMD in the US, EU and UK. The randomized, blinded, placebo and prednisone-controlled Phase 3 study of vamorolone in DMD demonstrated similar muscle functional benefits to prednisone with some lessening of side effects. While vamorolone treatment has similar adrenal suppression and likelihood of weight gain to prednisone, vamorolone does not interfere with normal linear growth and has a beneficial pattern of bone biomarkers. Vamorolone may also have a lower adverse influence on behavior than corticosteroids. Overall, the adverse effects typically associated with corticosteroids are less frequent and less severe with treatment with vamorolone. A randomized, controlled, blinded pilot study of vamorolone treatment of adults with BMD is underway with results expected in 2025. Preclinical mouse studies suggest that vamorolone or corticosteroids decrease expression of inhibitory microRNAs that have been shown to limit the expression of native, truncated dystrophin in patients with BMD. Future clinical studies and experience will shed light on a role for vamorolone in combination with other treatments developed for the medical management of patients with DMD and BMD.
SS.03.03
SS.03.03 - Advances in Imaging Techniques/MRI as an Outcome Measure
1Kennedy Krieger Institute, United States
Magnetic resonance imaging (MRI) based measurements are increasingly being employed as adjunct outcome measures in clinical trials of facioscapulohumeral muscular dystrophy (FSHD). We will discuss ways in which MRI-based measures can complement current clinical outcome measures and help overcome challenges that are specific to clinical trials of FSHD. The validation of MRI-based muscle imaging biomarkers has been met with multiple challenges, including time-intensive post-processing steps and the need to develop data analysis techniques capable of summarizing large quantities of imaging data. We will discuss ways in which FSHD researchers are addressing these challenges using artificial intelligence methodology.
SS.04.03
SS.04.03 - Challenges of Gene Therapy (Immune Response and Others)
1Sydney Children's Hospital, Australia
Gene therapy has the potential to provide transformative treatments of monogenic neuromuscular disorders. Various challenges are encountered in development, with safety and efficacy dependent on target tissue delivery within defined therapeutic windows, tolerability of protein dosage changes, and mitigation of immune responses. With several gene therapies using adeno-associated viral (AAV) vectors in clinical practice and many in pre-clinical and clinical development, multiple reports are emerging of severe and life-threatening inflammatory responses, including thrombotic microangiopathy, acute kidney injury due to atypical haemolytic uraemic syndrome, immune mediated myocarditis and hepatic toxicity. These highlight the need to understand the mechanisms and manage the risks produced by an immune response to AAV vectors. An overview of the pharmacokinetics of immune activation following AAV administration will be presented, including the innate immune response, antibody responses to the capsid and transgene, and cellular responses. The key implications that may impact the safety and efficacy of AAV gene therapies will be examined; i) risk factors associated with immunogenicity (product, manufacturing process, treatment, and patient-related factors), ii) immunosurveillance following infusions, iii) different prophylactic immunomodulatory regimens, and iv) interventions to manage immune mediated events. There is a need for unified reporting of treatment related adverse events and processes to improve safety.
SS.05.02
SS.05.02 - Oculopharyngeal Muscular Dystrophy and its Overlaps with Other Oculopharyngeal Myopathies
1Montreal Neurological Institute, McGill U.University, Canada
Oculopharyngeal muscular dystrophy (OPMD) was in the 1980’s the prototype muscular dystrophy with a diagnostic skeletal muscle intranuclear inclusions. The identification of the (GCN)/Polyalanine expansion in the PABPN1 gene in 1998 modified its diagnosis and since has helped identified a growing group of disorders with overlapping phenotypes and intranuclear inclusions that are not associated with the PABPN1 expansion. The objective is to review the growing field of oculopharyngeal myopathies as the discovery of new genes is changing the landscape of these late-onset diseases. We will first review the molecular basis of OPMD. This will allow us to summarize some of the important new insights into its pathophysiology and knowledge gaps still needed to be filled. We will then discuss OPMD’s phenotypic spectrum, since it opens the door to searches to uncover severity factors. By comparing the clinical, pathological and molecular overlap between OPMD and other oculopharyngeal myopathies we will underline how they may share some key pathological features. Lasty we will provide an overview of the clinical management of OPMD and upcoming treatments.
SS.07.04
SS.07.04 - Animal Model for a Hereditary Myopathy
1Children's Hospital Colorado, Aurora, USA
Congenital myopathies are a heterogeneous group of disorders that cause weakness, skeletal abnormalities, and breathing and feeding difficulties in infants and children. While disease mechanisms are well understood for some congenital myopathies they are poorly understood for others and there are no targeted therapies for congenital myopathies in routine clinical use. SELENON-related congenital myopathy is caused by changes in the gene SELENON, which encodes Selenoprotein N (SelN) and causes prominent hypotonia, axial and neck flexor weakness, respiratory weakness, rigid spine, and poor weight gain starting in infancy. SelN has been shown to be a redox enzyme that localizes to the endoplasmic reticulum/sarcoplasmic reticulum membrane and is expressed ubiquitously at low levels in adult tissues. The mechanisms through which loss of SelN leads to a myopathic phenotype have been difficult to elucidate, in part due to a lack of animal models for this disease. SelN has been reported to play a role in a variety of processes including muscle metabolism, redox homeostasis, excitation-contraction coupling, and satellite cell function. However, the primary role of SelN in muscle disease and SelN-related processes that could be targeted for therapeutic development remain unclear. Here, I highlight some of the challenges and successes in developing animal models and other tools for studying SELENON-related congenital myopathy and the insights they have provided for this disease.
SS.08.02
SS.08.02 - Car-T Therapy
1Fiona Stanley Hospital, Murdoch, Australia, 2University of Western Australia, Nedlands, Australia
The first chimeric antigen receptor (CAR) T-cell product was infused into a human patient in 2010, and first FDA approval of a CAR T-cell product was in 2017. Since then the landscape of CAR T-cell therapy has expanded rapidly. The premise of this treatment is immunotherapy, by ex vivo manipulation of T-cells to enable expression of a receptor for the desired antigen target, and infusion of these cells into the patient. These engineered T-cells are then able to expand and kill the targeted cells. There are now multiple CAR T-cell products available and FDA approved in haematological malignancies including acute lymphoblastic leukaemia, B-cell lymphomas and multiple myeloma. Could this potent immunotherapy be further indicated in non-malignant conditions where deep suppression of immune cells are required?
To consider this proposition, first we will review constructs of various CAR T-cell products. Different antigen receptors target different cells required for disease control, while modifications of co-stimulatory domains may impact efficacy or toxicity. Practical aspects in the process of administering CAR T-cells to patients involving leukapheresis, manufacture, lymphodepletion and infusion will be discussed. We will address safety aspects of therapy including cytokine release syndrome, immune effector cell-associated neurotoxicity syndrome, immunosuppression and cytopenias. These may influence patient suitability as recipients. Lastly, developments in CAR T-cell therapy in auto-immune disease including neuromuscular disorders will be explored with this revolutionary treatment potentially benefitting a broader range of patients.
SS.09.02
SS.09.02 - Ultrasound
1Fiona Stanley Hospital, Australia
There has been a resurgence of interest in neuromuscular ultrasound as a point of care diagnostic tool, owing to its accessibility, non invasive nature, ability to provide immediate patient feedback and real time imaging capabilities. The review comprehensively analyses the current literature on ultrasound's utility in neuromuscular disorders. We examine the interpretation of normal and pathological sonographic findings, evaluate various grading systems and their limitations, and compare quantitative and qualitative assessment methods' sensitivity and specificity. The review encompasses key ultrasound domains relevant to neuromuscular evaluation, assesses the diagnostic value of ultrasound across various conditions, and critically analyses its strengths and limitations. Despite its potential, widespread adoption of neuromuscular ultrasound faces challenges, including the need for operator expertise and standardised protocols. We explore these barriers and discuss future directions for research and clinical implementation. By synthesising current evidence and identifying areas for future investigation, the aim is to elucidate the role of point of care ultrasound in the diagnostic approach of neuromuscular diseases, contingent upon rigorous protocol standardisation and ongoing validation studies.
SS.09.03
SS.09.03 - MRI – Patterns in Muscular Dystrophy
1Kennedy Krieger Institute, United States
The growing literature on magnetic resonance imaging (MRI) in the muscular dystrophies has enhanced the field in several key ways. Muscle imaging allows more detailed disease characterization within individual muscles than can be ascertained through examination alone. The examination of muscles using different sequences has also provided insight into the pathophysiology of these disorders. The accumulation of imaging data across multiple different types of muscular dystrophy has allowed investigators to identify different patterns of muscle involvement and sparing within different disease types. We will review the patterns of muscle involvement reported in the literature and discuss the extent to which imaging can be used to diagnose various muscular dystrophies.
SS.09.04
SS.09.04 - Ultrasound of the Diaphragm and Respiratory Issues
1Mayo Clinic, United States
Diaphragm ultrasound is an excellent diagnostic tool in the work up of patients presenting with unexplained dyspnea or failure to wean from the ventilator. Diaphragm ultrasound is best used as a complement to electrophysiologic testing (phrenic nerve conduction studies and diaphragm needle EMG) in this setting.
Diaphragm ultrasound can be performed using either B mode or M mode. B mode ultrasound is performed using a linear probe, placed over an intercostal space in the zone of apposition (over the anterolateral chest wall region towards the costal margin). The diaphragm is identified as the layer of muscle deep to the intercostal muscles (the latter span two ribs) and is encased by a layer of hyperechoic connective tissue on either side of the muscle (the pleura and peritoneum). It is more easily visualized on the right side, due to the underlying liver providing an excellent acoustic window.
The diaphragm is first measured at rest, at the end of a quiet expiration, with electronic calipers placed immediately inside the two encasing layers of connective tissue. The patient is then asked to take a deep breath in, and the muscle is measured at the point of maximal thickening, before the lung encroaches into the field of view. The absolute thickness of the diaphragm can be used diagnostically (with a lower limit of normal of greater than 0.13 cm), and the thickening ratio (thickness at maximal inspiration divided by thickness at end expiration) can be calculated and should be at least 1.2. It is important that the patient takes a deep breath in, as the diaphragm does not thicken in many normal subjects with tidal breathing. In cases of severe dysfunction, paradoxical thickening may be seen, where the diaphragm actually becomes thinner during inspiration.
In M mode ultrasound, a curvilinear transducer is placed below the costal margin, directed cranially and posteriorly, to image the dome of the diaphragm as it moves back and forth, towards and away from the transducer during the respiratory cycle. When the diaphragm is weak, the degree of excursion will be reduced, and in the case of a paralyzed diaphragm, the muscle will actually move paradoxically, away from the transducer (up into the chest cavity) during inspiration. Normal values for diaphragm excursion are available in the literature, with some variability between sexes, but less than 2.5 cm of excursion is considered consistent with severe dysfunction.
Both B mode and M mode diaphragm ultrasound have been successfully used to predict weaning success in patients coming off mechanical ventilation. Diaphragm ultrasound has also been shown to have excellent sensitivity and specificity in the diagnosis of phenic neuropathy, may be abnormal in diaphragm myopathy, and is a very useful tool in increasing safety and accuracy of diaphragm needle EMG.
SS.10.04
SS.10.04 - What’s New in Lipid Storage Myopathies
1Neuromuscular Center and Department of Neurology, Qilu Hospital, Shandong University, Jinan, China
Lipid storage myopathy (LSM), pathologically defined by excessive accumulation of neutral lipid droplets in muscle fibers, represents a heterogeneous group of lipid metabolic disorders. Traditionally, disorders affecting carnitine and the carnitine transport system were identified as the primary causes of LSM. Recently, numerous cases of LSM have been reported worldwide. The most common cause is multiple acyl-CoA dehydrogenation defects (MADD) due to mutations in electron transfer flavoprotein (ETF) or electron transfer flavoprotein dehydrogenase (ETFDH), also known as ETF-ubiquinone oxidoreductase (ETF-QO). The second most common cause is neutral lipid storage disease with myopathy (NLSDM), associated with variants in the patatin-like phospholipase domain-containing 2 (PNPLA2) gene. More recently, pathogenic biallelic variants in the human coenzyme A synthase gene (COASY) have been identified in Chinese LSM patients with previously unknown genetic defects. Although rare, FAD-related LSM caused by defects in riboflavin transport or FAD synthesis has also been reported.
MADD, also known as glutaric aciduria type II, is characterized by dysmetabolism of fatty acids and amino acids. It presents with heterogeneous clinical phenotypes, including neonatal onset forms with (type I) or without (type II) congenital anomalies and a late-onset form (type III) that presents as LSM and occasionally accompanied by encephalopathy. Most patients with the late-onset myopathic form of MADD respond dramatically to riboflavin treatment (RR-MADD), as riboflavin supplementation may stabilize variant ETF-QO protein by restoring FAD homeostasis. To date, more than 800 cases of RR-MADD have been reported worldwide.
Since the PNPLA2 gene mutation was identified as the causative gene of NLSDM in 2007, nearly 90 cases have been reported. Phenotypes of NLSDM include asymptomatic hyperCKemia, pure skeletal myopathy, pure cardiomyopathy, and combined skeletal myopathy and cardiomyopathy. The distinct pathological hallmark for diagnosis is muscle fibers with excessive lipid droplets and rimmed vacuole formation. Currently, NLSDM is untreatable.
The COASY gene encodes a bifunctional enzyme containing 4’-phosphopantetheine adenyltransferase (PPAT) and dephospho-CoA kinase (DPCK) domains, which catalyze the final steps of de novo CoA biosynthesis. Biallelic COASY variants have been associated with severe neurodegenerative diseases. Interestingly, COASY variants were recently found to be a novel genetic cause of LSM in 16 Chinese patients, clinically mimicking RR-MADD.
LSM is the most common pathological phenotype of inherited lipid metabolic disorders. Given the heterogeneity of clinical presentations, LSM and other lipid metabolism defects are likely underdiagnosed. Supplementations of riboflavin, CoQ10, and carnitine, as well as low-fat diets, have demonstrated positive clinical effects for the majority of LSM cases. Early diagnosis through newborn screening combining MS/MS with genetic testing could help keep patients in the preclinical stage, preventing disease onset.
SS.11.03
SS.11.03 - Clinical Presentation and Investigation of Skeletal Muscle Channelopathies
1St George's University of London, United Kingdom
Skeletal muscle channelopathies include the periodic paralyses and non-dystrophic myotonias. They are rare genetic neuromuscular disorders that cause intermittent impairment of movement with generally normal muscle strength in between. They can be very challenging to diagnose. Genetic tests are diagnostic but suspicion of the disorder is first aroused by the history. Other investigations including blood tests, neurophysiology and MRI can have a supportive role or be useful in excluding differential diagnoses. Accurate genetic diagnosis is important for management, prognostication and family planning.
Securing a diagnosis can be very rewarding. Symptoms or paralysis and myotonia are not only disabling but can be frightening and embarrassing. Uncontrolled episodes can impact on education, socialisation, vocational opportunities, or independence. Many symptomatic treatments are available that often have transformative outcomes. Pain and fatigue can be under-recognised and more complex to manage. Educating patients and caregivers alongside teaching staff or employers is additionally beneficial to aid self-management as well as prevent assumptions and misunderstanding regarding perceived poor attendance or participation in physical activities. New and severe infantile presentations are also recognised that call for antenatal recommendations.
This talk will focus on the clinical presentations of muscle channelopathies. We will review the clinical utility of different investigations and how they can contribute to diagnosis.
SS.11.04
SS.11.04 - Treatment
1UT Southwestern Medical Center, United States
Muscle channelopathies are rare skeletal muscle ion disorders with marked phenotypic and genotypic heterogeneity. They are caused by mutations in genes encoding sodium channel (SCN4A), chloride channel (CLCN1), calcium channel (CACNA1S) or potassium channel (KCNJ2 & KCNJ18). Characteristic features include the episodic and fluctuating nature of symptoms, exacerbation by environmental factors, and frequently autosomal dominant inheritance. Symptoms start in early years, are lifelong, and affect quality of life. The phenotypic and genetic heterogeneity of these channelopathies present a challenge in diagnosis and management. For instance, SCN4A mutations can present as paramyotonia congenita, sodium channel myotonia, hyperkalemic periodic paralysis, or hypokalemic periodic paralysis. In contrast, non-dystrophic myotonia can occur due to mutations in the SCN4A or CLCN1 ion channels. Despite the rarity and heterogeneity of muscle channelopathies, there have been significant advances in the understanding of these disorders leading to expanded treatment options based on randomized clinical trials. However, more prospective studies are needed and should include long-term follow-up efficacy studies. Patients benefit from a dedicated multi-disciplinary approach in managing their condition. Lifestyle changes, dietary modifications, recognition and avoidance of triggers, and genetic counseling are important in managing the care of these patients.
SS.12.02
SS.12.02 - The Diagnosis and Differential Diagnosis of Ultra-Rare Fetal Akinesia Syndromes and Congenital Myopathies
1University Of Western Australia, Australia
Fetal akinesia is a broad term used to describe absent (or reduced, fetal hypokinesia) fetal movements, it can occur and be detected as early as the first trimester. Depending on the developmental age of onset of fetal hypokinesia or akinesia, a range of features can occur including muscle hypotrophy (or amyoplasia), contractures, pterygia, facial anomalies including micrognathia, pulmonary hypoplasia, diaphragmatic defects, short gut, fetal hydrops, and in utero growth restriction; pregnancy can also be complicated by polyhydramnios. Depending on the defining features at presentation, these cases can be diagnosed with arthrogryposis multiplex congenita (joint contractures in two or more body parts), distal arthrogryposis, fetal akinesia deformation sequence (FADS), lethal congenital contracture syndrome and multiple pterygium syndrome. The prevalence of arthrogryposis multiplex congenita is estimated at 1:3,000-5,000, with FADS representing a rare subset of these cases and a prevalence of 1:13,000. Variants in more than >400 genes are known to cause AMC, and it is increasing recognised that variants in genes encoding critical components of the neuromuscular system underlie a substantial proportion of fetal akinesia presentations. With unbiased screening approaches, including sequencing of comprehensive disease gene panels, exomes and genomes, novel genes and phenotypic expansions associated with known human disease genes have been uncovered in the setting of fetal akinesia. Autosomal-recessive titinopathy is the most frequent genetic cause of fetal akinesia. Variants in other neuromuscular genes are also frequently causative in the setting of fetal akinesia, including: BICD2, CHRNG, ECEL1, MAGEL2, NEB, RYR1 and TNNI2. Given the prevalence of recessive disorders within this cohort, a timely genetic diagnosis is critical to inform family planning. Around 50% remain undiagnosed following comprehensive diagnostic or research screening, thus there are likely many novel causative genes still to identify.
SS.12.03
SS.12.03 - The Diagnosis and Differential Diagnosis of Ultra-Rare Congenital Muscular Dystrophies
1University College London, United Kingdom
The classical congenital muscular dystrophies (CMD) are a group of relatively rare conditions characterized by early onset of muscle weakness, typically within the first 6 months of life, elevated creatine kinase and with dystrophic abnormalities on muscle biopsy. The major class of proteins responsible for CMD are those localized in the extracellular matrix. Indeed the most common variants (Ullrich CMD- due to collagen VI deficiency; LAMA2-related CMD, due to mutations in laminin alpha 2; and the long list of dystroglycanopathies, secondary to mutations in proteins involved in the glycosylation of alpha-dystroglycan) represent the most common CMD variants. Other less commonly involved proteins are localized in the nuclear envelope, or are implicated in regulation of oxidative stress.
In the last decade, the availability of next generation sequencing techniques has allowed to broaden the horizons related to genes very rarely involved in CMD, and / or in conditions with substantial clinic-pathological overlap with CMD. These genes encode for proteins involved in mitochondrial membrane stabilization, transcription regulation of developmental genes, in Golgi trafficking or are involve in specific metabolic functions in muscle- and often in brain.
In my presentation I will present the recently identified CMD genes that expand both the clinical spectrum of these conditions, and the understanding of the pathophysiology of muscle function.
SS.12.04
SS.12.04 - The Diagnosis and Differential Diagnosis of Ultra-Rare Limb Girdle Muscular Dystrophies
1John Walton Muscular Dystrophy Research Centre, Newcastle University, Newcastle upon Tyne, United Kingdom
At the time of abstract submission there are 34 different forms of limb girdle muscular dystrophy (LGMD) listed in OMIM, 29 autosomal recessive forms (LGMD R) and five autosomal dominant forms (LGMD D). More than 90% of patients with LGMD are affected by LGMD R and pathogenic variants in six genes are responsible for >50% of the diagnoses, with CAPN3 (LGMD R1) and DYSF (LGMD R2) being the two most common genes responsible for LGMD worldwide. The regional prevalence of specific sub-types of LGMD does also depend on founder mutations, and although LGMD R9 caused by pathogenic variants in FKRP is an ultra-rare form of LGMD in countries without Scandinavian ancestry, it is the most common form of LGMD in Scandinavia and regions with a strong Scandinavian ethnic background.
The number of newly identified genes responsible for LGMD has increased considerably with the introduction of massive parallel sequencing technologies and the establishment of large diagnostic consortia and networks that analysed huge sequencing datasets of patients with limb girdle weakness of unknown origin. The talk will focus on some of the ultra-rare forms of LGMD that nevertheless provide important insight into pathomechanisms leading to progressive limb girdle muscle weakness. Some of those diseases may only ever be diagnosed through the application of gene panels or whole exome/genome sequencing, whereas in others, investigations like muscle imaging and muscle biopsy analysis may give a clue of the underlying genetic diagnosis. For some of the ultra-rare forms of LGMD there are important treatment implications, including cardio-protective treatment, and it is therefore important to make sure that no one with an ultra-rare genetic disease is left behind by not being diagnosed.
SS.13.04
SS.13.04 - Risdiplam Treatment of SMA and Electrophysiological Response to Early and Late Treatment
1Sydney Children's Hospital, Australia
Risdiplam, an oral survival of motor neuron 2 (SMN2) pre-mRNA splicing modifier, is an approved treatment of spinal muscular atrophy (SMA). An overview of the evidence supporting its use across a wide range of severities will be presented, with SMN2 copy number, age, and disease severity at the time of treatment guiding prognostication of outcomes for individuals. In combination with SMN restoration, non SMN targets have been identified as a priority for active research, highlighting the need of biomarkers for improved disease monitoring to detect and measure treatment modified characteristics. In this presentation a range of neurophysiological measures and their contributions to assessing disease onset and progression and treatment response will be discussed, highlighting potential utility in diagnosis, prognosis and prediction of treatment response.
SS.14.02
SS.14.02 - Time to Event Trial Designs in ALS: How Useful Are They?
1University Medical Center Utrecht, Netherlands
Survival is a key clinical endpoint in Amyotrophic Lateral Sclerosis (ALS) clinical trials. Current regulatory guidelines require assessing survival time to characterize efficacy in pivotal settings. Measuring survival time, however, necessitates lengthy and large placebo-controlled trials, which are challenging both operationally and ethically due to the relatively low incidence of ALS and the grim prognosis of patients.
Fortunately, survival time, like other time-to-event outcomes, benefits from a favorable characteristic: statistical power to detect a treatment response is driven by the number of events rather than the number of patients. This can be leveraged to enhance clinical trial design. In this talk, we will introduce the event-driven or information-based design, demonstrating how better use of time can significantly improve the power of time-to-event outcomes.
A key consideration in the event-driven design is extending randomized follow-up for early-enrolled patients. While this increases statistical power, it also introduces uncertainty about the trial's duration and may result in extended placebo exposure for early enrolled patients. We will discuss mitigating these limitations through interim analyses, a hybrid approach with a maximum follow-up period for early-enrolled patients, or alternative event definitions. The event-driven design will be illustrated with a case study in ALS.
SS.14.03
SS.14.03 - The Utility of Neurotrophin P75 in ALS
1College of Medicine & Public Health/ Flinders Health & Medical Research Institute, Adelaide, SA, Aus, Adelaide, Bedford Park, Australia, 2Wicking Dementia Research Education Centre, University of Tasmania, Hobart, Australia, 3Flinders Medical Centre, and MND SA Clinic, Adelaide, Bedford Park, Australia, 4University of Miami Miller School of Medicine, Miami, USA, 5Queensland Brain Institute, University Of Queensland, Brisbane, Australia
Motor neuron disease (MND)/ amyotrophic lateral sclerrosis (ALS)/ motor is terminal within two to five years, affects more than 330,000 people globally, with 90% of cases arising sporadically¹. More than 60 Phase III clinical trials, costing billions of dollars, have failed partly due to heterogeneity in the patient population¹-³. Benchmarks such as validated biomarkers are needed to reduce heterogeneity by selecting those most likely to respond to treatment(s), and test efficacy, especially in Phase III trials4,2,5. Recently, serum neurofilament light (NfL) has shown potential as an MND biomarker6. However, to reduce heterogeneity more than one biomarker is needed, as NfL describes axonal loss in MND, but not other pathological processes such as motor neuron death, excitotoxicity, defects in RNA/DNA processing. mitochondria and immune dysfunction.
Our team has shown the common neurotrophin receptor (p75) is up regulated in MND/ALS and associated with motor neuron cell death7. The extracellular domain of p75 (p75ECD) is known to be cleaved from motor neurons as part of the apoptotic process and we found p75ECD up-regulated in the urine of MND/ALS patients8_10 and mice that model MND/ALS (SOD1G93A and TDP-43 rNLS8), when compared to healthy controls. We developed a sandwich enzyme-linked immunoassay (sELISA)10_11 for measuring urinary p75ECD and in observational studies showed urinary p75ECD as a biofluid progression biomarker of MND/ALS correlated to the revised ALS Functional Rating Scale 9_10. Urinary p75ECD has been used as an exploratory biomarker in a number of Phase I/II and II/III clinical trials, that include Monepantil, IC14, CNM-Au8, Fasudil, Tecfiderra and Triumeq.
Although we have promising data about urinary p75ECD further studies are needed to confirm urinary p75ECD usefulness for clinical trials of ALS/MND, and develop more sensitive assays to measure serum p75ECD. In an ongoing validation study being performed as part of the Clinical Research in ALS and Related Disorders for Therapeutic Development (CReATe) consortium, some 203 people with MND are being tested for urinary p75ECD, and results will be reported in this talk. To improve sensitivity and allow for serum p75ECD measurement we have been establishing a sensitive Single Molecule Array (SIMOA™) platform, using the same antibodies to p75ECD as we used for the sELISA. The p75ECD SIMOA assay has a limit of detection of 0.05pg/ml, compared to 25pg/ml for the sELISA. Ongoing work on serum p75ECD in people with MND/ALS and controls will be presented. In summary, urinary p75ECD shows utility as an MND/ALS biomarker, with more sensitive assays having the potential to further establish p75ECD measurement across biofluids.
1. Kiernan, et al. Nat Rev Neurol 2021;17:104-118.
2. Goyal, et al. Muscle Nerve 2020;62:156-166.
3. Shefner, et al. JAMA Neurol 2022;79:1312-1318.
4. Bendotti, et al. Amyotroph Lateral Scler Frontotemporal Degener 2020;21:485-495.
5. Benatar, et al. Muscle & Nerve 2016;53:169-182.
6. Benatar, et al. Ann Neurol 2024;95:211-216.
7. Smith, et al. J Comp Neurol 2015;523:1664-1682.
8. Shepheard, et al. Plos One 2014;9:e87398.
9. Shepheard, et al. Neurology 2017;88:1137-1143.
10. Shepheard, et al. Urinary neopterin: Eur J Neurol 2022;29:990-999.
SS.15.02
SS.15.02 - Biomarkers of Respiratory Dysfunction in ALS/MND: Role in Management and Clinical Trials
1Royal Prince Alfred Hospital, Australia
Chronic respiratory insufficiency in motor neurone disease (MND) is a major adverse prognostic factor and cause of morbidity and mortality, necessitating early diagnosis and intervention. Non-invasive ventilation (NIV) has been shown to both prolong and improve quality of life¹. However, the definition of chronic respiratory insufficiency in this population varies significantly, causing uncertainty about when NIV should commence. In addition, acceptance and adherence to NIV is multi-faceted and in itself is neither a simple nor objective measure of respiratory insufficiency. Respiratory assessment must also address suitability of other respiratory therapies such as cough augmentation, using careful correlation of symptoms and objective results.
The difficulty in defining this end-point is unsurprising in a disease where there is significant heterogeneity, not only in the clinical phenotype but in the genetics, pathophysiology and molecular mechanisms that have been ascribed to the condition². The fact that multiple assessments are used to collectively evaluate chronic respiratory insufficiency – symptoms, nocturnal monitoring, lung volumes, respiratory muscle strength and blood sampling - highlights that there is no one superior or conclusive test. There can also be dissent between tests, with abnormalities identified in only some tests. Adding difficulty, the validity of these measures are influenced by the disease itself, for example inability to form a lip seal with bulbar dysfunction, or to follow directions for volitional tests, or accurately report symptoms when fronto-temporal dementia co-exists. Retrospective assessments have shown that although guidelines for NIV initiation, comprising one symptom and one physiological parameter, were followed in the vast majority of cases (91%) in a well-established French MND centre, daytime hypercapnia was found in the majority (58%) of patients. In addition 10% were started in the context of acute respiratory distress³. This suggests late initiation of therapy, despite regular monitoring and guidelines being followed.
This review will highlight the current biomarkers used for routine monitoring and argue that the diagnosis of respiratory insufficiency should be assumed unless agreement in all domains of testing rule it out. This is because no test has enough negative predictive value alone and the risk of a late diagnosis may impact the acceptance and tolerance of therapy, not to mention quality or quantity of life. In addition, treating NIV commencement as a clinical step triggered by many permutations of clinical markers rather than an end-point or exclusion in clinical trials may reduce concern within the MND community about commencement of NIV before distressing late-stage symptoms are present.
1. Bourke SC et al. Effects of non-invasive ventilation on survival and quality of life in patients with amyotrophic lateral sclerosis: a randomised controlled trial. Lancet Neurol. Feb 2006;5(2):140-7. doi:10.1016/S1474-4422(05)70326-4
2. Tzeplaeff L et al. Unraveling the Heterogeneity of ALS-A Call to Redefine Patient Stratification for Better Outcomes in Clinical Trials. Cells. Mar 5 2024;13(5)doi:10.3390/cells13050452
3. Georges M et al. Initiation of non-invasive ventilation in amyotrophic lateral sclerosis and clinical practice guidelines: Single-centre, retrospective, descriptive study in a national reference centre. Amyotrophic Lateral Sclerosis and Frontotemporal Degeneration. 2017/01/02 2017;18(1-2):46-52. doi:10.1080/21678421.2016.1236817
SS.16.02
SS.16.02 - The Role of TDP-43 in ALS
1Nuffield Department Of Clinical Neurosciences, University Of Oxford, United Kingdom
Despite significant clinical and genetic heterogeneity, 97% of patients with amyotrophic lateral sclerosis have evidence of TDP-43 cytoplasmic aggregation and nuclear depletion in affected neurons at autopsy. The presence of genetic variants in TARDBP in a minority of ALS patients suggests a direct mechanistic relationship between altered TDP-43 function and ALS. TDP-43 has multiple functions, and it is not completely established which of these is critical to motor neuron integrity and most important for ALS pathogenesis. Alterations in the best characterised role of TDP-43 in splicing regulation leads to cryptic splice products which can be detected as potential biomarkers. TDP-43 is also a key regulator of stress granule assembly and is also present in axons. This indicates a broader role in RNA regulation in neurons beyond splicing regulation. The role of TDP-43 ‘aggregates’ in pathogenesis remain controversial, with much evidence suggesting that aggregation is downstream of pathways responsible for disease initiation. Although crude measurement of TDP-43 in blood or CSF is not a useful biomarker, recent work suggests that TDP-43 present in extracellular vesicles may be more specific. Finally, TDP-43 has been proposed as a ‘prion-like’ protein which could undergo aberrant phase transitions which promote aggregation and potential transcellular spread as a method of disease propagation.
SS.16.03
SS.16.03 - The Role of Stathmin-2 in ALS
1Murdoch University / Perron Institute, Australia, 2University of Notre Dame/Perron Institute, Australia
Stathmin-2 has become a focus of ALS research in recent years with the discovery of its dysregulation in sporadic ALS (sALS). A hallmark pathological feature in almost all sALS cases, as well as in a proportion of frontotemporal dementia and Alzheimer’s Disease cases, is the cytoplasmic mislocalisation and aggregation of TAR DNA-binding protein 43 (TDP-43). TDP-43 directly regulates Stathmin-2 expression by binding to its pre-mRNA to maintain normal splicing, with TDP-43 mislocalisation resulting in a reduction in Stathmin-2 expression.
Stathmin-2 is a microtubule-associated protein that is highly expressed in the nervous system. It is required for axon outgrowth and maintenance and is also involved in neuronal intracellular trafficking and neuroendocrine secretion. The first clinical trial testing an antisense oligonucleotide therapeutic to restore normal splicing and expression of Stathmin-2 in sALS patients is ongoing.
This presentation will highlight Stathmin-2 in the context of ALS research, including the key discovery of its involvement in ALS, its physiological roles in maintaining neuronal health, and its relevance to neurodegenerative disease. Genomic, cellular, and animal model findings will be presented, as well as the potential for Stathmin-2 to be a diagnostic and prognostic biomarker. In addition, evidence continues to accumulate indicating Stathmin-2 is a key therapeutic target for ALS. Finally, alternative therapeutic approaches that may also affect Stathmin-2 expression and function will be discussed.
SS.16.04
SS.16.04 - What Can the Motor Cortex Proteome Tell Us About ALS
1The University of Queensland, Australia
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease, defined by the presence of muscle weakness and progressive death of upper and lower motor neurones. ALS is thought to arise from a combination of genetic susceptibility and environmental exposure, through a multi-stage process. Multiple pathological processes have been implicated in the pathogenesis of ALS, including excitotoxicity, oxidative stress and aggregation of misfolded proteins within cells, along with deficiencies in dealing with misfolded proteins. There is substantial evidence that pathological changes in ALS are associated with changes in mitochondria. l morphology, bioenergetics and calcium homeostasis. There is also a role for neuro-inflammation with microglial activation, and a role for the adaptive immune system, with evidence of immune cell infiltrate and systemic inflammation. However, the ultimate mechanism of cell death is uncertain. The special vulnerability of motor neurons is poorly understood, but could be related to their size and high metabolic needs. More information is required about the cause of ALS and this requires study of well-documented human samples.
Mass spectrometry (MS) studies of the proteome offer the potential to identify biomarkers and provide new insights into perturbation of molecular pathways associated with ALS. The expression of proteins is the end-result of upstream variability in gene expression, effects of the environment through epigenetics, and genetic variability. There have been previous attempts to characterize the proteome of muscle and spinal cord of ALS patients.
Only one previous study investigated ALS protein profiles in human brain. That study used tissue from the frontal cortex of patients with frontal temporal dementia (FTD), ALS and ALS-FTD to characterize the genetic mechanisms underlying the ALS-FTD disease spectrum. However, in ALS, disease pathology primarily affects the motor neurones of the motor cortex and spinal cord.
We used a non-targeted quantitation MS approach called Sequential We used Sequential Window Acquisition of All Theoretical Mass Spectra (SWATH-MS) to characterize the proteomes of the motor cortex from ALS cases (n = 8) and control subjects (n = 8). A total of 1427 proteins were identified at a critical local false discovery rate < 5%; 187 of these exhibited significant expression differences between ALS cases and controls. Of these, 91 proteins were significantly upregulated and 96 proteins were significantly downregulated. Bioinformatics analysis revealed that these proteins are involved in molecular transport, protein trafficking, free radical scavenging, lipid metabolism, cell death and survival, nucleic acid metabolism, inflammatory response or amino acid metabolism and carbohydrate metabolism. Differentially expressed proteins were subjected to pathway analysis. This revealed abnormalities in pathways involving mitochondrial function, sirtuin signaling, oxidative phosphorylation, glycolysis, phagosome maturation, SNARE signaling, redox regulation and several others. Core analysis revealed mitochondrial dysfunction to be the top canonical pathway. The top-enriched networks involved JNK activation and inhibition of AKT signaling, suggesting that disruption of these signaling pathways could lead to demise of motor neurons in the ALS motor cortex.
SS.17.03
SS.17.03 - STING: Novel Inflammatory Pathway in ALS
1The Florey Institute Of Neuroscience And Mental Health, Australia
Cytoplasmic inclusions of aggregated TDP-43 are a disease hallmark in almost all patients with Amyotrophic Lateral Sclerosis (ALS). These protein aggregates are associated with inflammatory responses that play a key role in accelerating the progression of disease. Our previous work uncovered how these harmful responses are triggered due to aberrant build-up of TDP-43 (Yu et al. Cell 2020). This finding led to a shift in thinking internationally on how TDP-43 (1) disrupts mitochondria, (2) causes its DNA (mtDNA) release into the cytoplasm, (3) activates cGAS/STING inflammatory responses (i.e. type I interferons), leading to eventual degeneration of motor neurons. Excitingly, we found that the blockade of STING reduced neurodegeneration in a mouse model of mutant TDP-43, and even after symptoms were established. This revolutionary discovery was the feature of three editorials, including The New England Journal of Medicine, highlighting the importance of the innate immune system in ALS. Leveraging these newly established frameworks, we suggest that the cGAS/STING pathway is a critical pathomechanistic determinant that contributes to the propagation of TDP-43 pathology and activation of neurotoxic cascades. However, detailed cellular and molecular events underpinning cGAS/STING-mediated neurodegeneration due to TDP-43 abnormality are urgently needed prior to considerations about interference of this pathway in patients living with ALS.
To follow up, we showed that genetic deletion of the type I IFNα/β receptor subunit 1 (Ifnar1) mitigates motor deterioration and neurodegeneration in a transgenic ALS mouse model expressing mutant TDP-43 (p.A315T). The TDP-43-associated gliosis and peripheral monocyte infiltration can be also prevented. Surprisingly, in situ RNAscope and FACS analyses indicated that neuronal cells can also produce type I IFNs in addition to microglia in the brain. For this reason, we further hypothesised that these neuroinflammation events may be the secondary effect and whether neurodegeneration can occur within neurones. Indeed, we observed that TDP-43-induced IFN-I signalling correlated with exacerbated LDH release, axonal mitochondrial accumulation, and tubulin polyglutamylation in our SH-SY5Y models, as well as in iPSC-derived motor neurones from TDP-43-ALS patients. Importantly, this was associated with activation of the sterile alpha and TIR motif containing protein 1 (SARM1). These cell-autonomous degenerative cascades can be protected when IFNAR1 was deleted via CRISPR/Cas9 technology or using FDA-approved JAK1 small molecule inhibitors.
Emerging preclinical data support that cGAS-STING pathway is a compelling target for neurodegenerative diseases, including ALS. We now know that STING inflammation can trigger SARM1 activation, and thereby axonal degeneration in vitro and in vivo. Inhibiting axonal degeneration is key for neuroprotection. These findings will be foundational to advance our candidate therapeutics as a combination therapy with other approaches targeting TDP-43 pathology into new clinical trials.

SS.18.02
SS.18.02 - Tipping the Scales on MND
1The University of Queensland, Australia
Motor Neurone Disease (MND) is a complex neurodegenerative condition primarily characterised by motor deficits, leading to muscle weakness and paralysis. However, beyond these physical impairments, appetite loss has increasingly been recognised as a major contributor to early and rapid weight loss, which is closely linked to disease progression.
Research suggests that appetite loss may play a central role in this weight decline, driven by changes in peripheral and central pathways that regulate hunger and satiety. The complexity of appetite loss in MND is further compounded by the heterogeneity of the disease, presenting challenges in addressing weight loss across different patient groups. Metabolic dysfunction, impaired appetite signalling, and psychosocial factors all contribute to this issue, making it difficult to develop uniform strategies for disease management. As appetite dysregulation becomes more severe, it often leads to malnutrition, which can further accelerate the decline in physical function. This highlights the importance of personalised approaches to nutritional support in people with MND, as a single solution may not effectively address the diverse ways in which the disease impacts appetite and metabolism.
By understanding the mechanisms driving appetite loss and weight reduction, we can develop more targeted interventions aimed at slowing disease progression and improving the quality of life for those living with MND.
SS.18.03
SS.18.03 - The Role of Lipidomics in ALS: Pathogenic and Therapeutic Implications
1The Florey Institute Of Neuroscience And Mental Health, Australia
Increasing evidence suggests that a major factor shared between most ALS patients is disturbed metabolism. Several studies have found hypermetabolism in ALS patients, independent of their genetic status. Metabolic disturbances are also observed in mouse models of ALS. In the SOD1 mouse, researchers have shown increased glucose clearance/uptake, hypermetabolism and increased lipid clearance. With the development of omics studies such as metabolomics and lipidomics, more details have been discovered about the extent of metabolic and lipid dysregulation in ALS. Lipidomic analysis has been vastly evolving in the last decade. Where more lipids are becoming annotated and linked to cellular functions. Several lipidomic studies on ALS patient samples, blood and preclinical mouse data have been performed. The first lipidomic papers on ALS were in 2017, where Henriques and colleagues profiled the lipidomic profile of SOD1G86R mice and Blasco and colleagues profiled the cerebrospinal fluid of ALS patients. In the preclinical models, there was a significant change in sphingolipid profile, which was found to be druggable with promising results in preclinical mouse models. Recently, we have described extensive lipid restructuring in the spinal cord and muscle of symptomatic TDP-43Q331K mice. We then targeted these lipid changes with a glucocerebrosidase A2 inhibitor ambroxol. This lipid targeting resulted in enrichment of gangliosides around the neuromuscular junction, which we believe is preventing neuromuscular deterioration and thereby preventing disease progression in these mice. Furthermore, we recently discovered a unique signature of lipids in ALS. Here, we utilised lipidomic profiling to uncover blood-based signatures in mouse models of ALS as well as humans. We found that lipids significantly change over the disease course in murine C9, TDP and SOD models of ALS. We utilised this murine dataset as a diagnostic tool in a human dataset, which consisted of MND patients, controls and Parkinson’s disease (PD) patients. Due to the heterogeneity of MND, we decided to test lipids in sets of 3 lipids from distinct subclasses. We discovered a total of 160 lipids across the murine dataset that were significantly altered. This led to a total of 438,480 combinations to be tested in the human lipid dataset. Filtering of data was performed, and a total of 717 combinations remained. Next, we excluded combinations that performed well in control vs PD, leading to a reduction in the total number of combinations to 159. From here, the last filter of a positive correlation with disease outcome (higher odds of subject having MND) was used. A total of 18 combinations of lipids remained in our final dataset of MND specific biomarkers.
Together, this cumulative evidence suggests lipids play a pivotal role in ALS pathophysiology.
SS.18.04
SS.18.04 - Deciphering and Targeting Metabolic Dysfunction in ALS
1The University Of Queensland, Australia
Amyotrophic lateral sclerosis (ALS) is a complex neurodegenerative disease that is charactersed by the death of motor neurons in the brain and spinal cord. The progressive loss of these neurons leads to muscle wasting, paralysis and eventually death. Patients with ALS have a typical prognosis of 2-5 years after diagnosis.
The mechanisms that underpin the onset and progression of ALS remain under intense investigation. Nevertheless, the current consensus is that a combination of genetic and environmental risk factors contribute. In addition to genetics and environmental triggers, there is now significant evidence to show that alterations in energy balance can impact upon disease progression and prognosis. Indeed, studies reporting dysregulation in whole-body, tissue-specific, and cellular regulators of energy homeostasis indicate that these factors are key in the pathogenesis of ALS.
In the last two decades, considerable inroads have been made into understanding the impact of hypermetabolism on disease outcome, and how the targeting of pathways that lead to localised, and potentially contribute to whole-body, dysregulated metabolic homeostasis in ALS might be viable therapeutic strategies. In this presentation, I will discuss our studies in people living with ALS and in in vivo and in vitro models of the disease, and provide a summary of our approach to make sense of energy imbalance in ALS.
SS.19.02
SS.19.02 - Updates on ALS Diagnostic Criteria
1Neuroscience Research Australia, Australia
Amyotrophic lateral sclerosis (ALS) reflets involvement of both compartments of the human nervous system, spanning across the upper (UMN) and lower motor neuronal (LMN) systems. Without a diagnostic test, consensus criteria have developed, in part to promote patient recruitment for clinical trials. Previous consensus criteria developed algorithms that incorporated levels of diagnostic certainty from suspected, through to possible, probable and definite disease. To streamline the diagnosis of ALS, the World Federation of Neurology combined with key patient and clinical stakeholders at a consensus meeting on the Gold Coast, Australia in 2019. A collective understanding of ALS was established based around key tenets: that ALS represented a progressive disorder of the motor system; that while UMN signs were not always clinically evident, involvement of the LMN was more often apparent through clinical examination. In terms of diagnostic technologies, supportive evidence of LMN dysfunction has tended to be derived from electromyography and neuromuscular ultrasound. Supportive evidence of UMN dysfunction was more limited, relying on transcranial magnetic stimulation studies of the central motor nervous system, MRI, and neurofilament levels, although it was accepted that a diagnosis of ALS does not require these investigations.
Consensus was reached, whereby ALS was defined by the presence of:
1. Progressive motor impairment, documented by history or repeated clinical assessment, preceded by normal motor function.
2. Upper and lower motor neurone dysfunction in at least one body region (in the same body region if only one body region was involved), or lower motor neuron dysfunction in at least two body regions.
3. Investigation findings that excluded alternative disease processes.
Recent international cohort studies have validated and confirmed the clinical utility of the Gold Coast criteria.
SS.19.03
SS.19.03 - Diagnostic Utility of TMS in ALS
1University of Sydney, Australia
ALS is a progressive motor system disorder clinically diagnosed by the combination of upper motor neuron [UMN] and lower motor neuron [LMN] signs in a body region in the exclusion of mimic disorders. Clinical phenotype in ALS is variable and objective measures of UMN dysfunction would aid the reliable diagnosis of ALS.
Transcranial magnetic stimulation (TMS) is a neurophysiological technique that allows non-invasive and painless assessment of cortical function. Changes in many TMS measures have been described in ALS though the measure of cortical hyperexcitability, is the most consistent cortical change reported in ALS.
Threshold tracking TMS techniques of paired-pulse cortical stimulation, measure changes in motor evoked potential (MEP) threshold rather than amplitude and allow a reliable and rapid assessment of cortical function in ALS.
This presentation discusses the evidence for cortical hyperexcitability being an early feature in sporadic ALS preceding the onset of LMN dysfunction and possibly contributing to disease spread. It discusses the evidence for cortical hyperexcitability preceding the clinical onset of familial ALS. Further, the diagnostic utility of cortical hyperexcitability in ALS is discussed along with evidence of its evolution with clinical disease advancement and the relevance of cortical inexcitability.
In summary, this presentation discusses the evidence provided by TMS for cortical involvement at the earliest detectable stages of ALS, the utility of the technique for probing underlying pathophysiology and future directions for assessing cortical dysfunction in ALS.
SS.19.04
SS.19.04 - Utility of Muscle Ultrasound in ALS
1Brain And Nerve Research Center, Concord Clinical School, University Of Sydney, Australia
Amyotrophic Lateral Sclerosis (ALS) diagnosis is challenging due to its heterogeneity. Early diagnosis is crucial for patient comprehension, care access, and clinical trial participation. ALS is a clinical diagnosis, lacking a single diagnostic test, and relies on the demonstration of concurrent upper motor neuron (UMN) and lower motor neuron (LMN) dysfunction with disease progression.
Fasciculations, an early clinical sign and neurophysiological biomarker of LMN injury, are detectable in clinically strong muscles. Muscle ultrasound offers a more sensitive means of fasciculation detection than electromyography, particularly in bulbar muscles[1]. The presence of fasciculations on ultrasound in at least two muscles has been shown to be highly sensitive for the diagnosis of ALS[2]. This was confirmed by a retrospective series of 200 patients, using machine learning analysis[3]. Additionally, ultrasound detection of multifocal continuous fasciculations affecting multiple muscles was demonstrated to be highly diagnostic in a cohort of ALS and ALS mimic patients[4]. Muscle ultrasound can also detect other biomarkers of LMN dysfunction and degeneration. Increased fibro-fatty muscle replacement, for example, results in increased ultrasound echointensity, whilst muscle atrophy is measurable as reduced ultrasound muscle thickness[5]. The homogeneity of fibro-fatty replacement can also be measured using echovariation. However, diagnostic studies of these modalities are limited[6].
Ultrasound of bulbar and limb muscles was prospectively performed on all patients with suspected ALS referred to the Brain and Nerve Research Centre at Concord Hospital in Sydney Australia, between 2022 and 2023. In addition, clinical measures of disease severity and upper motor neuron (UMN) impairment were recorded. From a total of 94 patients initially suspected of ALS, 44 were diagnosed as ALS and 50 as disease mimics. ALS patients demonstrated a higher frequency and more generalised distribution of ultrasound fasciculations compared to mimics. The presence of ultrasound fasciculations in four or more muscles exhibited high diagnostic accuracy. Combining clinical biomarkers of UMN dysfunction with muscle ultrasound fasciculations increased the diagnostic utility, with an area under the curve (AUC) of 0.97 (95% CI 0.92-1.0), sensitivity 82% and specificity 94%. Abnormalities of muscle architecture, as reflected by ultrasound muscle thickness, echointensity, and echovariation, was of limited diagnostic utility in our cohort. This is not surprising given these features are non-specific, occurring in a wide variety of neuromuscular diseases.
In conclusion, muscle ultrasound improves ALS diagnosis, and is further enhanced when combined with clinical UMN signs.
1. Misawa S et al. Ultrasonographic detection of fasciculations markedly increases diagnostic sensitivity of ALS. Neurology 2011;77:1532-1537.
2. Tsuji Y et al. A muscle ultrasound score in the diagnosis of amyotrophic lateral sclerosis. Clin Neurophysiol 2017;128:1069-1074.
3. Fukushima K et al. Early diagnosis of amyotrophic lateral sclerosis based on fasciculations in muscle ultrasonography: A machine learning approach. Clin Neurophysiol 2022;140:136-144.
4. Ma J et al. Fasciculation score: a sensitive biomarker in amyotrophic lateral sclerosis. Neurol Sci 2021.
5. Pillen S et al. Skeletal muscle ultrasound: correlation between fibrous tissue and echo intensity. Ultrasound Med Biol 2009;35:443-446.
6. Arts IM et al. Muscle ultrasonography: a diagnostic tool for amyotrophic lateral sclerosis. Clin Neurophysiol 2012;123:1662-1667.
SS.21.02
SS.21.02 - The Emerging Role of Retrotransposons in Neurodegenerative Diseases
1University Of Liverpool, United Kingdom
Transposable elements (TEs) have been associated with neurological disorders as modulators of gene function, including but not limited to, changes in gene expression, alternate splicing, epigenetic modifiers and sources of cryptic exons. Through these actions, they can have pathogenic effects and have been implicated in a variety of diseases such Amyotrophic Lateral Sclerosis, Alzheimer’s and Parkinson’s disease. These TE’s are highly polymorphic within the human genome, both for presence/absence, as well as within their primary sequence. There are growing numbers of examples of mono-genic forms of disease caused by the presence of a single TE which disrupts the normal processing of a key gene. One of the most cited examples of this is a SINE-VNTR-Alu (SVA) insertion within intron 32 of the gene TAF1 which is causative of X-linked dystonia parkinsonism (XDP). In that example, the SVA element leads to partial intron 32 retention and decreased TAF1 expression leading to the disease phenotype. Our work expands the role of retrotransposons in disease, by exploring how more common elements in our genome can confer risk within complex disorders. It is likely that the majority of retrotransposons work in consort as cumulative risk factors, spread across multiple elements, which give rise to disease through increased risk over the course of our lifetime. We have demonstrated that an SVA element within the MAPT locus (which has strong links to multiple neurodegenerative diseases), termed SVA_67, is significantly associated with differential gene expression of several genes within the locus. We were able to show this using bioinformatic approaches, which utilised the Target ALS and Parkinson’s Progressive Markers Initiative (PPMI) datasets, as well as in vitro cell models of CRISPR modified SVA_67 knockout HEK293 cell lines. In my talk, I will discuss the current approaches and links between TEs and movement disorders, focusing primarily on neurodegeneration, as well as our current projects aimed at deconvoluting the pathways by which retrotransposons are linked to disease onset, progression and severity. A key finding is that the regulatory properties of a specific locus are modified by the polymorphism of the TE primary sequence, in addition to its presence or absence in our genome. The former is hypothesised to reflect differential interactions of sequence specific DNA binding proteins within the TE which can impact gene function.
SS.21.03
SS.21.03 - A Genome-Wide Screen for the Exonisation Of SVA Elements in MND
Abigail Pfaff1,
1Murdoch University, Australia
The hominid-specific retrotransposon SINE-VNTR-Alu (SVA) is a composite element that has contributed to the genetic variation between individuals and influenced genomic structure and function. SVAs are involved in modulating gene expression and splicing patterns, altering mRNA levels and sequences, and have been associated with the development of disease. We evaluated the genome-wide effects of SVAs present in the reference genome on transcript sequence and expression in the CNS of individuals with and without the neurodegenerative disorder Amyotrophic Lateral Sclerosis (ALS). This study identified SVAs in the exons of 179 known transcripts, several of which were expressed in a tissue-specific manner, as well as 92 novel exonisation events occurring in the motor cortex. An analysis of 65 reference genome SVAs polymorphic for their presence/absence in the ALS consortium cohort did not identify any elements that were significantly associated with disease status, age at onset, and survival. However, there were transcripts, such as transferrin and HLA-A, that were differentially expressed between those with or without disease, and expression levels were associated with the genotype of proximal SVAs. We have also shown that SVAs modify the course of other neurodegenerative diseases. This study demonstrates the functional consequences of several SVA elements altering mRNA splicing patterns and expression levels in tissues of the CNS. Therefore, SVAs have a remarkable impact on the pathogenesis of the MND and are significant targets for genomic-based drug development.
SS.21.04
SS.21.04 - Dissecting the Heterogeneity of ALS Using Large-Scale Genomics
1Department of Biostatistics and Health Informatics, King's College London, United Kingdom, 2Perron Institute for Neurological and Translational Science, Perth, Australia
SS.22.03
SS.22.03 - Evaluation of Lower Motor Neurons in ALS
1Teikyo University, Japan
Amyotrophic lateral sclerosis (ALS) affects both lower motor neurons (LMN) and upper motor neurons (UMN). LMN involvements can be evaluated either by clinical signs or electrophysiological examinations. Clinical signs suggesting LMN involvements include atrophy, weakness, and fasciculations. However, atrophy or weakness can occur even by UMN involvements or by myopathy. Fasciculations are considered to be more specific to LMN impairment. Contraction fasciculations that correspond to voluntary contractions of a motor unit (MU) enlarged due to reinnervation should be differentiated from true fasciculations that are spontaneous firing of an MU and are more specific to ALS although both suggest LMN involvements. Practically, however, nonspecific twitching or ordinary voluntary contractions may be often misdiagnosed as fasciculations by visual assessment.
The LMN involvement in ALS has been conventionally evaluated by needle EMG. Fibrillation potentials and positive sharp waves suggest denervation, but they are not specific to LMN impairment. They can be observed in myopathy or even in central weakness. Fasciculation potentials (FPs) are more specific to ALS. They are not observed in myopathy or central weakness, and rather rare in other neurogenic disorders than ALS or demyelinating neuropathy. Strict differentiation of FPs from voluntary motor unit potentials (MUPs) is crucial and should be done based on the firing rhythm. Regarding voluntary activities, reduced recruitment, high-amplitude and long-duration MUPs, and polyphasic and/or unstable MUPs are listed as findings suggesting LMN impairment. However, high-amplitude, and even long-duration, MUPs can be observed in chronic myopathy. Polyphasic MUPs are frequently observed in myopathy. Unstable MUPs are characteristic to ALS, but not specific. Reduced recruitment is specific to neurogenic change, but this should not be confused with “reduced interference” that may occur due to poor activation of central origin. In existing diagnostic criteria, presence of both spontaneous activities and voluntary changes are required to diagnose LMN involvement. I personally think, however, that widespread FPs are sufficiently specific to ALS and can be used as a strong evidence of ALS.
In clinical practice, signs specific to ALS or non-ALS would be useful to make a definitive diagnosis. We have reported that the decremental responses (Decr) in repetitive nerve stimulation of the trapezius muscle and spontaneous activities of needle EMG in the trapezius muscle are specific to ALS in comparison with cervical spondylotic amyotrophy. We recently reported that the absence of Decr in the trapezius and deltoid together with the absence of spontaneous activities in the trapezius muscle in a patient with upper-limb onset symptoms are sufficiently specific to non-ALS.
For a surrogate marker that can evaluate disease progression and treatment effect, repeated examinations and quantitation are mandatory. Needle EMG is not suited for this purpose. Motor unit number estimate (MUNE) and MUNIX have been proposed as such neurophysiological markers. Simple compound muscle action potential (CMAP) may be used for this purpose, but rather low reproducibility was its limitation. We reported that the far-field potential of ulnar or tibial CMAP achieved a high reproducibility and proposed as a promising surrogate marker.
SS.22.04
SS.22.04 - Assessment of Motor Neurons in SMA and SBMA: Diagnostic and Outcome Biomarkers
1University Of Malaya, Malaysia
Spinal muscular atrophy (SMA) and spinal-bulbar muscular atrophy (SBMA) are rare genetic diseases that lead to the degeneration of motor neurons. Each has a set of characteristic clinical features but can often be misdiagnosed as amyotrophic lateral sclerosis (ALS). There are numerous biomarkers that can facilitate in the diagnosis, including electrophysiological and genetic testing. The last decade has seen the development of new disease-modifying drugs in SMN gene expression, revolutionising SMA treatment. With the rise in clinical trials of potential therapeutic agents, early and accurate assessment of motor neurons become increasingly important not just in the diagnosis but also to guide treatment and monitor disease activity in SMA and SBMA.
SS.24.03
SS.24.03 - Amyloid Autonomic Neuropathy
1University Of Minnesota, United States
Amyloidosis is characterized by deposition of insoluble beta-pleated amyloid sheets within various tissues leading to progressive organ failure. Autonomic neuropathy occurs when amyloid fibrils deposit in peripheral nerves and affect small-diameter nerve fibers resulting in autonomic impairment. It typically coexists with sensory-predominant peripheral neuropathy. Autonomic neuropathy is most common in systemic light chain amyloidosis (AL) and hereditary transthyretin amyloidosis (ATTRv). The risks of developing autonomic impairment in ATTRv vary among different mutations; patients with Val30Met mutations have higher prevalence of autonomic dysfunction and have the shortest time from disease onset to the first autonomic symptom when compared to other mutations. In addition, patients with late-onset ATTRv from non-endemic areas have less frequent and milder autonomic involvement compared to early-onset endemic ATTRv. The most common autonomic symptoms in amyloidosis are orthostatic hypotension and gastrointestinal dysfunction. Orthostatic hypotension in amyloid neuropathy is attributable to impaired norepinephrine release from sympathetic postganglionic neurons but it could also be a consequence of heart failure in the setting of amyloid cardiomyopathy or volume depletion due to diarrhea and poor oral intake. Similarly, amyloid involvement of the enteric nervous system results in altered motility and defective secretory function but diarrhea and gastroparesis can also be a consequence of direct amyloid deposition in the gastrointestinal mucosa. Secretomotor symptoms (e.g. dry eyes and dry mouth) from impaired cholinergic innervation to lacrimal and salivary gland and urinary retention from pelvic parasympathetic failure can emerge later in the disease course. Evaluation of autonomic impairment in patients with amyloidosis include careful history taking for multiorgan autonomic symptoms, physical examination to assess for coexisting somatic neuropathy, and autonomic function testing to characterize the severity and distribution of autonomic impairment. Abnormal postganglionic sympathetic sudomotor impairment, reduced heart rate variation to deep breathing, and orthostatic hypotension are seen in 74%, 69%, and 36% of ATTRv patients respectively. Autonomic impairment in amyloidosis is typically widespread and severe with median total composite autonomic severity score of 7 (out of 10). The presence of autonomic neuropathy is an important prognostic feature and associate with poorer survival outcome in both AL and ATTRv. AL patients with both cardiomyopathy and autonomic neuropathy have shorter survival than those with cardiomyopathy alone. ATTRv patients with autonomic impairment consistently report worse quality of life than those without. Autologous stem cell transplantation (ASCT) is considered a mainstay in treatment of AL. AL patients with autonomic neuropathy can undergo ASCT with relative safety but there are increased risks of peritransplant atrial fibrillation and sepsis that need to be considered. Treatment of ATTRv has been revolutionized by novel gene therapy and TTR stabilizers in the past recent years. Clinical trials of these novel therapies have reported stabilization and even improvement of autonomic impairment in ATTRv patients. Symptomatic management of autonomic symptoms in amyloidosis is similar to those of other autonomic neuropathies. However, special considerations are required in patients with concurrent neurogenic orthostatic hypotension and cardiomyopathy as volume-expansion therapies (fludrocortisone and salt intake) should be minimize and higher dosage of vasoconstrictor agents may be needed.
SS.24.04
SS.24.04 - Diabetic Autonomic Neuropathies
1Mayo Clinic, United States
Diabetic neuropathy is a known complication of diabetes mellitus. Large-fiber sensorimotor, small-fiber somatic, and autonomic nerve involvement can occur in isolation or in combination when the corresponding fibers are damaged and their function impaired. Diabetic autonomic neuropathy may be present in up to 50% of cases and its risk may increase with suboptimal diabetic control and duration of disease. Autonomic neuropathy may also be a prominent feature of other forms of diabetic neuropathy including treatment induced neuropathy of diabetes (TIND) and diabetic lumbosacral radiculoplexus neuropathy (DLRPN). The symptoms of diabetic autonomic neuropathy (DAN) are numerous given the varied functionality of the autonomic nervous system manifesting as dysfunction of the sudomotor, cardiovagal, cardiovascular adrenergic, secretory, gastrointestinal (GI), and genitourinary (GU) systems. The detection of DAN relies on careful clinical evaluations (neurological and autonomic history and examination including orthostatic vital signs) and the appropriate utilization of autonomic function testing, autonomic reflex screen (ARS) and thermoregulatory sweat test (TST); and other small fiber assessments (e.g., skin punch biopsies for epidermal nerve fiber density). The ARS is a battery of standardized tests that assess multiple aspects of the autonomic nervous system. It is composed of quantitative sudomotor axon reflex test (postganglionic sympathetic cholinergic), hear rate response to deep breathing and Valsalva maneuver (cardiovagal), and blood pressure response to Valsalva maneuver and head up tilt (cardiovascular adrenergic). Together with the TST, which evaluates central and peripheral sudomotor pathways, the ARS allows for the diagnosis, pattern characterization, and severity determination of DAN. Early signs of DAN include reduction in expected HRDB and distal sweat output on QSART or TST. However, the pattern and severity of DAN are protean ranging from distal sudomotor neuropathy to generalized autonomic failure together with autonomic ganglionopathy and afferent baroreflex failure. Clinically, these may pose significant morbidity, as in the case of neurogenic orthostatic hypotension (OH) and gastroparesis, and increased mortality as in cardiac autonomic neuropathy. The management of DAN includes judicious treatment of hyperglycemia in addition to targeted symptomatic relief for OH, constipation, and urinary dysfunction for instance. Conservative and pharmacologic measures may be implemented (e.g., to support blood volume and vasoconstriction in OH and improve peristaltic motion in delayed intestinal emptying) which can improve the quality of life in patients with DAN and reduce the risk of further complications such as falls and malnutrition. Future research in DAN may help further our understanding of its pathophysiology; optimize diabetic care to reduce the risk of neuropathy; develop disease modifying and neuroprotective treatments; and expand the awareness and availability of reliable autonomic testing.
SS.25.04
SS.25.04 - Optimal use of Ultrasound in Neuropathy
1Mayo Clinic, United States
Ultrasound imaging of nerve in isolation or in conjunction with electrodiagnostic testing is a rapidly evolving field, with many potential applications. In contrast to electrodiagnosis, high-resolution ultrasound in nerve disease provides anatomic information about the nerve itself and surrounding structures. It can be used to identify many types of nerve pathology including focal entrapment, nerve transection, neuroma, nerve tumor, intraneural ganglion, and more diffuse involvement of the nerve as seen in multifocal motor neuropathy with conduction block, hereditary motor sensory neuropathy, and acute and chronic inflammatory demyelinating polyradiculoneuropathy.
Structural lesions causing focal neuropathies can be identified, such as fascial bands, anomalous muscles, hematomas, pseudoaneurysms, lipomas, fibromas, and hemangiomas. Ultrasound is useful in identifying anatomic variants such as a bifid median nerve or persistent median artery, which may have implications for surgical treatment. The dynamic aspect of ultrasound can be used to advantage in certain settings, such as diagnosing a subluxing ulnar nerve, and simultaneously evaluating for a snapping head of the medial triceps. Dynamic entrapment related to anomalous muscle or compressive structures such as fascial bands can be visualized, allowing the clinician to more confidently implicate the lesion as a cause of entrapment.
When entrapped or inflamed, nerves show changes in mobility, shape, and echotexture, likely related to intraneural edema, venous congestion, and/or fibrosis. Entrapped nerves typically show focal enlargement, although in cases of severe compression or longstanding demyelinating disease the nerve may be atrophied. At a site of focal compression, the nerve may be flattened with fusiform hypoechoic (i.e. dark) swelling just proximal, but in other situations the nerve will be swollen right at the site of entrapment.
Inflammatory hyperemia within the nerve is evident on ultrasound as increased signal on color Doppler imaging, and qualitative changes in echogenicity, hyperemia, mobility, and fascicular pattern are often present. Cross-sectional area is the most reliable measure for sonographic diagnosis of nerve pathology currently, and it can be helpful to use the patient as their own control when evaluating for focal or unilateral pathology, especially when coexisting pathology such as peripheral neuropathy or radiculopathy co-exist.
The advantages of ultrasound over electrodiagnosis include cost, noninvasive nature, and ability to identify underlying causes of entrapment neuropathies. However, in order to promote more widespread integration of neuromuscular ultrasound into the electrodiagnostic laboratory setting and in isolation as a standalone diagnostic tool in certain cases, there is a need for ongoing research.
SS.26.02
SS.26.02 - Charcot Marie Tooth Disease (CMT) Trial Readiness
1UCL Queen Square Institute of Neurology, London, UK
Most forms of Charcot Marie Tooth disease (CMT) are slowly progressive but are often life-long conditions with high morbidity and affecting quality of life. Some forms especially some of the early onset forms are severe with early loss of ambulation and with respiratory muscle involvement. There are a few ongoing clinical trials in CMT and many promising genetic therapies in advanced pre clinical development.
A major challenge in CMT is the design of clinical trials. Even though there are comprehensive CMT specific clinical scales such as the CMT neuropathy score (CMTNS) which measures severity these have been shown to lack responsiveness over the 1-2 years duration needed for a typical clinical trial. Newer functional outcome measures such as the CMT-FOM for adults have been developed but this also shows lack of responsiveness over 1 -2 years. CMTPeds which has been developed for children performs better.
Bridgeable biomarkers which are responsive over 1-2 years but which predict clinical change over a longer period are ideally needed in CMT. MRI lower limb fat fraction has proven to be the most responsive outcome measure to date in multiple forms of CMT including CMT1A in adults and children, CMT1B, CMT2A, CMT1X and HSN1. Blood biomarkers including NEFL and TMPRSS5 have also been studied in CMT.
In this talk, I will discuss the challenges of clinical trial design in CMT and provide an update on the recent clinical outcome measure and biomarker developments.
SS.26.03
SS.26.03 - Other Inherited (HSAN, HAS, HMN, dHMN, SFN..)
1ANZAC Research Institute Sydney Local Health District, Sydney, Australia, 2Faculty of Medicine and Health University of Sydney, Sydney, Australia, 3Samsung Medical Centre, Sungkyunkwan, University School of Medicine, Seoul, South Korea, 4The Royal Children's Hospital, Melbourne, Australia
The journey from genetic diagnosis to disease clinical trials forms the framework for precision medicine. In this presentation, aspects of this journey will be presented through the lens of solving genetically challenging families and the development of treatments for ultra-rare (less than 30 families affected worldwide) forms of CMT and related neuropathies.
A pre-requisite for patient clinical trials is a genetic diagnosis for families. Despite having identified over 100 genes causing CMT and related neuropathies, up to 40% of families remain genetically unsolved after excluding genome wide coding mutations. For these unsolved families, interrogating the non-coding or “dark genome” for mutations is now a high priority. An overview of multi-omics (long read sequencing and transcriptome guided gene prioritization) will be discussed in combination with the availability of a “complete” human reference genome and increased genetic diversity data for variant filtering.
When families are genetically solved, developing effective therapies requires an understanding of disease mechanisms and using this knowledge to investigate therapeutic molecules in patient pre-clinical models. We have implemented “drug re-purposing strategies” for an ultra-rare form of CMT. Our team discovered CMTX6, a form of X-linked CMT caused by point mutations in the pyruvate dehydrogenase kinase type 3 enzyme (PDK3). To date, only two additional CMTX6 families have been reported. We have screened 1840 FDA-approved bioactive compounds on a 3-phase approach that includes the use of CMTX6 derived skin fibroblasts, patient-derived iPSC motor neurons (iPSC-MNCMTX6) and an in vivo C. elegans model overexpressing the human PDK3R158H mutation. Several promising candidate drugs have been identified that may be suitable for treating this ultra-rare form of CMT.
SS.26.04
SS.26.04 - ATTRv Neuropathy
1Department of Neurology and Neurophysiology and St Vincent's Amyloidosis Centre, St Vincent's Hospital, Darlinghurst, Australia, 2Westmead Amyloidosis Centre, Westmead Hospital, Westmead, Australia, 3Brain and Mind Centre, Faculty of Medicine and Health, Sydney University, Camperdown, Australia
Hereditary transthyretin amyloidosis (ATTRv, v for variant) is a severe, adult-onset, autosomal dominant, inherited systemic disease predominantly affecting the peripheral and autonomic nervous system, heart, kidney and the eyes. ATTRv is caused by over 130 mutations in the transthyretin gene, leading to dissociation, misfolding and extracellular deposition of amyloid fibrils in multiple organs including the peripheral nervous system. Transthyretin has a physiological role as a transporter of Vitamin A and thyroid hormone and is predominantly made by the liver with smaller quantities produced by the retinal epithelium and the choroid plexus.
Typically, the neuropathy associated with ATTRv is characterised by a rapidly progressive and disabling sensorimotor axonal neuropathy with early small fibre and autonomic involvement. Carpal tunnel syndrome and cardiac dysfunction frequently coexist as part of the ATTRv phenotype. Although awareness of ATTRv polyneuropathy (ATTRv-PN) among neurologists has increased, there remains significant delays in diagnosis, resulting in accrued disability for patients.
Timely and definitive diagnosis is important given the emergence of effective therapies which have revolutionised the management of ATTR. Transthyretin protein stabilisers diflunisal, tafamidis and acoramidis, can delay the progression of the disease, if treated early in the course. Additionally, TTR gene silencing medications, patisiran and inotersen, and second-generation silencers vutrisiran and eplontersen, have resulted in up to 80% reduction in TTR production leading to stabilisation or improvement of peripheral neuropathy and cardiac dysfunction, as well as improvement in quality of life and functional outcomes. Gene editing strategies using CRISPR-Cas9 technology have demonstrated safe and persistent reduction in TTR concentrations after a single infusion in a Phase 1 study, with Phase 2/3 studies ongoing. Novel strategies, including monoclonal antibodies designed to deplete amyloid deposits, are under investigation.
Advances in these therapeutics have revolutionised outcomes for patients with ATTRv, improving quality of life and survival. However, significant unmet needs remain, including the role of combination therapies, optimal timing of treatment commencement, the potential role of treatments in delaying disease onset and therapeutic strategies in individuals not responding or progressing despite treatment. Furthermore, current management strategies do not modify occular or CNS TTR production, and as such late occurrence of the disease in these compartments is becoming more prevalent. As such, novel treatment strategies or optimised CNS and ocular penetrance of existing treatments are required to treat these emerging manifestations.
SS.27.02
SS.27.02 - Systemic vs Non-systemic Vasculitic Neuropathy
1Medical College Of Wisconsin, United States
Vasculitis is a clinicopathologic entity featuring inflammatory damage to vessels and ischemic injury to involved tissues. Systemic vasculitides affecting small vessels commonly produce neuropathies, but in some individuals, vasculitis is confined to peripheral nerves over long-term follow-up, a condition designated nonsystemic vasculitic neuropathy (NSVN). Names and definitions were updated in the 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides. Polyarteritis nodosa, eosinophilic granulomatosis and polyangiitis, microscopic polyangiitis, rheumatoid vasculitis, and cryoglobulinemic vasculitis are the systemic disorders having the strongest associations with neuropathy. In unselected series, NSVN (26%) is the most frequently encountered VN. Subtypes of NSVN include Wartenberg migratory sensory neuropathy, postsurgical inflammatory neuropathy, and diabetic radiculoplexus neuropathy (RPN). Nondiabetic NSVN with proximal involvement is sometimes termed nondiabetic lumbar RPN. A 2019 study showed the annual incidence of lumbar RPN in Minnesota to be 4.16/100,000 (2.79 diabetic/1.27 nondiabetic). This study was the first to determine the incidence of any VN and suggested that NSVN is the commonest inflammatory neuropathy. Pooling data from 57 series/case reports since 1985 yields a mean age of onset 60.0±14.8 years. Males and females are equally affected. Tempo of the neuropathy is acute (≤ 1 month) in 17%, subacute (1-3 months) in 11%, and chronic in 72%. While most patients exhibit a stepwise course, 40-45% slowly progress from onset. Patients usually develop motor and sensory deficits, but ∼15% have purely sensory signs/symptoms. Pure motor or autonomic presentations are rare. Most NSVNs are painful (80%). Three patterns of clinical involvement have been distinguished: multifocal neuropathy (MFN), asymmetric polyneuropathy, and distal symmetric polyneuropathy (DSPN). Reported frequencies of these phenotypes are highly variable due to their lack of standardized definitions. In the largest series, prevalence in NSVN has varied widely: MFN 10–78%, asymmetric polyneuropathy 18–85%, and DSPN 2–41%. Combining data from all reports yields 46% asymmetric polyneuropathy, 38% MFN, and 16% DSPN. To improve standardization, new definitions were proposed in 2017. A Peripheral Nerve Society task force published consensus recommendations on classification, diagnosis, and treatment of NSVN in 2010. New diagnostic criteria for VN and NSVN were developed by the Brighton Collaboration in 2017. Evaluation of patients with suspected VN includes EMG, laboratory studies, imaging, and nerve/muscle biopsy. An ultrasonographic pattern of multifocal proximal nerve enlargement was 94% sensitive and 88% specific for VN in one study. In patients lacking vasculitis in nerve biopsy, clinically probable VN can be diagnosed by recourse to predictive clinicopathologic findings. NSVN is distinguished from systemic VNs by the absence of clinical, laboratory, or pathologic evidence of other-organ involvement; ANCAs; cryoglobulins; sedimentation rate ≥ 100 mm/hour; and medical condition/drug predisposing to systemic vasculitis. VNs in ANCA-associated vasculitides are treated in accordance with consensus guidelines. No randomized controlled trial of therapy has been reported for NSVN, but observational evidence suggests that NSVN might be better controlled by corticosteroids combined with an immunosuppressive agent than with corticosteroids alone. Treated NSVN rarely spreads to other organs, but ∼40% of patients relapse. Long-term outcome is favorable, but chronic pain is common.
SS.28.02
SS.28.02 - Mechanisms of Diabetic Neuropathy
1Flinders Medical Centre, Adelaide, Australia, 2Royal Adelaide Hospital, Adelaide, Australia, 3University of Adelaide, Adelaide, Australia
The mechanisms underpinning the development of diabetic neuropathy remain incompletely elucidated and represents a combination of factors . This presentation will summarise the current knowledge regarding these contributory mechanisms resulting in nerve injury in diabetic neuropathy. Major contributors include acyl carnitine accumulation, excessive reactive oxygen species (ROS) production, advanced glycation end products (AGE) accumulation, alteration of gene expression and the development of an inflammatory state due to pro-inflammatory cytokine release.
SS.28.04
SS.28.04 - Epidemiology and Risk Factors for Diabetic Neuropathy
1University Of Michigan, United States
Diabetic neuropathy affects about one third of patients with diabetes. In type 1 diabetes, neuropathy often comes many years after onset. In type 2 diabetes, neuropathy is often diagnosed at the same time or shortly after diabetes diagnosis although this is highly variable. Risk factors for diabetic neuropathy include obesity, dyslipidemia, and hypertension. Diabetic neuropathy is frequently underdiagnosed particularly in under-resourced populations. These populations demonstrate a high prevalence of the metabolic syndrome with poor control of metabolic risk factors. In the United States, Black and Hispanic patients are also more likely to have neuropathy
SS.29.02
SS.29.02 - Mechanisms, Predisposing Factors, Identifying at Risk Patients
1University Of Sydney, Australia
Chemotherapy-induced peripheral neuropathy (CIPN) is a prominent toxicity of commonly used cancer therapies including microtubule targeting agents such as taxanes and vinca alkaloids, platinum agents, proteasome inhibitors and antibody-drug conjugates. CIPN produces predominantly sensory neuropathy, leading to deficits in function and balance and producing long-lasting disability and reduced quality of life. However, there remain gaps in our ability to understand mechanisms causing CIPN, and limited prevention and treatment options. This presentation will provide an overview of CIPN mechanisms and risk factors, including recent evidence of genetic and metabolic risk factors and neurophysiologic and protein biomarkers of CIPN. This will focus on the importance of axonal degeneration pathways in paclitaxel-induced neurotoxicity including genetic predisposition to axonal degeneration and evidence that the serum biomarker of axonal damage, neurofilament light chain, provides an early indicator of clinical neurotoxicity in vivo. Metabolic risk factors which may outline differential susceptibility to neuropathy will also be outlined. In order to understand the impact of long-term neurotoxicity on cancer survivors, optimal CIPN assessment tools and strategies to improve assessment, rehabilitation and treatment in clinical practice will be considered. Ultimately, these findings will highlight how best we can support cancer survivors with CIPN to improve quality of life and reduce the burden of long-term neuropathy.
SS.30.02
SS.30.02 - Nonsystemic Vasculitic Neuropathy: Updated Peripheral Nerve Society Guideline on Classification, Diagnosis, Investigation, and Immunosuppressive Therapy
1Medical College Of Wisconsin, United States, 2King's College Hospital, United Kingdom
Vasculitis confined to peripheral nerves over long-term follow-up is designated nonsystemic vasculitic neuropathy (NSVN). While rare, NSVN (including its lumbar radiculoplexus subtype) appears to be one of the most common inflammatory neuropathies. It is underrecognized because nerve biopsy is needed for diagnosis. While considered treatable, no randomized controlled trials have been performed. The Peripheral Nerve Society (PNS) evidence-driven consensus guideline on classification, diagnosis, investigation, and immunosuppressive therapy of NSVN was published in 2010. In the consensus classification scheme for vasculitic neuropathy, nondiabetic radiculoplexus neuropathy and some cases of Wartenberg migrant sensory neuropathy were included as subtypes of NSVN. Diabetic radiculoplexus neuropathy was categorized as a separate form of single-organ vasculitis. The consensus definition of pathologically definite vasculitic neuropathy required that vessel wall inflammation be accompanied by vascular damage. In patients lacking nerve biopsy evidence of vascular damage, diagnostic criteria for pathologically probable vasculitic neuropathy included several predictors of definite vasculitic neuropathy: vascular deposits of IgM, C3, or fibrinogen; hemosiderin deposits; asymmetric/multifocal nerve fiber loss; prominent active axonal degeneration; and myofiber necrosis, regeneration, or infarcts in concomitant muscle biopsy. To diagnose clinically probable vasculitic neuropathy in patients without nerve biopsy proof of definite vasculitis, the most typical phenotypic features were used, including sensory or sensory-motor involvement, asymmetric/multifocal pattern, lower-limb predominance, distal-predominance, pain, acute relapsing course, and axonal electrodiagnostic features. Criteria favoring systemic rather than nonsystemic VN were clinical, laboratory, or pathologic evidence of other-organ involvement; ANCAs; cryoglobulins; sedimentation rate ≥ 100 mm/hour; and medical condition/drug predisposing to systemic vasculitis. Good practice point recommendations on treatment included corticosteroid monotherapy for ≥ 6 months in most patients; combination therapy with cyclophosphamide, azathioprine, or methotrexate plus corticosteroids for patients with rapidly progressive NSVN and those refractory to corticosteroids; cyclophosphamide for severe NSVN; and maintenance therapy with azathioprine or methotrexate for 18-24 months in patients achieving remission with combination therapy. In the intervening 14 years, new evidence on classification of the ANCA-associated systemic vasculitides and various subtypes of NSVN, clinical features of NSVN (new series from new countries), clinical and histopathological diagnosis of vasculitic neuropathy, differential diagnosis of vasculitic and other multifocal neuropathies, and treatment of NSVN and – to a much greater extent – ANCA-associated systemic vasculitides (e.g., rituximab for induction and maintenance of remission and lower cumulative doses of prednisone) has appeared, as have the 2012 Revised International Chapel Hill Consensus Conference Nomenclature of Vasculitides and 2017 Brighton case definition of vasculitic neuropathy. To adapt this new evidence, improve the sensitivity and specificity of the clinicopathological diagnostic criteria, incorporate new subtypes of NSVN, more fully address diabetic radiculoplexus neuropathy, and implement superior methodology, a new PNS-endorsed task force has initiated work on a revision of this guideline.
SS.32.02
SS.32.02 - Innovative Trial Design
1University Of Minnesota, United States
Chronic inflammatory demyelinating polyneuropathy (CIDP) is a rare but clinically and immunobiologically heterogeneous disorder. These features present unique challenges for CIDP clinical trials. First, the rarity of the disease necessitates that recruitment for large phase 3 CIDP trials be spread across several dozens of sites in order to enroll several hundreds of patients. Although each individual site may have a low recruitment burden, the model is inefficient and places strains on the enrolling network – both of which may impact the success of the trial. Second, considering the absence of diagnostic and disease activity biomarkers, two common obstacles for successful CIDP clinical trials include identification of a diagnostically accurate population of patients and avoidance of including patients with immunologically inactive disease. Third, trials have been limited in their ability to enroll balanced populations of patients that are newly diagnosed/treatment naive and those patients that are already on immunotherapy. Given the rarity of CIDP, the treatment naive group is especially problematic for clinical trial enrollment but in many respects a cleaner population to investigate where the effects of subject expectation and conditioning are less pronounced than trials that predominately include treatment experienced participants. Fourth, CIDP outcome measures incompletely capture improvement (or deterioration) in all patients with CIDP and are not specific to changes attributable to CIDP. Fifth, many of the definitions used in CIDP clinical trials have not been standardized. For example, “treatment-naive” and “treatment-refractory” may mean something different in two separate trials.
In order to mitigate these obstacles modern clinical trials have employed several features with varying degrees of success. Expert adjudication panels are now commonly used to confirm diagnosis; and IVIG/corticosteroid withdrawal procedures are used to determine disease activity status. In order to expedite enrollment most trials have focused on treatment experienced patients and have adopted a placebo withdrawal design to look at relapse rate in patients randomized to investigation drug vs placebo. Some trials have placed a focus on enrollment stratification by treatment history, with variable levels of attention paid to treatment naive patients. Most trials have utilized the adjusted INCAT score as the primary outcome measure, however highlighting key secondary outcomes that complement the primary are also recognized as valuable. The variable definitions used across CIDP remains a point of controversy and confusion.
The objectives of this discussion are to highlight the challenges of CIDP clinical trials and to outline steps that have been taken to minimize the limitations. To illustration these issues, trial designs from active and recently completed CIDP trials will be presented, and contrasted to CIDP trials that have been completed over the last 2 decades. By doing so, this discussion hopes to call attention to the unmet needs of CIDP clinical trials and promotes a dialog that aims to find a better way forward.
SS.33.02
SS.33.02 - Over 40 Years of Clinical Trials in Myositis: What have we Learned?
1Brigham and Womens Hospital / Harvard Medical School, United States
Many advances in myositis clinical have occurred over the past 40+ years. In 1975, Bohan and Peter published an article in the NEJM that distinguished two major forms of inflammatory myopathy (i.e., polymyositis and dermatomyositis). The only difference distinguishing these was a rash in patients with dermatomyositis. Although the first person reported as “inclusion body myositis” was made in 1971, this patient almost certainly did not have inclusion body myositis but rather a hereditary inclusion body myopathy, likely GNE myopathy, given onset in 20s of progressive foot drop. Subsequent, cases of what we now know to be IBM were described but thought to be rare (mainly because of incomplete physical examinations and the inexperience of myopathologists given this was only recently recognized. Berch Griggs and I wrote an editorial (Unicorns, dragons, polymyositis, and other mythological beasts. Neurology 2003 12;61:288-9) in regard to an important article by van der Meulen and colleagues (Polymyositis: an overdiagnosed entity. Neurology 2003;61316-21). We got grief from many of our colleagues for our belief that polymyositis was ultra rare and frequently misdiagnosed. Some 20 years later we have been proven correct. Many were also skeptical about the existence of an autoimmune necrotizing myopathy, some triggered by statins, until the HMGCR antibody was discovered in many of these causes. Nowadays, we know that most cases of what were previously diagnosed as polymyositis actually have IBM, IMNM, ASyS, or a hereditary myopathy with inflammatory cell infiltrates.
An important advancement in the IIM treatment trials is the recognition of the different forms of myositis and that the pathogenic bases differ. As such, the response to different immunotherapies to each type of myositis be different. In the past, clinical trials lumped subjects with different forms of myositis together and assessed a treatment proven to be effective in transplantations or other autoimmune conditions. Thus, it is important to do a trial of patients with a specific form of IIM or that the participants should be stratified according to the type of IIM they have. The reliability of accurate diagnosis is still unfortunately poor. This adjudication committees of experts in the field are needed, particularly when the studies include patients with “polymyositis” or a diagnosis is based on muscle biopsy results.
Another major issue in clinical trials is what primary outcome measure should be used. This need not be the same for each type of IIM. Various outcomes have been employed, including the Myositis Disease Activity Assessment Took, the Total Improvement Score, manual muscle testing, quantitative muscle strength testing (fixed or handheld dynamometry), the Cutaneous Dermatomyositis Disease Area and Severity Index Activity score, functional testing (e.g., timed gait- timed up and go,10 meter walk test, 6 minute walk test), various questionaires (e.g. IBMFRS and SIFA for IBM), and imaging (e.g., MRI, DEXA). Each of these have their drawbacks and need to be understood. The reliability and responsiveness to each measure according to the specific IIM need to be known to adequately power a study.
SS.33.05
SS.33.05 - Innovative Trial Designs: Early Endpoints, Basket-Trials and Adaptive Platforms
1Johns Hopkins University School Of Medicine, United States
Clinical trials in Idiopathic Inflammatory Myopathies (IIM) have significantly evolved in the past 5 years. There has been an exponential growth of clinical trials in IIM and in fact, has led to an FDA approved drug for dermatomyositis. While this has led to much enthusiasm for unlocking the path for novel therapeutics in IIM, it has also lead to constraints in recruitment in a rare disease such as IIM. There is an urgent need to develop innovative trial designs such as surrogate and earlier endpoints and adaptive or platform trials in IIM. In this session, the benefits of such novel trial design will be discussed and applied to the current therapeutic landscape of IIM.
SS.34.03
SS.34.03 - Clinical Presentations and Treatment of Igg4 Paranodopathies
1Translational Neuroscience Research Center, Graduate School of Medicine, International University of Health and Welfare, Fukuoka City, Japan
Autoantibodies against proteins expressed around the nodes of Ranvier, such as neurofascin 155 (NF155), contactin-1 (CNTN1), contactin-associated protein 1 (CASPR1), and leucine-rich repeat LGI family member 4 (LGI4), have been identified in a subset of patients with chronic inflammatory demyelinating polyneuropathy (CIDP). Compared with seronegative CIDP patients, those with anti-nodal/paranodal antibodies often show unique features, including deep sensory impairment leading to sensory ataxia, tremor, distal dominant motor weakness and atrophy, extremely high cerebrospinal fluid protein concentrations, and peripheral nerve hypertrophy as shown by magnetic resonance neurography and ultrasonography. Of note, CIDP patients with IgG4 anti-nodal/paranodal antibodies frequently show poor response to intravenous immunoglobulin (IVIg) and require additional treatments with corticosteroids and immunosuppressants. Based on these unique features, CIDP with these antibodies is now collectively termed autoimmune nodopathy (AN). Given that the IgG subclass of these antibodies is predominantly IgG4, the characteristic features of AN are assumed to be attributable to the special properties of IgG4. NF155+ AN typically shows juvenile and early adulthood onset while both CNTN1+ AN and LGI4+ AN preferentially show aged onset. NF155+ AN is characterized by high frequency of optic nerve involvement and cranial nerve hypertrophy. Both CNTN1+ AN and LGI4+ AN frequently show acute/subacute onset, masquerading Guillain-Barre syndrome. Concomitant membranous nephropathy causing nephrotic syndrome is characteristic of CNTN1+ AN. We have established AN/CIDP biobank and registry in Japan, collecting 566 cases, of which 208 cases (38%) were positive for anti-nodal/paranodal antibodies. Among the seropositive patients, anti-NF155 antibody-positive (NF155+) AN occupied 77%, followed by CNTN1+ AN (12%), CASPR1+ AN (9%) and LGI4+ AN (3%). Next generation sequencing of the HLA genes revealed that NF155+ AN patients harbored either HLA-DRB1*15:01-DRB5*01:01-DQA1*0102-DQB1*06:02 or HLA-A24:02-B52:01-C12:02-DRB1*15:02-DRB5*01:02-DQA1*01:03-DQB1*06:01. Because the latter haplotype is relatively frequently seen in Japanese, NF155+ AN may be more prevalent in Japanese than European descendants. Intraneural injection of NF155+ AN and LGI4+ AN patients’ IgG into mouse sciatic nerves induced shrinkage and mislocation of CASPR and Nav immunostaining while the alteration patterns of the nodes were different between both IgG, suggesting distinct actions of these anti-paranodal antibodies.
SS.35.02
SS.35.02 - Tacrolimus in Myasthenia Gravis
1Department of Neurology, Yonsei University College Of Medicine, Seoul, Republic of Korea
Myasthenia gravis is an autoimmune disorder affecting neuromuscular junction, characterized by fluctuating muscle weakness and fatigability. In terms of immunopathogenesis, myasthenia gravis is caused by T cell-dependent, B cell-mediated autoimmune response leading to production of autoantibodies against the nicotinic acetylcholine receptor, muscle-specific kinase (MuSK) or other components of the post-synaptic membrane of the neuromuscular junction. T cells play an important role in the production of the pathogenic autoantibodies. Antigen-presenting cells present peptide fragments of nicotinic acetylcholine receptor or MuSK to T cells. The activated autoreactive T cells initiate B cell activation and differentiation, generating acetylcholine receptor or MuSK specific B cells. The conventional treatment approach for myasthenia gravis has relied on acetylcholinesterase inhibitors, corticosteroids, immunosuppressants, and thymectomy. Although these therapeutic options have shown good efficacy in improving myasthenia gravis related symptoms in most patients, about 15% of patients with myasthenia gravis are refractory to the conventional treatment.
Tacrolimus, also known as FK506, is a potent immunosuppressive agent primarily used to prevent organ transplant rejection and to treat various autoimmune diseases, including myasthenia gravis. Tacrolimus binds to the intracellular protein FK506-binding protein-12 (FKBP-12), forming a tacrolimus-FKBP-12 complex, which inhibits the activity of calcineurin. By inhibiting calcineurin, tacrolimus suppresses the transcription of IL-2 and other cytokines, leading to reduced T-cell activation and proliferation. Tacrolimus indirectly affects B cells by suppressing T-helper cell function, which is necessary for B cell activation and antibody production. This results in decreased production of autoantibodies, which are pathogenic in diseases like myasthenia gravis. In addition, tacrolimus modulates the ryanodine receptor, the calcium release channel of sarcoplasmic reticulum, and increase calcium release from skeletal muscle sarcoplasmic reticulum, which may improve excitation–contraction coupling in myasthenia gravis.
Since successful treatment of MG with tacrolimus was reported in the early 2000s, it has been used to treat myasthenia gravis. A number of observational studies support the use of tacrolimus in patients with myasthenia gravis and tacrolimus is recommended in several myasthenia gravis treatment guidelines. Although available randomized controlled trials failed to show efficacy of tacrolimus in patients with myasthenia gravis, there was criticism that the design of these randomized controlled trials was inadequate to address the question. Based on the results of the studies so far, tacrolimus may have a beneficial role in reducing corticosteroid burden and myasthenic symptom severity in the patients with refractory myasthenia gravis as well as those newly diagnosed. Long-term use of tacrolimus in the patients with myasthenia gravis seems to be safe. To confirm the efficacy and safety of tacrolimus, additional large-scale studies of high quality are needed.
SS.35.03
SS.35.03 - Rituximab
1University Of Oxford, United Kingdom
Rituximab (RTX) is a human/murine chimeric monoclonal antibody directed against the CD20 protein on developing B-cells, leading to B-cell depletion. Its off-label use in the treatment of refractory myasthenia gravis (MG) has increased over the past decade [1]. The proposed mechanism of action and evolving landscape of RTX treatment in myasthenia gravis will be summarised.
RTX is associated with improved clinical status and decreased prednisolone use in the refractory MG cohort. MuSK+ MG patients appear to experience a greater clinical response compared with AChR+ MG, but the literature is observational and largely retrospective. Two recent randomised-controlled trials studying RTX in AChR+ MG demonstrated conflicting results. The BeatMG Study [2] found RTX to be safe and well tolerated but there were no differences in the primary steroid-sparing outcome compared to placebo. The RINOMAX trial [3] demonstrated a significant difference in the proportion of patients with minimal disease manifestations and low doses of corticosteroids, favouring RTX. Clinical outcomes and key differences between these studies will be discussed.
There is no consensus on the dosing schedule of RTX in MG, with older studies adopting a B cell lymphoma treatment protocol comprising of 4 weekly infusions of 375mg/m2 or 1gm given 2 weeks apart, with an interval between cycles of 6 months. Given the pathophysiological disparities between haematological disorders and autoimmune diseases, along with concerns for infective risks, low and very-low doses of RTX have been employed more recently with success. In addition, the cost renders it prohibitive in certain healthcare systems and reduced dosages increases the accessibility. The literature on differing RTX treatment regimens globally, efficacy data and safety profile are introduced.
In the era of biologics with rapidly emerging alternative high efficacy therapies, the role of RTX in AChR+ MG is undefined. The speaker will discuss its role in MG therapeutics personalised to the individual patient, and the current movement towards RTX initiation early in the MG disease course as a disease modifying therapy.
1. Delate T, Hansen ML, Gutierrez AC, Le KN. Indications for Rituximab Use in an Integrated Health Care Delivery System. J Manag Care Spec Pharm. 2020 Jul;26(7):832-838.
2. Nowak RJ, Coffey CS, Goldstein JM, Dimachkie MM, Benatar M, Kissel JT, Wolfe GI, Burns TM, Freimer ML, Nations S, Granit V, Smith AG, Richman DP, Ciafaloni E, Al-Lozi MT, Sams LA, Quan D, Ubogu E, Pearson B, Sharma A, Yankey JW, Uribe L, Shy M, Amato AA, Conwit R, O'Connor KC, Hafler DA, Cudkowicz ME, Barohn RJ; NeuroNEXT NN103 BeatMG Study Team. Phase 2 Trial of Rituximab in Acetylcholine Receptor Antibody-Positive Generalized Myasthenia Gravis: The BeatMG Study. Neurology. 2022 Jan 25;98(4):e376-e389.
3. Piehl F, Eriksson-Dufva A, Budzianowska A, Feresiadou A, Hansson W, Hietala MA, Håkansson I, Johansson R, Jons D, Kmezic I, Lindberg C, Lindh J, Lundin F, Nygren I, Punga AR, Press R, Samuelsson K, Sundström P, Wickberg O, Brauner S, Frisell T. Efficacy and Safety of Rituximab for New-Onset Generalized Myasthenia Gravis: The RINOMAX Randomized Clinical Trial. JAMA Neurol. 2022 Nov 1;79(11):1105-1112.
SS.37.02
SS.37.02 - The Role of the Thymus in Autoimmunity
1George Washington University, WASHINGTON, United States
The thymus functions as the site of T cell maturation, where immature T cells undergo both positive and negative selection to produce highly specialized effector cells that recognize pathogens. Thymic epithelial cells in both the cortex and medullary regions are crucial to this selection process. Thymus atrophy, thymic involution, occurs with age when and the key factors that are involved with thymopoiesis are reduced. However, abnormalities in the thymus, such as thymic hyperplasia and thymomas occur in patients with myasthenia gravis (MG), shedding light on the autoimmune process that defines the break of tolerance of CD4+ T cells. Early-onset MG patients demonstrate follicular hyperplasia with an increase in lymphoid follicles in the medulla and perivascular spaces. The presence of an increase in B cells and germinal centers, associate with increased numbers of high endothelial venules, indicates an active immune response. This response leads to the generation of long-lived plasma cells that produce antibodies against the acetylcholine receptor (AChR). In thymoma, the epithelial tumors contain cellular composition of immature thymocytes, predisposing patients to autoimmune diseases, with MG being the most common of those displayed. Thymoma-associated MG patients have thymomas that lymphoid follicles surrounded by high endothelial venules, however the generation of autoantibody-producing plasma cells is considered to be external to the thymus. The autoimmune regulatory protein AIRE, responsible for expression of self-epitopes to allow for negative selection of autoreactive T cells, is absent in nearly all thymomas, regardless of the presence of MG. This absence allows for the break in tolerance during the negative selection and the decrease in the maturation of T regulatory cells. Additionally, the histological subtypes of thymomas may vary in their functional pathway to autoimmunity. Autoantibodies produced in thymoma-associated MG are against the AChR, but also muscle autoantigens such as titin, the ryanodine receptor and neurofilament protein. Understanding the key factors that drive loss of tolerance in thymic hyperplasia and thymoma will enable identification of targets for therapeutic development.
SS.37.03
SS.37.03 - Complement in Myasthenia Gravis
1George Washington University, United States
A critical component in the pathophysiology of myasthenia gravis (MG) with antibodies directed to the acetylcholine receptor (AChR) is the involvement of the complement system. Autoantibodies target the AChRs at the neuromuscular junction (NMJ) and activate the complement cascade by binding C1q complex. The activation leads to the final formation of the membrane attack complex (MAC), which results in the shedding of AChR-rich postsynaptic membrane into the synaptic cleft, as well as the loss of synaptic folds and voltage-gated Na channels. The damage to the NMJ impairs signal transmission from the nerves to the muscles and thereby causing muscle weakness and fatigue. Therapeutic interventions for MG often aim to inhibit complement activation at the C5 level, as blocking the formation of the MAC can reduce tissue damage and improve clinical symptoms. Eculizumab, a monoclonal antibody that inhibits C5, and Zilucoplan, a peptide that inhibits the cleavage of C5, have shown efficacy in reducing symptoms and improving quality of life in many MG patients. However, some patients do not respond adequately to complement inhibition, highlighting the complexity of the disease and the involvement of other pathological mechanisms beyond complement-mediated damage. Additionally, levels of C3a and C5a have been shown to be elevated in MG patients. The potent anaphylatoxins act as chemoattractants, signaling for the recruitment of immune cells such as neutrophils, eosinophils, and macrophages to the site of the NMJ. Once at the NMJ, these immune cells can become activated and release a variety of cytokines and inflammatory mediators that exacerbate local inflammation and tissue injury. Anaphylatoxins also play roles in the activation of immune cells. One important mechanism is their action on their receptors (C3aR and C5aR) expressed on immune cells that modulate cell activation. In addition to their role in innate response, anaphylatoxins regulate the adaptive response either directly or indirectly through B-cell and T-cell activation. The complexity of the complement system in an autoimmune disease underscores the necessity to understand the off targets in any complement directed therapeutic.
SS.39.02
SS.39.02 - Juvenile MG
1Northwestern Feinberg School Of Medicine, United States
Disease-causing autoimmune attack on presynaptic and postsynaptic targets within the neuromuscular junction are less frequent in children and adolescents than in adults. Lambert Eaton Myasthenic Syndrome (LEMS) is a rare disorder in which VGCC antibodies interfere presynaptically with acetylcholine release. Only 23 pediatric cases (3-16 years of age) have been reported in the literature by 2024. Differences from adult LEMS include a higher incidence of oculobulbar weakness (58%) and lower incidences of underlying malignancy (16%) and dysautonomia (10%). Myasthenia gravis (MG) due to antibodies against post synaptic NMJ targets is more common but still a rare disorder occurring at an incidence of 1:100,000. Children with acetylcholine receptor (AchR) antibodies develop symptoms relatively equivalently in prepubertal and postpubertal years. In prepubertal years, male/female ratio is even, ocular symptoms are more common as presenting symptoms and spontaneous remission rate is higher. Post pubertal onset has a clinical profile more similar to that observed in adults except that the incidence of underlying thymoma or other malignancy is lower (only 10 cases of myasthenia associated with thymoma in children reported in the literature by 2022). MuSK antibody-associated MG is more frequent in children of Asian and African heritage. Symptoms are predominantly facial, bulbar and respiratory with milder limb weakness. Treatment goals for all types of autoimmune neuromuscular junction (NMJ) disorders are to reach minimal manifesting symptoms with no mortality. 3,4-diaminopyridine phosphate in LEMS and pyridostigmine in MG are valuable treatments to provide transient improvements in strength. The recent introduction of biologics to these disorders has greatly improved treatment for children and adolescents in whom steroid-sparing agents are more important due to the vulnerability of children to side effects of chronic corticosteroid therapy. Diagnostic tools and treatment options will be discussed.
Toxins are infrequent but important causes of acquired NM dysfunction in children. Botulism (via ingested toxin or wound contamination and growth of botulinum organism) are relatively uncommon in most areas of the world. Infantile botulism due to ingestion of botulinum spores by infants with rapid colonization and toxin production due to slow gut motility is a recognized (and likely underdiagnosed) problem in the United States with 60-100 cases reported to the CDC annually. While a 2021 report in the literature summarizing several decades of recognized cases in Canada and Europe identified only 1-2 cases per year in those countries, infantile botulism is serious, treatable and likely underdiagnosed worldwide. Tick paralysis (with over 70 species secreting a toxin causing paralysis) is another example of toxic NMJ dysfunction occurring worldwide in children.
SS.39.03
SS.39.03 - Effects of Age and Sex
1National Neuroscience Institute, Singapore
Myasthenia gravis is one of the commonest and most recognizable autoimmune neuromuscular disorder. There is increasing evidence on the vast heterogeneity of the underlying autoimmune mechanisms and possible genetic susceptibility leading to the disease process. Equally important are biological variables that have been shown to be of importance in the disease patho-mechanism, manifestations and management. Age and sex are by far the most implicated variables. Myasthenia gravis has a bimodal distribution with females accounting for most patients with early onset myasthenia gravis and males dominating in those with a late-onset disease. This increased incidence in females is observed in both acetylcholine receptor antibody and muscle specific tyrosine kinase antibody sero-positivity and are possibly related to hormonal and genetic factors. Thymic pathology is in the form of hyperplasia with widespread germinal follicles. Disease manifestations have also been shown to be more marked in females. This is reflected both in physician derived compositive myasthenia gravis score and in patient reported quality of life scores. As most of these patients are in the child-bearing age group, several factors need to be considered while instituting immunosuppressive medications. Most of the steroid sparing agents are contraindicated in pregnancy and biological agents like Rituximab are to be avoided during the last trimester of pregnancy. Additional precautions also must be taken during the peripartum and postpartum period. Late onset myasthenia gravis (more than 50 years) and very-late onset myasthenia gravis (more than 65 years) are more common in males. The incidence of late onset myasthenia gravis has been increasing, and diagnosis can be delayed in this age group in view of the presence of other comorbidities that share similar clinical manifestations. Immunologically and clinically late onset myasthenia gravis differs from early-onset myasthenia gravis. The mechanisms of autosensitization to the acetylcholine receptors in late onset myasthenia gravis is presumed to be due to immune dysregulation from immunosenescence or other unexplained mechanisms. Thymic pathology is one of thymoma or thymic atrophy. Management decisions in this category of patients are subject to a different set of variables. As patients with late onset myasthenia gravis have an increased incidence of other co-morbidities caution must be instituted while selecting immunosuppressive medications, with closer monitoring required to assess for development of complications. In an era where personalized and precision medicine are gaining importance, age and sex are presently the foremost variables to be considered while deciding on investigations and management pathways. This will continue to remain so even after other factors like ethnicity, more advanced immunological and genetic profiling are also considered when deciding the treatment protocol.
SS.41.02
SS.41.02 - Thymectomy in Myasthenia Gravis
1KUMC, United States
Since the 1939 report by Dr. Alfred Blalock’s 1939 of dramatic improvement in following excision of a thymic tumor in a patient with myasthenia gravis (MG), benefits of thymectomy in non-thymomatous MG were until 2016 based on retrospective case series. In thymomatous MG, the tumor and thymic tissue must be removed in patients with lower surgical risk profile or irradiated in MG with higher surgical morbidity predictors. Tumor histologic grade, excision margins, and presence of distal spread guide treatment decisions regarding post-operative radiation, chemotherapy, and monitoring. While delayed improvement in myasthenic symptoms after thymoma resection may or may not occur, MG patients at risk for peri-operative myasthenic crisis (persistent bulbar, respiratory, or limb weakness) should have MG therapies optimized and may benefit from pre-operative plasma exchange. In addition to its long-accepted role in thymomatous MG, thymectomy has become the standard of care for many adults with non-thymomatous generalized MG. Wolfe et al reported in 2016 the landmark international, randomized, rater-blinded clinical trial in adults with recently diagnosed acetylcholine receptor (AChR) antibody positive generalized MG. Patients were randomized to receive either extended transsternal thymectomy plus prednisone or medical management with prednisone. The beneficial effects of thymectomy became evident within the first 3 to 4 months. Over a 3-year follow-up period, the thymectomy group performed better on the time-weighted average QMG score and on time-weighted alternate-day prednisone dose requirement. The thymectomy group also had fewer patients requiring additional immunosuppression, fewer adverse events, and fewer admissions for myasthenic crises. The trial of thymectomy in MG provides Class I evidence supporting its safety and efficacy in many adults with AChR antibody positive non-thymomatous generalized MG. Even though the thymectomy trial utilized the sternal-splitting approach, less invasive procedures are now accepted for maximal thymic tissue removal. The 2020 update of the International Consensus Guidance for Management of MG recommends thymectomy be considered early on in the course of non-thymomatous, generalized AChR antibody positive MG patients aged 18 to 50 years. It should also be “strongly considered” in generalized AChR antibody positive MG patients if they fail to respond to an initial adequate trial of immunotherapy or have intolerable side effects. Class I evidence in support of thymectomy is lacking in those AChR antibody negative generalized MG. Though evidence does not support thymectomy in those with MuSK, low-density lipoprotein receptor–related protein 4, or agrin antibodies, the 2020 update indicates that thymectomy “may be considered” in generalized AChR antibody negative MG in cases of failure to respond adequately to or intolerable adverse effects to immunosuppressive therapy. Unresolved questions persist regarding the efficacy of thymectomy in ocular MG (OMG). The 2020 update indicates that thymectomy “may be offered” to patients with AChR antibody non-thymomatous OMG who do not respond to acetylcholinesterase inhibitors or immunosuppressive agents or who have contraindications or prefer not to take immunosuppressants. Though data regarding thymectomy for Juvenile MG are limited to retrospective case series, it seems to suggest that patients who underwent thymectomy may improve in disease severity and have rare post-operative complications.
SS.41.03
SS.41.03 - Thymectomy for Myasthenia Gravis: Analyzing Usage Patterns and Dispelling Safety Concern
1George Washington University, United States
The application of thymectomy as a treatment for myasthenia gravis (MG) has been a contentious issue for nearly a century. Alfred Blalock performed the first thymectomies in a small group of patients in the late 1930s, sparking ongoing debate about its efficacy. Variability in how expert clinicians applied thymectomy further fueled this controversy. In 2000, an evidence-based practice parameter concluded that the benefit of thymectomy in non-thymomatous autoimmune MG could not be definitively determined due to methodological inconsistencies and heterogeneous study populations.
The subsequent decade witnessed a decline in thymectomy procedures in the United States. However, the landscape changed in 2016 when the MGTX Trial demonstrated that thymectomy combined with prednisone was superior to prednisone alone in acetylcholine receptor (AChR) antibody-positive MG. Following this, various organizations endorsed thymectomy for AChR antibody-positive MG patients younger than 50 years.
Despite the introduction of new knowledge, clinical practice often lags in its adoption. An evaluation of the National Surgical Quality Improvement Program (NSQIP) database revealed that the annual rate of thymectomy increased by only 1% in the three years following 2016, a change that was not statistically significant. Further analysis using the Cosmos data repository established by Epic Systems Corporation, which includes de-identified EHR data from over 250 healthcare organizations and over 234 million patients, indicated that the rate of thymectomy for all MG patients increased from 1% in 2016 to 3% in 2023.
Thymectomy remains an underutilized therapy for MG, even though it has low morbidity and near-zero mortality. In August 2023, a high-profile article raised concerns about an increased risk of cancer and autoimmune disease among patients who had undergone thymus removal, including those with and without MG. This publication has led to anecdotal reports of patients hesitating to undergo thymectomy, even in the presence of a thymoma. Given the slow adoption of thymectomy despite demonstrated efficacy, the neuromuscular community must develop systems to enhance its utilization. Addressing this issue is critical to improving outcomes for MG patients.
SS.42.02
SS.42.02 - Should we use Statins in Patients with Neuromuscular Diseases?
1Hadassah-hebrew University Medical Center, Israel
Hyperlipidemia can be found in patients with hereditary myopathies as part of their 'normal aging' or in some specific conditions at risk. Using statins, the common cholesterol reducing medications becomes a dilemma in such myopathies. Statins' side effects including myalgia, myopathy and rhabdomyolysis typically may deter physicians from prescribing these medications. A literature review of statins' side effects in hereditary myopathies does not provide clear evidence about the true risk of these drugs. Setting criteria to assess the reports on statins-induced side effects in various hereditary myopathies suggests that only few disorders present high risk (MELAS and possibly other mitochondrial myopathies, Myotonic dystrophy type 2, and McArdle's disease). Possible solutions to the dilemma of statins usage in hereditary myopathies (other medications, trial with close follow up) will be discussed.
SS.43.02
SS.43.02 - The Rare Disease Research Network for Myasthenia Gravis, MGNet
1George Washington University, United States
MGNet is one of 20 consortia within the Rare Diseases Clinical Research Network (RDCRN), supported by the National Institutes of Health. The RDCRN's mission is to foster collaboration among research centers to enhance biomarker development, improve clinical trial readiness, and train the next generation of rare disease scientists.
Established in 2019, MGNet received critical leadership and financial support from patient advocacy groups, including the MG Foundation of America and Conquer MG. This consortium of US-based centers is conducting a natural history study led by Richard Nowak and Vern Juel with an associated biobank housed at Duke University, actively following 300 MG patients over two years. Peripheral blood monocytes, sera, and plasma are collected every six months. Investigators in and outside academia are welcome to apply for use of the biospecimen bank and form active collaborations with MGNet investigators. Presently, serum and plasma are being used to assess autoreactome signatures and SARS-COV-2 serology. Two separate projects focused on biomarker discovery are led by Kevin O'Connor at Yale University and Linda Kusner at George Washington University. The MGNet biostatistical core is led by Drs. Inmaculada Aban and Gary Cutter at the University of Alabama at Birmingham.
MGNet has funded pilot studies and supported MGNet Scholars across the US and Europe, with financial backing from Argenx and UCB Pharma. MGNet investigators actively collaborate with industry partners BioSensics and Care Constitution. The presentation will highlight MGNet's scientific contributions to the field of myasthenia gravis, the administrative challenges of managing a multi-institutional and international organization, and the vision for MGNet's future development.
SS.43.03
SS.43.03 - MGBase: A Global, Observational Registry for Real-World Outcomes Research in Myasthenia Gravis
1Eastern Health Clinical School, Monash University, Box Hill, Australia, 2Dept of Neurology, Royal Melbourne Hospital, Parkville, Australia, 3Dept of Neurosciences, Monash University, Prahran, Australia, 4Dept Neurology, Alfred Hospital, Prahran, Australia, 5Division of Neurology, Dept of Medicine, University Health Network and University of Toronto, Toronto, Canada, 6University of Alabama Birmingham, Dept of Biostatistics, Birmingham, USA, 7Dept of Neurology and Rehabilitation Medicine, George Washington University, Washington DC, USA, 8Division of Neurology, Dept of Medicine, University of Cape Town, Cape Town, South Africa, 9Neurology Research Group, Neuroscience Institute, University of Cape Town, Cape Town, South Africa, 10Dept of Neurology, Concord Hospital, Concord, Australia
MGBase is a global observational registry for myasthenia gravis (MG) designed to promote collaborative outcomes research. The registry has been built alongside the successful MSBase registry which has allowed the utilisation of existing registry infrastructure. Like MSBase, the MGBase registry is owned and operated by the MSBase Foundation, an independent, not-for-profit company governed by a Global Board of Directors that advises and supports the registry. Funding for MGBase has been provided by the MSBase Foundation together with fundraising efforts from the Myasthenia Alliance Australia, contributions from pharmaceutical companies and philanthropic donations.
The data fields and minimum dataset for MGBase were selected by a panel of international MG experts. The Myasthenia Gravis Composite (MGC) and myasthenia gravis- activities of daily living (MG-ADL) were chosen as the disease-specific outcome measures included in the minimum dataset. Additional optional disease-specific outcome measures include the quantitative myasthenia gravis (QMG) and myasthenia gravis quality of life 15 revised (MGQOL15r). Data is collected on patient demographics, disease characteristics, investigations, treatments (including prednisolone doses), thymectomy, comorbidities and safety. An international MGBase scientific leadership group provides oversight for the scientific direction of the registry.
The MGBase registry has been designed for use during routine outpatient clinic consultations. Data is entered in real-time via a purpose-designed electronic data entry system. Patient identifiable information is only stored at the local level. Deidentified and encrypted data is regularly uploaded to the MGBase registry where participating centres can request access to the global dataset for investigator-driven studies.
MGBase was officially launched in October 2021. As of 23rd June 2024, MGBase had 678 patient records from 18 clinics across 9 countries. Interim analysis will be presented including patient demographics, disease characteristics, treatment patterns and preliminary outcomes.
SS.43.04
SS.43.04 - Myasthenia Gravis in Australia: a Cause of Significant Morbidity
1The University of Queensland, Brisbane, Australia, 2University of Sydney, Australia
Myasthenia gravis (MG) is a severe neuromuscular disease that has poor community recognition. The incidence of MG in Australia is uncertain. There has been one study of prescriptions for pyridostigmine that gave some data about prevalence. There have been two surveys of the clinical features of MG in Australia. These provide information about clinical features and therapies, and disability. In addition, the first survey contained a section for patients to respond to questions in their own words. 90% of patients indicated that MG had affected their work. In 37% this was due to weakness and fatigue and in 15% this was due to ocular symptoms. 56% of patients considered that their disease had been provoked by stress. These data will be presented.
SS.44.03
SS.44.03 - Handling Fatigue in MG
1Princess Alexandra Hospital, Brisbane, Australia, 2University of Queensland, Brisbane, Australia
Myasthenia gravis (MG) is an autoimmune disease mediated by auto-antibodies against protein targets within the neuromuscular junction. Whilst by its nature MG causes neuromuscular fatigue, patients frequently complain of cognitive fatigue as well. This central fatigue appears to be more frequent in MG than with other neuromuscular conditions, with prevalence figures between 44 and 82% being reported.
Impact of cognitive fatigue on the well-being of patients is high and often more bothersome than the physical disabilities associated with neuromuscular weakness. The cause for this high prevalence of fatigue in MG is unclear; CNS effects of AChR antibodies have been hypothesized, but not proven.
Whilst fatigue severity does appear to correlate with myasthenia motor symptoms, differentiation with other conditions is not straightforward. Exclusion of other co-morbid causes of fatigue, such as psychiatric disease, sleep disorders and metabolic or systemic disorders, is important.
Different therapeutic approaches to fatigue management will be discussed.
SS.44.04
SS.44.04 - Cell Based Autoantibodies
1University Of Oxford, United Kingdom
We have learnt much from establishing diagnostic antibody tests for MG. The first assays were based on radioimmunoprecipitation (RIA) of 125I-alpha-bungarotoxin labelled human muscle AChR. This assay remains ones of the best for diagnosis, but many clinical laboratories cannot use radioactivity and there is clearly a move towards non-radioactive methods. The enzyme linked immunosorbent assays (ELISAs) are preferred by many laboratories but the results are more variable in terms of specificity and sensitivity.
The first cell-based assays (CBAs) were established in order to measure antibodies that only bound to clustered AChRs. The idea was that there might be antibodies in well-defined clinical MG that could not bind monovalently to the AChR in solution, but might bind divalently and therefore more strongly to adjacent AChRs expressed as clusters on the surface of a cell membrane. Human embryonic kidney (HEK) cells were transfected with plasmids encoding the four subunits of adult AChR plus rapsyn which clusters the AChRs at the NMJ. Detecting antibody binding with a red fluorescent anti-human IgG, red clusters could be seen on the live cells. This ‘clustered AChR’ assay has been successful in demonstrating antibodies to AChRs in around 20% of patients previously negative by RIA or ELISA. Similarly, using HEK cells expressing muscle specific kinase (MuSK), MuSK antibodies are detected at slightly higher rates than by RIA alone. CBAs can also be used to distinguish between antibodies binding to adult and fetal AChRs, in mothers whose offspring are at risk of joint contractures and other developmental problems, and to increase positivity in patients with purely ocular MG.
An alternative to the visual detection described here is to use fluorescence-activated cell sorters (FACS) to detect, objectively, the binding of antibodies. Of course, the use of live transfected for either method requires laboratory expertise, and live cells cannot be transported to other labs restricting their use. However, carefully fixed cells are now commercially available for AChR and MuSK antibody tests, and perform almost as well as the live CBAs in systematic comparisons. The cells are expressed on tiny chips. For instance, four chips with adult AChR, fetal AChR or MuSK (and one control cells with no antigen expressed) can be placed together within a well on the glass or plastic slide. Only a small amount of serum is required because of the low volume of the well. As the antigen-expressing cells are pre-mixed with cells expressing no additional antigen, those that bind the antibody can stand out clearly against a negative background. Moreover, use of a relatively high serum to antigen ratio in these tests may be an advantage; in theory, rare antibodies will bind more strongly to each antigen-expressing cell if there are few of them on the chip, increasing detection of low antibody levels. Unfortunately, a CBA for calcium channel antibodies has not yet been established for LEMS, and some of the newer antibodies are only detected in specialist centres. Nevertheless, commercial CBAs are now used worldwide for diagnosis of almost all autoimmune neurological disorders.
SS.45.04
SS.45.04 - Agents that Worsen/Reveal MG
1Griffith University, Sunshine Coast, Australia, 2Sunshine Coast Hospital and Health Service, Birtinya, Australia
Myasthenia gravis is a disorder involving impaired neuromuscular junction transmission, manifesting with variable and fatigable muscle weakness. Historically, a large number of medications and other substances have carried warnings about the risk of worsening the disease and its symptoms. This talk explores the variety of agents implicated in worsening myasthenic control and critically assesses which are likely to have a significant risk versus those that are confounded by the natural variable nature of the disease (and thus false association). Patients and clinicians alike benefit from a rigorous understanding of which agents' warnings to take seriously, such that the person with myasthenia can avoid agents that may be dangerous but also is not unnecessarily deprived of effective treatment options.
SS.46.03
SS.46.03 - FcRn Antagonists
1Department of Neurology, National Institute of Mental Health and Neuro Sciences, Bangalore, India, 2Ellen & Martin Prosserman Centre for Neuromuscular Diseases, Toronto General Hospital, University Health Network, University of Toronto, Toronto, Canada
Advancements in our understanding of the immunopathology of myasthenia gravis (MG) and the progress in drug development have ushered in an era of targeted immune therapies, offering the potential for more effective and better-tolerated treatment options. Several novel molecules have been approved for the treatment of MG in recent years, among which are the class of neonatal Fc receptor (FcRn) antagonists that have a unique mechanism of action.
The circulating pathogenic IgG subclass of antibodies plays a pivotal role in the immunopathology of MG. One method through which conventional treatment techniques, such as large volume plasma exchange, mitigate disease activity is by reducing IgG levels. The extended half-life of IgG, compared to other immunoglobulins in the circulation, is maintained by the protective action of neonatal Fc (FcRn) receptors. These MHC class I molecules bind to and shield IgG from lysosomal catabolism. FcRn antagonists, by blocking the recycling action of FcRn, induce a rapid and marked reduction in IgG levels, earning them the epithet of ‘medical plasma exchangers.’ Several phase III trials have demonstrated the efficacy and safety of FcRn antagonists. Efgartigimod and rozanolixizumab have received US FDA approval for use in seropositive MG, while nipocalimab and batoclimab also hold much promise. Being selective for IgG recycling, FcRn inhibition has not been associated with significant loss of IgM, IgA, or IgE. With the added advantage of being well tolerated and having relatively mild side effects such as headache, nasopharyngitis, diarrhoea, and asymptomatic reductions in monocyte counts observed in clinical trials, FcRn inhibitors offer a valuable therapeutic option for patients with MG.
However, the long-term effects and benefits of FcRn antagonists are still being assessed, and several nuanced considerations and questions arise when comparing them to upstream targeting therapies. The rapid drop in IgG levels is sustained especially after multiple doses, but the level of pathogenic IgG eventually rises back upon discontinuation of FcRn, as the memory B cells and long-lived plasma cells, which are the primary drivers perpetuating MG immunopathology, remain untouched. However, a rebound increase in IgG and thus a clinical backlash after discontinuation have not been noted so far in trials. Questions have arisen regarding the differential impact on IgG subclasses, noting a lower reduction of IgG4 levels compared to IgG1 and IgG3. This disparity raises questions about the efficacy of FcRn antagonists in treating MuSK MG but has not been sufficiently explored. As a distinction is not made, the loss of both pathogenic and non-pathogenic antibodies and the long-term impact of this FcRn-induced immunodeficiency state remain currently unanswered. With the increasing use of monoclonal antibodies in autoimmune diseases, the interactions between FcRn inhibitors and other classes of biological agents and their impact on vaccine efficacy are currently unclear. Unlike the 'broad-spectrum' conventional immunosuppressants and biological agents that act upstream in MG immunology, FcRn antagonists may only theoretically be below par in terms of mitigating MG immunopathology but have proven clinically efficacious. Further research and direct clinical comparisons among agents will be necessary to unravel these complexities.
TC.01.03
TC.01.03 - Rhabdomyolysis- Metabolic Myopathies and Exertional
1Hadassah-hebrew University Medical Center, Australia
The clinical syndrome of rhabdomyolysis is based on the 'classical' triad of myalgia, muscle weakness and pigmenturia. For this lecture it will be defined as an acute clinical disorde5 with markedly elevated serum CK. The common general triggers of rhabdomyolysis are: exertion, external heat and body temperature, fasting and dehydration, drugs and muscle trauma. The causes of rhabdomyolysis are divided into two general groups: genetic tendency (metabolic myopathies and dystrophies) and acquired. The main complications of rhabdomyolysis are acute kidney failure and electrolyte imbalance. There is no consensus on treatment guidelines, but the main therapeutic modes are high fluid load (if renal status allows) and alkalization of urine.
The talk will present the management and diagnostic issues with clinical cases and their dilemmas to enhance the relevance between the theory and clinical practice.
TC.02.03
TC.02.03 - Painful Neuropathy
1University Of Michigan, United States
Pain is a common symptom of neuropathy with one third of patients with diabetic neuropathy experiencing pain. The risks factors for painful neuropathy are similar to those for non-painful neuropathy. Treatment of painful diabetic neuropathy is similar to the treatment of pain from other neuropathies including post herpetic neuralgia, trigeminal neuropathy, idiopathic neuropathy, and chemotherapy induced neuropathy. Treatment of neuropathic pain from neuropathy is similar to the treatment of neuropathic pain from a variety of causes including stroke and multiple sclerosis. Further, treatment of neuropathic pain is similar to the treatment of nociplastic pain.
TC.02.04
TC.02.04 - Evaluation and Management of Myopathic Syndromes - Distal Muscle Weakness
1Tampere Neuromuscular Center, Finland
Distal muscle weakness can be caused by neurogenic or myopathic disorders. The distal myopathies are genetic primary muscle disorders with progressive loss of muscle tissue causing at onset prominent weakness in hands and/or feet. Some may progress later to involve proximal muscles while others remain mainly restricted to distal weakness. The age of onset and the histological findings are extremely variable. High throughput sequencing has further expanded the long list of genes associated with a distal muscular dystrophy. Currently, some 20 genes (ACTN2, CAV3, CRYAB, DNAJB6, DNM2, FLNC, HNRNPA1, HSPB8, KHLH9, LDB3, MATR3, MB, MYOT, PLIN4, TIA1, VCP, NOTCH2NLC, LRP12, GIPS1) have been associated with an autosomal dominant form and four genes (ADSSL, ANO5, DYSF, GNE) are known causes of autosomal recessive forms. Disease-causing variants in five genes (DES, MYH7, NEB, RYR1 and TTN) result in either dominant or recessive distal muscular dystrophy and we have recently identified the first X-linked form. An even more complex digenic mechanism has also been clarified; rare pathogenic mutations in SQSTM1, previously identified with Paget bone disease, cause a distal dystrophy when inherited in combination with a common polymorphism in TIA1.
TC.03.02
TC.03.02 - NGS as First Test in Evaluation of Myopathy?
1University of Western Australia, Australia
The aim of clinician and medical scientist working in partnership is to obtain an accurate diagnosis for the patient and their family. The question is the most efficient way to do this, while doing as little invasive harm to the patient as possible, especially if the patient is a neonate. At what point in the diagnostic process after clinical examination should the current gold standard DNA test be performed? In 1987, the only myopathy gene we had to analyse was the DMD gene associated with Duchenne and Becker muscular dystrophy. Even then however definitive laboratory DNA testing changed the diagnosis of some patients from Kugelberg-Welander spinal muscular atrophy to Becker muscular dystrophy. Today, in 2024, the gold standard molecular diagnostic test is one or other form of next generation sequencing (NGS). If the NGS test works and produces a definitive molecular diagnosis, no further testing such as muscle biopsy is required. Having the molecular diagnosis also allows definitive cascade testing of relatives including identifying the carriers of recessive disease and for example at what point in a Duchenne family the DMD variant arose. Having the molecular diagnosis is also essential for preimplantation or prenatal diagnosis and for presymptomatic diagnosis in late onset disease such as C9Orf72 or SOD1 motor neuron disease. It is crucial to remember that NGS diagnostic tests have limitations – they do not detect everything, and different sequencing methods are differently successful in identifying different types of pathogenic variants. You need to know exactly what NGS test was performed by the laboratory you sent the sample to, so that you have a chance to understand what might have been missed. Standard short-read NGS is not good at identifying tandem repeat disorders. If your patient has DM1 an NGS test won’t detect that or facioscapulohumeral muscular dystrophy. I have seen plenty such cases. New bioinformatics are helping overcome the repeat expansion issue. Exome sequencing is less successful at detecting deletions and duplications than targeted panel testing since the exome sequencing performed by diagnostic laboratories generally does not have the depth of coverage targeted panel sequencing provides. The clinician has to order the right test for the patient. Even the right test can give surprising results in “diagnosis by sequencing” where the laboratory identifies a known pathogenic variant in a gene associated with a totally different disease to the clinical diagnosis. In every such case, the molecular diagnosis, not the clinical diagnosis, has been correct. Exome sequencing and targeted panels will not detect deep intronic or intergenic pathogenic variants. If the clinician can then, through muscle biopsy and immunohistochemistry, pinpoint the gene that should have the pathogenic variant, the laboratory may perform other testing: genome or long-read sequencing, or optical genomic mapping to obtain a definitive diagnosis. Many patients will nevertheless remain without a molecular diagnosis because of variants of uncertain significance, undetected structural variants, or because they do not in fact have a genetic disease – a scenario increasingly likely with patient age.
TC.03.03
TC.03.03 - Repeat Expansion Disorders In NMD
1University Of Auckland, New Zealand
Next Generation gene panels or whole exome sequencing have become the default first test of choice of diagnostic testing for most neurologists when confronted with neuromusuclar conditions. However, some of the most common conditions will be missed with this approach. Expansion repeat disorders need to be considered even before requesting a panel. At the rare end, when common condtions, point variatns and small indels have been excluded there are rare conditions that need to be considered. In this workshop we will consider both these groups of diseases, when and how to investigate them.
TC.03.04
TC.03.04 - ALS Genetics- Why Do They Matter
1Centre for Molecular Medicine and Innovative Therapeutics, Murdoch University, Perth, Australia, 2Perron Institute for Neurological and Translational Science, Perth, Australia
Amyotrophic lateral sclerosis (ALS) is a fatal neurodegenerative disease characterised by the loss of motor neurons in the brain and spinal cord leading to muscle weakness, atrophy and paralysis. The development of ALS is an interaction of genetic and environmental factors, and heritability is estimated at 40-60%. More than 30 genes have been associated with ALS, however an estimated 30-40% of the heritability has yet to be identified. This ‘missing heritability’ may lie in genetic variants not often investigated such as repeat sequences and structural variants. The genetic component of ALS is complex and characterised by monogenic and oligogenic inheritance, incomplete gene penetrance, pleiotropic variants and different variants in a single gene displaying specific effects on disease progression. Understanding the genetic architecture of ALS is important to provide information to individuals identified as carriers of disease-causing variants and to identify pathogenic mechanisms and potential therapeutic targets. Gene-based therapies targeting genes harbouring ALS-causing variants, such as SOD1, C9orf72 and FUS, are currently being developed with several clinical trials underway and is of critical relevance for the treatment of ALS. The current knowledge of the genetic landscape of ALS and its implications will be discussed.
TC.03.05
TC.03.05 - Clues to Acquired vs. Hereditary Neuropathy in a Single Patient
1Neuromuscular Reference Center, Dept of Neurology, Hôpital Universitaire de Bruxelles, Université Libre de Bruxelles, Belgium
The early and reliable distinguishing between acquired neuropathies (AN) and hereditary neuropathies (HN) is an essential issue to adequately orientate the diagnostic process between ad hoc investigations related to acquired etiologies and targeted genetic analyses or next generation sequencing approaches.
Despite inconstantly informative, the systematic collection of family history data remains an essential milestone in the diagnostic pathway for HN while in AN external factors or underlying conditions should be looked for.
The age of onset provides important clues. HN generally manifest in childhood or adolescence, though some forms can appear in adulthood or in elderly. AN tend to develop later in life and may be linked to many factors such as diabetes, drugs intake, toxic exposures, infections, or autoimmune diseases. Progression patterns differ as well; HN progress gradually and insidiously over many years, while AN can present with rapid onset and progression, especially in autoimmune, infectious, or toxic conditions.
Symptom distribution and features are other key factors. HN typically exhibit a length-dependent distal symmetric pattern, while AN can present with asymmetric involvement, multifocal patterns, or include proximal weakness. Initially motor symptoms and muscle atrophy will be predominant in HN while sensory symptoms are more often seen in the early stages of AN.
Associated systemic features often accompany AN. For instance, diabetic neuropathy is linked with other signs of diabetes, autoimmune neuropathies coexist with systemic autoimmune symptoms, and infectious neuropathies might show signs of systemic infection or inflammation. In contrast, apart elevated creatine kinase level, HN usually do not present with abnormal routine blood tests.
Electrodiagnostic assessment including nerve conduction studies and needle electromyography are invaluable for differentiation. HN typically show uniform and symmetric involvement, indicating a long-standing diffuse process. AN can display asymmetric conduction blocks or temporal dispersion, indicating focal demyelination, or asymmetric reduced amplitudes suggestive of non-homogenous axonal degeneration.
Laboratory tests can reveal underlying causes of AN. Elevated blood glucose levels in diabetic neuropathy, positive serological markers in autoimmune neuropathies, low vitamin levels in nutritional neuropathies, or abnormal liver function tests in alcoholic neuropathy are examples.
Peripheral nerve pathology examination is indicated and can be helpful in selected cases. For instance, amyloid depositions being seen in hereditary amyloidosis and inflammatory infiltrates or vasculitis features seen in autoimmune neuropathies are essential to ascertain the diagnosis.
Response to treatments can also provide diagnostic clues such as in acquired immune-mediated neuropathies improving with immunosuppressive therapy or diabetic neuropathy stabilizing with strict glucose level control.
Distinguishing acquired from hereditary peripheral neuropathies involves a comprehensive collection of data including a thorough evaluation of family history, age of onset, progression pattern, symptom distribution and features, clinical examination findings, electrodiagnostic characteristics, associated systemic features and response to past treatments. This rigorous assessment will lead clinician to order tailored ancillary tests such as laboratory tests, nerve pathology or genetic investigations. Mastering these diagnostic clues is crucial for neurologists to ensure fast and accurate diagnosis for the optimal patient care. Engage in this course to enhance your diagnostic skills and elevate your efficiency in managing peripheral neuropathies.
TC.04.02
TC.04.02 - What are the Mechanisms Leading To GBS
1Brigham and Womens Hospital / Harvard Medical School, United States
GBS has an estimated annual incidence ranging from 0.9 to 4 per 100,000. Approximately 60%–75% of patients with GBS have a history of a recent infection within the 8 weeks prior to the onset of the neuropathy. Campylobacter jejuni is most commonly identified, with roughly 30% of patients having serologic evidence of recent infection. Serologic evidence of recent Epstein–Barr virus or Mycoplasma pneumoniae infection are found in 5%–10% of patients. Other infectious agents associated with GBS include influenza, hepatitis A, B, C, and E, Zika virus, and human immunodeficiency virus. Vaccinations have at times been associated with GBS, most notably the swine flu vaccine in the 1970s. By contrast, the risk of GBS following influenza infection is estimated to be 17 cases per 1 million infections. A possible infrequent link with SARS-CoV-2 infection and mRNA vaccination has emerged.
IgG autoantibodies against GM1 or GD1a are strongly associated with AMAN and AMSAN. IgG anti-GQ1b antibodies, which cross-react with GT1a, are strongly associated with the Miller Fisher syndrome. Cellular immunity likely plays a role in the pathogenesis of GBS, given the T-cells and macrophages apparent in the nerves, markers of T-cell activation (e.g., soluble interleukin-2 receptor and interferon-γ) in the serum, and the resemblance to experimental allergic neuritis. Further, injection of serum from patients with AIDP into nerves of animal models induces complement-dependent demyelination and conduction block.
The nature of the responsible epitope(s) is not known but probably is a glycolipid. Unlike AMAN, which has been more clearly linked with autoantibodies targeting the GM1 ganglioside, autoantibodies against gangliosides, neurofascins, or otherwise are very rarely detected in AIDP. Molecular similarity between myelin epitope(s) and glycolipids expressed on Campylobacterand other infectious agents may be the underlying trigger for the immune attack. Antibodies directed against these infectious agents may cross-react with specific antigens on the Schwann cell because of this molecular mimicry. These autoantibodies may bind to the Schwann cells and then activate the complement cascade, leading to lysis of myelin sheaths. Inflammatory cells are subsequently recruited to complete the demyelinating process.
Autoantibodies targeting neurofascin-155, neurofascin-140, neurofascin-186, contactin-1 (CNTN-1), and contactin-associated protein-1 (Caspr-1) have been implicated in cases of autoimmune neuropathy. The fre¬quency of these antibodies among patients diagnosed with “CIDP” has ranged from 1% to 18%. Patients harboring these autoantibodies can have unique clinical features not typically seen with CIDP or other variants. Patients are fre¬quently refractory to standard treatments for CIDP (corticosteroids and IVIg), but instead appear to respond uniquely well to rituximab. The 2021 EAN/PNS Guidelines explicitly excluded the nodopathies and paranodopathies from the CIDP umbrella, creating a new disease carve-out. Neurofascin-140 and -186 help cluster voltage-gated sodium channels within the nodes of Ranvier. Potassium channels are clustered in the juxtaparanodal portion of the axons. Neurofascin-155 is expressed by Schwann cells at the paranodes. It affixes these myelin loops to the axon via binding to CNTN-1 and Caspr-1, which form a complex together on the axonal cell membrane
TC.04.03
TC.04.03 - Breakthrough Treatments Exist for TTR, What Is Next?
1National Taiwan University Hospital, Taiwan
Hereditary transthyretin amyloidosis (ATTRv) with polyneuropathy was considered a relentless neurodegenerative disorder of the peripheral nervous system with amyloid deposition as the pathognomonic diagnosis. There are already more than 140 mutations of transthyretin (TTR) reported. Mutant TTRs easily become monomeric form from the native tetrameric status. Over the last decade, there has been a revolution in treatments including TTR stabilizer (tafamidis and diflunisal) and gene therapy of RMAi, antisense, and CRISPR. The introduction of these treatments has tremendously reduced the progression of the disease by stabilizing TTR or reducing the production of TTR. This lecture will focus on the effects of these new technology-based therapies and the emerging challenge.
TC.04.04
TC.04.04 - Genes to Know and Treatments to Come
1Department Of Human Genetics, University Of Miami Miller School Of Medicine, United States
Neuromuscular disease genetics has been very successful in characterizing monogenic causes and underlying molecular mechanisms, such that we are entering an age of therapeutics for the very first time form most NMD phenotypes. Several major challenges and strategic tasks are beaconing obvious for the field, including: 1. To address the still considerable diagnostic gap of 30-50% of patients, 2. To develop strategies that deal with variants of uncertain significance as gene specific treatments are becoming available, and 3. To accelerate the development of gene specific therapies, ideally using platform technologies. This presentation will outline these opportunities, illustrated with remarkable recent examples of success, and discuss the technological changes that are coming to the field of NMD.
TC.06.03
TC.06.03 - Inflammation in Muscle Biopsy - Myositis or Not
1Brigham and Womens Hospital / Harvard Medical School, United States
There are five major categories of idiopathic inflammatory myopathy (IIM): dermatomyositis (DM), polymyositis (PM), antisynthetase syndrome (ASyS), immune-mediated necrotizing myopathy (IMNM), and inclusion body myositis (IBM), which are clinically, histologically, and pathogenically distinct. However, the category of PM is increasingly diminishing as most cases are now known to be IBM, ASyS, and IMNM. Clinical, laboratory (e.g., antibodies, CK levels), and histopathological features and can distinguish these from each other. Inflammatory myopathies may occur in isolation or in association with cancer, various connective diseases (overlap syndromes), and autoantibodies. Other less common myositides include granulomatous/giant cell/sarcoid myositis, eosinophilic myositis, myositis associated with infections), and complications of check-point inhibitors that myositis, myocarditis, and
MG used in the treatment of various malignancies. Muscle biopsies in patients with myasthenia gravis, although typically not done, can show lymphorrhages without frank clinical myositis. Additionally, sporadic late onset nemaline myopathy (SLONM) can have inflammatory infiltrates on muscle biopsies and is speculated to be autoimmune in nature but is not classified per se as an “inflammatory myopathy”.
Furthermore, inflammatory infiltrates on a muscle biopsy do not necessarily imply an autoimmune etiology for the myopathy in question. In this regard, various muscular dystrophies (e.g., congenital, facioscapulohumeral, calpainopathies, dysferlinopathies, anoctaminopathies, etc.) may be associated with inflammation and can be misdiagnosed as PM. Likewise, rhabdomyolysis with prominent inflammatory infiltrate invading necrotic fibers are a hallmark of IMNM but are also see in toxic myopathies (e.g., statin myotoxicity) and metabolic myopathies (e.g., McArdle disease, CPT2 deficiency). Again, other clinical, laboratory, and histopathological features are important in distinguishing these disorders from an autoimmune myositis.
On the other end of the spectrum is the patient with muscle weakness, laboratory features of muscle damage (e.g., elevated muscle enzymes, myopathic electromyography, abnormal skeletal muscle MRI) that have a dystrophic appearing muscle biopsy without muscle inflammatory cell infiltrate on muscle biopsy. These patients can be misdiagnosed as having an untreatable limb-girdle muscular dystrophy, but they may have IMNM that is amenable to immunotherapy.
Despite what we know or think we know even the most experience clinician comes across challenging cases in which the nature of the myopathy is not clear. In such cases, a trial of immunotherapy may be warranted. However, aggressive to treat such patients is a difficult decision one needs to make with the patient and family, weighing risks versus benefits.
TC.06.04
TC.06.04 - Storage Disorders
1University of Gothenburg, Sweden
The main fuels for muscle energy production are fatty acids and glucose. Defects in their metabolism can cause glycogen or lipid storage myopathies, characterized by the abnormal accumulation of glycogen or fat in muscle fibers.
Muscle glycogen storage diseases manifest as either exercise intolerance or fixed muscle weakness, although both can occur in the same disease. Glycogen storage myopathies with fixed muscle weakness are often characterized by the storage of polyglucosan, an abnormally structured, less-branched form of glycogen. Examples include branching enzyme (GBE1) deficiency, glycogenin-1 (GYG1) deficiency, and RBCK1 (RBCK1) deficiency. Polyglucosan storage diseases can also exhibit accumulation of various proteins, such as desmin, which can lead to misdiagnosis as desminopathy. In Pompe disease (GAA), fixed muscle weakness is also a typical feature, but the abnormal glycogen storage occurs primarily in lysosomes, making Pompe disease fundamentally different from other muscle glycogen storage diseases (GSDs). McArdle disease (PYGM) typically manifests as exercise intolerance, with symptoms including muscle fatigue, cramps, and rhabdomyolysis. The storage material in McArdle disease consists of normal glycogen located in the cytoplasm, similar to normal glycogen. In the rare glycogen storage myopathy 10, caused by phosphoglycerate mutase (PGAM2) deficiency, the presence of tubular aggregates may provide a diagnostic clue.
Lipid storage myopathies are rare diseases caused by defects in the import or metabolism of fatty acids in mitochondria. Massive lipid storage can also result from a deficiency of the cytosolic enzyme adipose triglyceride lipase (PNPLA2). Clinically, lipid storage diseases present as either fixed, slowly progressive muscle weakness or metabolic crises induced by exercise, fasting or infections, which may cause rhabdomyolysis. These diseases are characterized by enlarged size and increased numbers of the small lipid droplets in muscle fibers, which in severe cases coalesce into large lipid lakes observable by light microscopy. In diagnosing lipid storage myopathies, investigating the acyl-carnitine profile in blood by mass spectrometry is crucial, as different enzyme defects exhibit characteristic profiles. Carnitine may be effective in treating carnitine deficiency and riboflavin may be effective in treating multiple acyl-CoA dehydrogenase deficiency (MADD). Genetic investigations are therefore important; however, drug-induced lipid storage myopathy may be more frequent than genetic causes in adults.
TC.07.05
TC.07.05 - Muscle and Nerve Biopsy – Do We Still Need it in the Era of Ngs?
1Neuromuscular Center, Department Neurosciences,Azienda Ospedaliera Di Padova, Italy
Many neuromuscular diagnoses are based on muscle/nerve pathology findings. In recent years, extensive genetic analysis by NGS has been increasingly used, however, muscle/nerve pathology remains essential for both diagnostic and prognostic purposes.
INFLAMMATORY MYOPATHIES
Autoantibody testing for myositis has been extensively used, and muscle biopsies are less commonly performed, muscle biopsies are done in cases of autoimmune myositis that could be misdiagnosed as muscular dystrophy. Checkpoint inhibitors used in melanoma and cancer can cause inflammatory myopathies.
METABOLIC MYOPATHIES
The detection of enzyme defects in glycogenosis and metabolic disorders such as primary and secondary carnitine deficiency, coenzyme Q deficiency, and polyglucosan body diseases, sets up diagnosis, allowing treatment. Glycogenosis type 2 (GSD2) is caused by the deficiency of the lysosomal enzyme acid α-glucosidase with a spectrum of clinical phenotypes that range from the infantile Pompe Disease (PD) to childhood, juvenile forms, and progressive adult form, named "late-onset" Pompe disease (LOPD). Biopsy structure is severely affected in PD, whereas the degree of vacuolization is extremely variable in biopsies of LOPD. Some vacuoles in LOPD present caveolin or other sarcolemmal protein staining, indicating their complex structure. Since 2006 Enzyme Replacement Therapy (ERT) has been available, several trials in GSD2 with various types of Neo-GAA and chaperones have been done as well as trials with Next Generation ERT. The biopsy for GSD2 is of great importance for a correct diagnosis and is still the golden standard for ERT.
TOXIC VACUOLAR MYOPATHIES
Muscle biopsies' utility is provided from toxic-metabolic cases such as cocaine myopathy, cocaine users have rhabdomyolysis. Many rhabdomyolytic episodes are not predictable, making biopsy evaluation useful to demonstrate its pathology (Figure illustrates necrotic myofibers invaded by macrophages), likewise in colchicine or statin myopathies.
MITOCHONDRIAL MYOPATHIES/ NEUROPATHIES
In mitochondrial myopathies or neuropathies (MELAS; MINGIE; SANDO etc.), due to the cooperation of two genomes nuclear and mitochondrial, nerve/muscle biopsies for histopathological, ultrastructural, and biochemical measurement of respiratory chain complexes or mitochondrial nuclear-driven disorders are needed.
MUSCULAR DYSTROPHIES
Biopsy allows the identification of BMD by western blotting and of female carriers by immunohistochemistry. Various LGMD types can be identified by immunohistochemistry or western blotting, muscle pathology should be proactively considered when a single gene presents multiple phenotypes.
CRITICAL ILLNESS MYOPATHY/NEUROPATHY
Critical Illness Myopathy can be diagnosed by immunohistochemistry and ultrastructure demonstrating myosin filament loss.
NERVE BIOPSY INDICATIONS
Indications for nerve biopsy have changed over time since alternative diagnostic modalities have reduced their use. These include neuroimaging techniques such as magnetic resonance neurography and nerve ultrasound. A nerve biopsy may be performed to identify nerve degeneration, and inflammatory neuropathy, and confirm specific diagnoses such as Chronic Inflammatory Demyelinating Polyradiculoneuropathy (CIDP). A wide spectrum of therapies is now available for acquired neuropathies including CIDP and its variants, multifocal motor neuropathy, and vasculitic neuropathy. A biopsy might be required in amyloid neuropathies and familial ATTR amyloid polyneuropathy, currently becoming a treatable disease. Nerve biopsy in diseases such as pure neuritic leprosy, neurosarcoidosis, and neurolymphomatosis enables a definitive diagnosis.
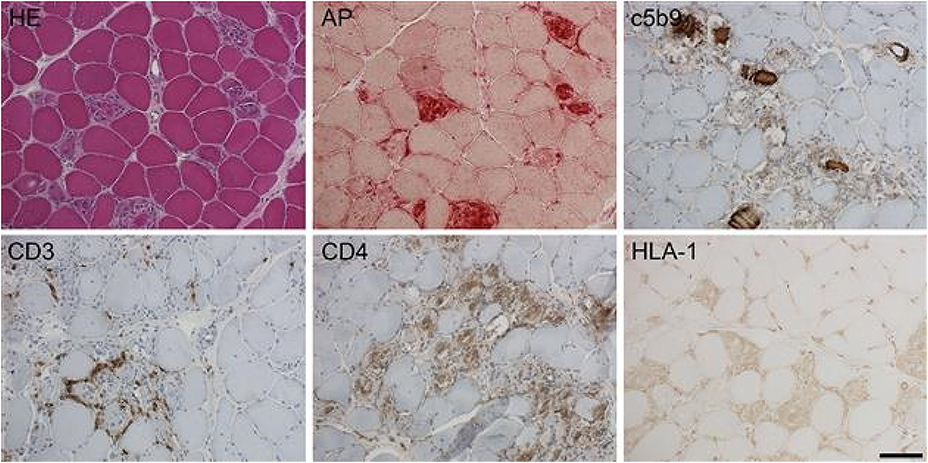
TC.08.02
TC.08.02 - What are the Mechanisms Leading to ALS
1Neuroscience Research Australia, Australia
Recent evidence has emerged concerning unique pathophysiological processes linked to the development of amyotrophic lateral sclerosis (ALS), including glutamate-mediated excitotoxicity, which has resulted in development of novel diagnostic investigations and uncovered potential therapeutic targets. Advances in genetics, particularly the C9orf72 gene, have radically changed the pathological mind-set, such that ALS forms part of a continuum with other primary neurodegenerative disorders, including frontotemporal dementia. The mechanisms underlying neurodegeneration in ALS are multifactorial and operate through inter-related molecular and genetic pathways. Specifically, neurodegeneration in ALS likely results from a complex interaction of glutamate excitoxicity, generation of free radicals, cytoplasmic protein aggregates, SOD1 enzymes, combined with mitochondrial dysfunction, and disruption of axonal transport processes through accumulation of neurofilament intracellular aggregates. Separately, the development of the human motor system might potentially be perturbed during childhood by increased exposure to childhood infections, as occurs in families with young children. Various environmental risk factors for ALS have also been suggested, including a lifetime of intensive sport or physical exertion and active service in the military. Mutations in TARDBP and FUS result in formation of intracellular aggregates, which are harmful to neurons. Activation of microglia results in secretion of proinflammatory cytokines, resulting in further toxicity. Ultimately, motor neuron degeneration occurs through activation of calcium-dependent enzymatic pathways. The hallmark of the TDP-43 pathological signature of sporadic ALS is restricted to cortical areas as well as to subcortical nuclei that are under the direct control of corticofugal projections. This provides anatomical support that the origins of the TDP-43 pathology reside in the cerebral cortex itself, secondarily in corticofugal fibres and the subcortical targets with which they make monosynaptic connections. The latter feature explains the multisystem degeneration that characterises ALS. Consideration of ALS as a primary neurodegenerative disorder of the human brain may incorporate concepts of prion-like spread at synaptic terminals of corticofugal axons. Further, such a concept could explain the recognised widespread imaging abnormalities of the ALS neocortex and the accepted relationship between ALS and frontotemporal dementia.
TC.08.05
TC.08.05 - How Should we Treat Metabolic Neuropathy?
1University Of Michigan, United States
Metabolic neuropathy is secondary to underlying diabetes, obesity, dyslipidemia, and hypertension. These factors comprise the metabolic syndrome. Treatment of this common condition should focus on each of these individual components. Diabetes management is fast evolving including newer treatments such as SGLT2 inhibitors and GLP-1RA. Obesity treatment includes dietary weight loss, surgical weight loss, and medication weight loss. Dyslipidemia and hypertension are known cardiovascular risk factors that have many effective agents. Treatment of metabolic factors has the potential to improve cardiovascular, cognitive, and neuropathy outcomes.
TC.09.04
TC.09.04 - The Differential Diagnosis of Limb Girdle Weakness
1John Walton Muscular Dystrophy Research Centre, Newcastle University, Newcastle Upon Tyne, United Kingdom
Weakness of the proximal limb muscles, especially the shoulder and pelvic girdle muscles, is one of the first manifestations of many genetic neuromuscular diseases. Most of those diseases are primary muscle diseases, but diseases of the anterior horn cell and the neuromuscular junction can also present with onset of weakness in the limb girdle muscles. The first problem patients will often encounter is the difficulty to walk upstairs independently, without holding on to a rail. Generally, proximal muscle weakness will gradually progress and can also affect the truncal, respiratory and at later stages even the distal muscles, although the latter ones are surprisingly well preserved in some limb girdle syndromes. It is not well understood why certain primary muscle diseases show a preference for pathology of the proximal muscles whereas others manifest with distal weakness, even though the responsible genes are ubiquitously expressed in skeletal muscle. Limb girdle weakness is fairly unspecific, and an accurate diagnosis will depend on additional clinical features and nowadays be most commonly established by genetic sequencing via gene panels, exome or whole genome sequencing.
When it comes to the differential diagnosis of inherited diseases with limb girdle weakness, the heterogenous group of limb girdle muscular dystrophies (LGMD) needs to be considered, keeping in mind that patients with Becker muscular dystrophy and spinal muscular atrophy type 3 (SMA3) can be very difficult to distinguish from LGMD. One should always make sure that treatable diseases like SMA3 and Pompe disease form part of the differential diagnosis. As several LGMD also develop cardiomyopathy and patients with myofibrillar myopathies and Emery-Dreifuss muscular dystrophy can also present with proximal weakness and cardiac involvement, cardiac assessments form an integral part of the diagnostic workup of patients with limb girdle weakness. Despite the fact that most genetic neuromuscular diseases presenting with limb girdle weakness can now be diagnosed with massive parallel sequencing technologies, the interpretation of the results and supportive investigations like muscle imaging and muscle biopsy analysis requires the input of a neuromuscular specialist.
Abstract Submissions
OS.01.03
OS.01.03 - Effect Size Analysis: Cipaglucosidase Alfa+Miglustat versus Alglucosidase Alfa in ERT-Experienced Adults with Late-Onset Pompe Disease
1PARC Research Clinic, Royal Adelaide Hospital, Adelaide, Australia, 2University of Florida, Gainesville, USA, 3Department of Neurology, University Hospitals Leuven, and Department of Neurosciences, Laboratory for Muscle Diseases and Neuropathies, KU Leuven, Leuven Brain Institute (LBI), Leuven, Belgium, 4Department of Neurology, University of Pittsburgh School of Medicine, and VA Pittsburgh Healthcare System, Pittsburgh, USA, 5John Walton Muscular Dystrophy Research Centre, Newcastle University, Newcastle upon Tyne, UK, 6Neuromuscular Disorders Unit, Neurology Department, Hospital de la Santa Creu I Sant Pau, Barcelona, Spain, 7Centro de Investigacíon en Red en Enfermedades Raras (CIBERER), Barcelona, Spain, 8Department of Neurology, University of Kansas Medical Center, Kansas City, USA, 9Duke University Medical Center, Durham, USA, 10Department of Neurology & Rehabilitation Medicine, University of Cincinnati College of Medicine, Cincinnati, USA, 11Salford Royal NHS Foundation Trust, Salford, UK, 12ERN-NMD Center for Neuromuscular Disorders of Messina, Department of Clinical and Experimental Medicine, University of Messina, Messina, Italy, 13Amicus Therapeutics, Inc., Princeton, USA, 14Department of Neurology, University of California, Irvine, USA, 15Friedrich-Baur-Institute at the Department of Neurology, LMU University Hospital, LMU Munich, Munich, Germany
OS.01.04
OS.01.04 - Safety and Efficacy of Delandistrogene Moxeparvovec versus Placebo in DMD: Phase 3 EMBARK Primary Results
Dr. Jerry R. Mendell1,2,
1Center for Gene Therapy, Nationwide Children’s Hospital, Columbus, USA, 2The Ohio State University, Columbus, USA, 3The Dubowitz Neuromuscular Centre, NIHR Great Ormond Street Hospital Biomedical Research Centre, Great Ormond Street Institute of Child Health and Institute of Neurology, University College London, & Great Ormond Street Hospital Trust, London, UK, 4UC Davis Health, Sacramento, USA, 5Pediatric Neurology Institute, Catholic University and Nemo Pediatrico, Fondazione Policlinico Gemelli IRCCS, Rome, USA, 6University of Rochester Medical Center, Rochester, USA, 7Translational Medical Center, National Center of Neurology and Psychiatry, Kodaira, Japan, 8Department of Pediatrics, University of Florida, Gainesville, USA, 9Neuromuscular Unit, Neuropaediatrics Department, Hospital Sant Joan de Déu, Fundacion Sant Joan de Déu, CIBERER – ISC III, Barcelona, Spain, 10Children’s Hospital of the King’s Daughters, Norfolk, USA, 11Department of Pediatric Neurology, Center for Neuromuscular Disorders in Children and Adolescents, University Clinic Essen, University of Duisburg-Essen, Essen, Germany, 12Department of Pediatrics, Division of Neurology, University of Arkansas for Medical Sciences, Arkansas Children’s Hospital, Little Rock, USA, 13Department of Neurology, Washington University in St Louis, St Louis, USA, 14F. Hoffmann-La Roche Ltd, Basel, Switzerland, 15Roche Products Ltd, Welwyn Garden City, UK, 16Sarepta Therapeutics, Inc., Cambridge, USA
Delandistrogene moxeparvovec, a recombinant adeno-associated virus rhesus isolate serotype 74 (rAAVrh74)-based gene transfer therapy designed to address the absence of functional dystrophin by delivering a transgene encoding engineered micro-dystrophin, is approved in the USA, UAE, Qatar, Kuwait, Bahrain and Oman for the treatment of ambulatory paediatric patients aged 4 through 5 years with Duchenne muscular dystrophy (DMD) with a confirmed mutation in the DMD gene (as of March 2024).
We report findings from Part 1 (Week 52) of the 2-part EMBARK trial (NCT05096221).
At Week 52 (N=125), the primary endpoint was not statistically significant— the least-squares mean between-group difference for delandistrogene moxeparvovec (n=63) versus placebo (n=61) was 0.65 points (P=0.2441). Between-group differences for key secondary functional endpoints of time to rise (–0.64 seconds; P=0.0025) and 10-metre walk/run (–0.42 seconds; P=0.0048) favoured the treatment group (nominal P-values). Other secondary functional endpoints of stride velocity 95th centile (0.10 metres/second; P=0.0402) and time to ascend 4 steps (–0.36 seconds; P=0.0412) also favoured the treatment group (nominal). A pre-specified global statistical test on a composite of functional endpoints supported treatment benefit (P=0.0044). Week 12 biopsies in a subset of patients (n=31) showed delandistrogene moxeparvovec micro-dystrophin expression in the treated group by western blot analysis. Mean creatine kinase levels decreased from baseline to Week 52 with delandistrogene moxeparvovec versus placebo (between-group difference, –4,343.59 U/L; P=0.0002, nominal). Treatment-related treatment-emergent adverse events (AEs) included vomiting (54.0%), nausea (31.7%) and decreased appetite (27.0%), and no study discontinuations, deaths or complement-mediated AEs were observed.
Based on the totality of functional assessments including the timed function tests, treatment with delandistrogene moxeparvovec indicates beneficial modification of the disease trajectory. The safety profile was manageable and consistent with prior experience.
OS.01.05
OS.01.05 - Safety and Efficacy of DYNE-101 in Adults with DM1: Phase 1/2 ACHIEVE Trial Data
1Neurogenetics Clinic Centre for Brain Research, University of Auckland, Auckland, New Zealand, 2Institut de Myologie, Paris, France, 3John Walton Muscular Dystrophy Research Centre, Newcastle University, Newcastle-Upon-Tyne, United Kingdom, 4Muscle Disease Unit, Northern Care Alliance NHS Foundation Trust, Manchester Academic Health Science Centre, Manchester, United Kingdom, 5Fondazione Policlinico Universitario A. Gemelli, Rome, Italy, 6Friedrich-Baur-Institute, Dep. of Neurology LMU Clinics, Ludwig-Maximilians University, Munich, Germany, 7University College London Hospitals, London, United Kingdom, 8Dyne Therapeutics, Waltham, United States, 9Centro Clinico NEMO, University of Milan, Milan, Italy, 10Radboud University Medical Center, Nijmegen, Netherlands
DM1 is a spliceopathy caused by expansion of CUG repeats in the DMPK RNA that leads to multisystem clinical manifestations. DYNE-101, an investigational therapeutic for treatment of DM1, consists of a TfR1-binding Fab conjugated to an ASO designed against mutant nuclear DMPK RNA to correct splicing. The safety and efficacy of DYNE-101 in adults (18-49 years old) are being investigated in the Phase 1/2 ACHIEVE trial (NCT05481879). For this analysis, 16 participants had efficacy data through 6 months (6M) of follow-up in the 1.8 mg/kg (approximate ASO) dose cohort and 16 participants had biopsy data through 3M of follow-up in the 3.4 mg/kg dose cohort of the multiple ascending dose (MAD) portion of ACHIEVE. Participants were randomized to receive DYNE-101 (n=6) or placebo (n=4) every 4 weeks (Q4W), or two doses of DYNE-101 followed by placebo for the remainder of the MAD period (n=6). Safety and tolerability are based on 45 participants enrolled in ACHIEVE as of the data cut-off date. At 3M, 1.8 and 3.4 mg/kg DYNE-101 showed mean 10.0 ng/g and 21.5 ng/g ASO concentration, mean 25% and 40% DMPK knockdown, and mean 13% and 19% splicing correction from baseline, respectively. At 6M, 1.8 mg/kg DYNE-101 showed mean 16% DMPK knockdown, +7% CASI change from baseline, 3.8-second improvement in myotonia, and improvement in MDHI, a patient-reported outcome. 3/5 evaluable participants had splicing correction at 3M, persisting through 6M. In the 3.4 mg/kg cohort, 5/5 evaluable participants had splicing correction at 3M. DYNE-101 had a favorable safety profile as of the data cut-off date, with mostly mild or moderate TEAEs and no clinically meaningful changes in kidney and liver parameters or treatment-emergent anemia. The initial data with DYNE-101 demonstrated dose-dependent DMPK knockdown and splicing correction with durable effect. Improvement in myotonia was also observed at the lowest dose tested. The data support the continued development of DYNE-101 for the treatment of DM1.
OS.01.06
OS.01.06 - Developing Allele-Selective Antisense Oligonucleotide Therapeutics for ACTA1-Related Myopathy
1Harry Perkins Institute of Medical Research, QEII Medical Centre, Nedlands, Australia, 2Centre for Medical Research, University of Western Australia, QEII Medical Centre, Nedlands, Australia, 3APHP, Centre de Référence de Pathologie Neuromusculaire Nord-Est-Ile-de-France, Henri Mondor Hospital, Créteil, France, 4Université Paris Est, INSERM, IMRB, Créteil, France, 5Sorbonne Université, Myology Institute, Neuromuscular Morphology Unit, Center for Research in Myology, GH Pitié-Salpêtrière, Paris, France, 6Centre de Référence de Pathologie Neuromusculaire Paris-Est, GHU Pitié-Salpêtrière, Assistance Publique-Hôpitaux de Paris, Paris, France, 7Health Futures Institute, Murdoch University, Murdoch, Australia
Skeletal muscle alpha actin (ACTA1) is the predominant actin isoform expressed in post-natal skeletal muscle. ACTA1 is the principal component of the thin filament and interacts with myosin in the thick filament to generate the force of muscle contraction. More than 340 pathogenic or likely pathogenic variants have been reported in ACTA1, and these are associated with 20 distinct phenotypes. The vast majority (>90%) of these pathogenic/likely pathogenic variants are dominant (many arising de novo) and elicit a ‘poison protein’ effect that prevents the normal structure and/or function of the sarcomere, leading to a decrease in muscle contractile force. Although dominant pathogenic ACTA1 variants cause a wide spectrum of disease severities, most patients present with severe infantile nemaline myopathy (CMYP2C; OMIM#620278) at birth and pass away during childhood. With no approved therapies available to patients, there is an urgent unmet need to develop life-saving treatments.
Various lines of evidence from both patients and mouse models indicate that reducing the abundance of the pathogenic protein (relative to wild-type protein) is likely to have a therapeutic benefit. Furthermore, carriers of ACTA1 null variants are asymptomatic, indicating that a single wild-type copy of ACTA1 is sufficient for normal muscle function. Therefore, therapies that knock down the pathogenic ACTA1 mRNA and protein are likely to confer a therapeutic benefit in patients.
Here, we set out to establish a pipeline to develop and screen allele-selective oligonucleotide therapeutics for dominant ACTA1-related myopathy. We have established three independent patient-derived induced pluripotent stem cell (iPSC) lines that we have differentiated into myogenic progenitor cells. These myogenic progenitors can be further differentiated to multinucleated myotubes expressing a range of myogenic markers, including key sarcomeric components (actin and myosin) and myogenic transcription factors (myogenin) by Day 4 of differentiation.
As there are many pathogenic ACTA1 variants, treatments should ideally act independently of the individual patient variant. As such, we used phased genotyping to identify single nucleotide polymorphisms (SNPs) common in the general population (MAF >0.1) that occur in cis with each patient’s pathogenic variant. These SNPs were subsequently used as the target site for the design of allele-selective antisense oligonucleotides (AS-ASOs) that could knock down pathogenic mRNA. Cultured patient-derived myotubes were treated with AS-ASOs at a range of doses (0.25 – 1 µM). After four days of treatment, the efficacy and selectivity of each AS-ASO was quantified and toxicity was assessed using a cell viability assay. Our data provide the first evidence for the feasibility of this approach at the ACTA1 locus. Further, we have successfully developed a workflow for the design and screening of AS-ASO treatments for congenital myopathies in patient-derived myotubes.
OS.01.07
OS.01.07 - Clinical Outcome Study for Dysferlinopathy (LGMDR2/2B):Ten Years of Natural History Data and Implications for Management
1John Walton Muscular Dystrophy Research Centre, Newcastle Upon Tyne, United Kingdom, 2COS Consortium funded by the Jain Foundation, Seattle, USA, 3Center for Translational Science, Division of Biostatistics and Study Methodology, Children’s National Health System, Washington, USA, 4Department of Neurology and Neurological Sciences, Stanford University, Stanford, USA, 5Nationwide Children’s Hospital, Columbus, USA, 6Biodonostia Health Research Institute, Neurology Service, Donostia University Hospital, Donostia-San Sebastian, Spain, 7Department of Neurology, Columbia University Irving Medical Center, New York, USA, 8Department of Neurology, Washington University School of Medicine, St Louis, USA, 9Copenhagen Neuromuscular Center, Department of Neurology, Rigshospitalet, University of Copenhagen, Copenhagen, Denmark, 10Carolinas MDA Care Center, Atrium Health, Charlotte, USA, 11Department of Neurology, National Center of Neurology and Psychiatry, Tokyo, Japan, 12Laboratory of Molecular Neurology, Pusan National University Yangsan Hospital, Yangsan, Republic of Korea, 13Department of Neurology, MDA ALS and Neuromuscular Center, University of California, Irvine, USA, 14Institut de Myologie, Groupe Hospitalier Pitié-Salpêtrière, Paris, France, 15Department of Neuroscience, University of Padova, Padova, Italy, 16Departamento de Neurología y Neurocirugía Clínica, Clínica Dávila, Santiago, Chile, 17Neuromuscular Disorders Unit, Neurology Department, Hospital de la Santa Creu i Sant Pau, Barcelona, Spain, 18Neuromuscular Unit, Department of Neurology, Hospital U. Virgen del Rocío/Instituto de Biomedicina de Sevilla, Seville, Spain, 19Jain Foundation, Seattle, USA
COSII aims to validate the findings from COSI in an expanded cohort, and introduced a care discussion with patients, as access to multidisciplinary clinical care was highly variable across trial sites. Care guidelines for LGMDR2 are under development.
OS.01.08
OS.01.08 - ReInForce: A Bicentric, Randomized, Double Blind, Placebo-Controlled Study to Evaluate Satralizumab in FSHD1
1Ottawa Hospital Research Institute, Ottawa, Canada, 2Peripheral Nervous System and Muscle Department, University Côte d’Azur, CHU Nice, Nice, France, 3University Cote d’Azur, Inserm, CNRS, Institute for Research on Cancer and Aging of Nice (IRCAN), Nice, France, 4Roche Products Ltd, Welwyn Garden City, UK, 5F. Hoffmann-La Roche Ltd, Basel, Switzerland
Facioscapulohumeral muscular dystrophy (FSHD) is marked by progressive muscle weakness in facial, shoulder girdle, upper arms, lower limbs, and abdominal muscles, causing considerable morbidity and decreasing quality of life. There are currently no approved therapies for FSHD. The primary form, FSHD1, is linked to harmful overactivity of the DUX4 gene, resulting in muscle atrophy and weakness. Studies indicate that abnormal DUX4 expression triggers inflammatory processes in the initial stages of the disease. Patients with FSHD1 had increased inflammatory and reduced anti-inflammatory cytokines levels, indicating chronic inflammation. IL-6 levels strongly correlate with clinical severity in patients, and with functional scores in patients and FSHD-like mouse models.
Here we present the study design of ReInForce (NCT06222827), a bicentric, randomized, double-blind, placebo-controlled, Phase 2 study to investigate the safety and efficacy of satralizumab, an IL-6 receptor inhibitor, in adults with FSHD1. Patients (N=40) will receive 120 mg satralizumab or placebo subcutaneously at Weeks 0, 2, 4 and then every 4 weeks until Week 48. The 48 weeks of the Double-Blind period will be followed by a 48-week Open Label period. The study will evaluate efficacy by assessing changes in muscle composition and function, as well as measures of clinical disease progression.
Given the pathological relevance of inflammation in FSHD, and correlation of IL-6 levels with disease severity, satralizumab may reduce muscle and systemic inflammation, thereby reducing fibrofatty degeneration in FSHD.
OS.02.02
OS.02.02 - Whole-Body Muscle Magnetic Resonance Imaging in 81 Patients with Spinal and Bulbar Muscular Atrophy
1Department of Neurology, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Republic of Korea, 2Department of Immunology, Kyungpook National University, Daegu, Republic of Korea, 3Department of Neurology, Kyungpook National University Chilgok Hospital, Daegu, Republic of Korea, 4Department of Neurology, Dongguk University College of Medicine, Dongguk University Gyeongju Hospital, Gyeongju, Republic of Korea, 5Department of Neurology, Dongsan Hospital, Keimyung University School of Medicine, Daegu, Republic of Korea
OS.02.03
OS.02.03 - The Role of LINE-1 Retrotransposons in Amyotrophic Lateral Sclerosis
1Centre for Molecular Medicine and Innovative Therapeutics, Murdoch University, Perth, Australia, 2Perron Institute for Neurological and Translational Science, Perth, Australia
An individual’s genetics contributes to their risk of developing amyotrophic lateral sclerosis (ALS), however there is still a large proportion of the heritability of ALS, particularly in sporadic cases, to be understood. Part of this missing heritability may lie in complex variants yet to be evaluated. Active transposable elements drive inter-individual variability, and the long interspersed element 1 (L1) retrotransposons are a source of polymorphic insertions in the population. There are at least 29 instances of disease caused by a L1 insertion, generally through loss of function mutation or aberrant splicing. The majority of L1 insertions in the human genome are no longer able to retrotranspose, however to date 276 retrotransposition competent (RC) L1s have been reported. Many RC-L1s are polymorphic for their presence/absence, therefore each individual will have a different number and complement of RC-L1s. These elements have been hypothesised to be involved in disease processes through multiple mechanisms such as somatic mutation by retrotransposition, the triggering of neuroinflammation via the interferon pathway and DNA damage from the endonuclease activity of ORF2 protein. We hypothesise that L1s could influence disease development either through their effects on endogenous genes or due to the properties that enable them to retrotranspose. Therefore, using whole genome sequencing data from the New York Genome Center (NYGC) ALS consortium we characterised L1 variation genome wide and then focused on the subset of RC-L1s.
In 4393 whole genomes from the NYGC ALS consortium we identified 205 reference and 2598 non-reference polymorphic L1 elements. Association analysis was performed in European individuals who has been diagnosed with ALS or ALS and other neurological disorder (ALSND) (n=2653) and non-neurological controls (NNC) (n=320). There were no individual L1 elements associated with disease, however we did identify an increased risk of ALS/ALSND in individuals with a higher number of RC-L1 in their genomes. We compared the total number of present alleles of 102 polymorphic RC-L1s found in this cohort using linear regression, identifying a significant increase in the number of these elements in ALS/ALSND genomes (p=6.02x10-5). The number of present alleles in NNC genomes ranged from 36-60 and in ALS/ALSND from 34-67 and having ≥51 RC-L1 present alleles showed the strongest association with ALS/ALSND (p=6.73x10-5, OR=1.14 (1.07-1.21)). Focusing on a smaller group of RC-L1s that were the most highly active in terms of retrotransposition also identified an increased number of these elements ALS/ALSND genomes (p=0.009).
Analysis of individual L1s and their association with age at onset and survival in the NYGC ALS consortium identified a L1 whose presence was significantly associated with a lower age at onset (52.7 years) compared to homozygous absent individuals (59.2 years) (padj=0.009, est=-6.54, SE=1.51). The genomic burden of RC-L1s was not associated with age at onset of disease or survival.
Our study has identified novel genetic factors for both disease risk and age at onset in ALS providing further evidence for the role of L1 retrotransposons in neurodegenerative disease.
OS.02.04
OS.02.04 - Characterising Disease Heterogeneity in Sporadic ALS Patient iPSC-Derived Motor Neurons
1Medical School, University Of Western Australia, Crawley, Australia, 2Centre for Molecular Medicine and Innovative Therapeutics, Murdoch University, Murdoch, Australia, 3Perron Institute for Neurological and Translational Science, Centre for Neuromuscular and Neurological Disorders, The University of Western Australia, Nedlands, Australia
OS.02.05
OS.02.05 - Development of a Novel SOD1 Antisense Therapy for SOD1-Linked and Sporadic ALS
1Centre for Molecular Medicine and Innovative Therapeutics, Murdoch University, Perth, Australia, 2Perron Institute for Neurological and Translational Science, Perth, Australia, 3Black Swan Pharmaceuticals, Durham, United States, 4Florey Institute of Neuroscience and Mental Health, Melbourne, Australia, 5Department of Neurology, Duke University, Durham, United States
SOD1-PMO treatment led to an 11% increase in survival of SOD1G93A mice following a single dose (p<0.0001), resulting in improved motor function, as measured by Rotarod performance and high-limb strength. Notably, the SOD1-PMO has an excellent safety profile, with no safety concerns demonstrated with high dosages, and toxicology studies supporting up to 60-fold human equivalent dosing on current SOD1 suppression ASO therapies.
1. Miller, T.M., et al., N Engl J Med. 2022;387(12):1099-110;
2. Forsberg, K., et al., PLoS One, 2010. 5(7)
OS.02.06
OS.02.06 - ALPS Index in Amyotrophic Lateral Sclerosis: Clinical and Structural Correlation
1National Taiwan University Hospital, Taipei, Taiwan, 2National Taiwan University Hospital Bei-Hu Branch, Taipei, Taiwan, 3College of Medicine, National Taiwan University, Taipei, Taiwan
OS.02.07
OS.02.07 - Branchpoints as potential targets of exon-skipping therapies for genetic disorders
1Department of Drug Discovery Medicine, Kyoto University Graduate School of Medicine, Kyoto, Japan, 2Department of Developmental Biology and Functional Genomics, Ehime University Graduate School of Medicine, Ehime, Japan, 3Center for Anatomical, Pathological, and Forensic Medical Researches, Kyoto University Graduate School of Medicine, Kyoto, Japan, 4Department of Neuromuscular Research, National Institute of Neuroscience, National Center of Neurology and Psychiatry, Tokyo, Japan, 5Department of Mental Retardation and Birth Defect Research, National Institute of Neurology, National Center of Neurology and Psychiatry, Tokyo, Japan, 6Department of Neurology, Kyoto University Graduate School of Medicine, Kyoto, Japan
Fukutin (FKTN) c.647+2084G>T creates a pseudo-exon with a premature stop codon, which causes Fukuyama congenital muscular dystrophy (FCMD). We aimed to ameliorate aberrant splicing of FKTN caused by this variant. We screened compounds focusing on splicing regulation using the c.647+2084G>T splicing reporter and discovered that the branchpoint, which is essential for splicing reactions, could be a potential therapeutic target. To confirm the effectiveness of branchpoints as targets for exon skipping, we designed branchpoint-targeted antisense oligonucleotides (BP-AONs). This restored normal FKTN mRNA and protein production in FCMD patient myotubes. We identified a functional branchpoint by detecting splicing intermediates and creating branchpoint mutations in the FKTN reporter gene; this branchpoint was non-redundant and sufficiently blocked by BP-AONs. Next, a BP-AON was designed for a different FCMD-causing variant, which induces pathogenic exon-trapping by a common SINE-VNTR-Alu-type retrotransposon. Notably, this BP-AON also restored normal FKTN mRNA and protein production in FCMD patient myotubes. Our findings suggest that branchpoints could be potential targets in exon-skipping therapeutic strategies for genetic disorders.
OS.03.03
OS.03.03 - Safety and Efficacy of DYNE-251 in Males with DMD Mutations Amenable to Exon 51 Skipping
1Children's Hospital at Westmead, Westmead, Australia, 2London Health Sciences Centre, London, Canada, 3Neuromuscular Reference Center UZ Gent, Gent, Belgium, 4University Hospitals Leuven, Leuven, Belgium, 5Nationwide Children's Hospital, Columbus, United States, 6Rare Disease Research, LLC, Atlanta, United States, 7University of California Los Angeles, Los Angeles, United States, 8Dyne Therapeutics, Waltham, United States, 9Royal Victoria Infirmary, Newcastle University, Newcastle-Upon-Tyne, United Kingdom
Duchenne muscular dystrophy (DMD) is caused by absence of functional dystrophin protein. Approved PMO therapies induce exon skipping to restore the DMD mRNA reading frame leading to the production of truncated, functional dystrophin, but their potential is limited by poor muscle delivery. DYNE-251, an investigational therapeutic for DMD, consists of an exon 51-skipping PMO conjugated to a TfR1-targeting Fab to deliver increased levels of PMO to muscles. The safety and efficacy of DYNE-251 in ambulant and non-ambulant males aged 4-16 with DMD mutations amenable to exon 51 skipping are being studied in the Phase 1/2 DELIVER trial (NCT05524883).
In the MAD portion of DELIVER, participants are randomized to receive DYNE-251 or placebo Q4W or Q8W for 6 months across 7 PMO dose levels up to 40 mg/kg. For analysis of exon skipping and dystrophin data from the 5 mg/kg cohort, 4 participants received DYNE-251 and 2 received placebo. Safety and tolerability are based on 37 participants enrolled in DELIVER as of the data cut-off date.
At 6 months, 5 mg/kg DYNE-251 showed a mean 657 ng/g PMO concentration in muscle and mean absolute exon skipping level of 0.90% (0.80% difference from baseline). Mean absolute dystrophin level, measured by Western blot, increased from 0.60% at baseline to 0.88% of normal at 6 months, and the mean level of dystrophin positive fibers (PDPF) increased from 2.4% at baseline to 22.2% at 6 months. As of the data cut-off date, DYNE-251 demonstrated a favorable safety profile with mostly mild or moderate TEAEs. There was no treatment-emergent anemia or clinically meaningful changes in kidney parameters or electrolytes.
Based on these initial data, DYNE-251 had a favorable safety profile and reached levels of dystrophin expression, exon skipping, and PDPF at 6 months that exceeded levels reported at the same time point in prior clinical trials evaluating the standard of care PMO. The data support the continued development of DYNE-251 for DMD.
OS.03.04
OS.03.04 - Safety and Efficacy of Rituximab in Myasthenia Gravis Patients Resistant to the Standard Treatment
1Isfahan University Of Medical Sciences, Isfahan, Iran (Islamic Republic of)
Myasthenia gravis (MG) is an autoimmune neuromuscular disease characterized by muscle weakness. The role of the antibodies has been identified in the pathogenesis of the disease. Rituximab is an anti-CD20 agent used for various autoimmune and rheumatologic diseases. During this prospective study, 53 MG patients were registered in Al-Zahra University Hospital, Isfahan, Iran. Thirteen patients fit the inclusion criteria and enrolled. Each patient received 1000mg rituximab on day 0, day 14, and after six months. In a year follow-up period, the physical and functional status of the patients were measured every three months. Statistical analysis revealed that primary outcome measures (MGFA, MGC, MG-QoL-15, and MG-ADL scores) were significantly different in 5 follow-up time points. Reported infusion reactions, adverse events (AEs) and serious adverse events (SAEs) were minimal and easily manageable. There was no need for discontinuing infusion in the patients. In conclusion, our results showed Rituximab efficacy and safety in refractory MG patients. As a recommendation of the authors, further studies are necessary for setting up guidelines.
OS.03.05
OS.03.05 - Safety and Efficacy of Chronic Weekly Rozanolixizumab Treatment in Patients with Generalised Myasthenia Gravis (MG0004)
1University Health Network, Toronto, Canada, 2Precision Neurology of Neuromuscular Diseases, Department of Neurology, University of Lübeck, Lübeck, Germany, 3MDA ALS and Neuromuscular Center, University of California, Irvine, Irvine, USA, 4Department of Neuroimmunology and Neuromuscular Diseases, Fondazione IRCCS, Istituto Nazionale Neurologico Carlo Besta, Milan, Italy, 5Université Côte d’Azur, Peripheral Nervous System & Muscle Department, Pasteur 2 Hospital, Centre Hospitalier Universitaire de Nice, Nice, France, 6Department of Neurology, Hanamaki General Hospital, Hanamaki, Japan, 7Department of Neurology, University of South Florida Morsani College of Medicine, Tampa, USA, 8UCB Pharma, Monheim, Germany, 9UCB Pharma, Slough, UK, 10Department of Neurology, Rigshospitalet, University of Copenhagen, Copenhagen, Denmark
OS.03.06
OS.03.06 - Response Rates with Zilucoplan in Generalised Myasthenia Gravis: 120-Week Interim Analysis of RAISE-XT
1Department of Neurology, University Of Chapel Hill, North Carolina, Chapel Hill, USA, 2Department of Neurology, The Ohio State University Wexner Medical Center, Columbus, USA, 3Clinical Research Unit, The Montreal Neurological Institute, Montreal, Canada, 4Academic Neuroscience Unit, Sheffield Teaching Hospitals Foundation Trust, Sheffield, UK, 5Sheffield Institute for Translational Neurosciences (SITRAN), University of Sheffield, Sheffield, UK, 6Department of Neurology, Oslo University Hospital, Oslo, Norway, 7Department of Neuroimmunology and Neuromuscular Diseases, Fondazione IRCCS, Istituto Nazionale Neurologico Carlo Besta, Milan, Italy, 8Department of Neurology, Hanamaki General Hospital, Hanamaki, Japan, 9Department of Neurology, University of South Florida Morsani College of Medicine, Tampa, USA, 10Department of Neurology, University of Washington Medical Center, Seattle, USA, 11UCB Pharma, Monheim, Germany, 12UCB Pharma, Cambridge, USA, 13UCB Pharma, Slough, United Kingdom, 14UCB Pharma, Brussels, Belgium, 15Nuffield Department of Clinical Neurosciences, University of Oxford, Oxford, UK
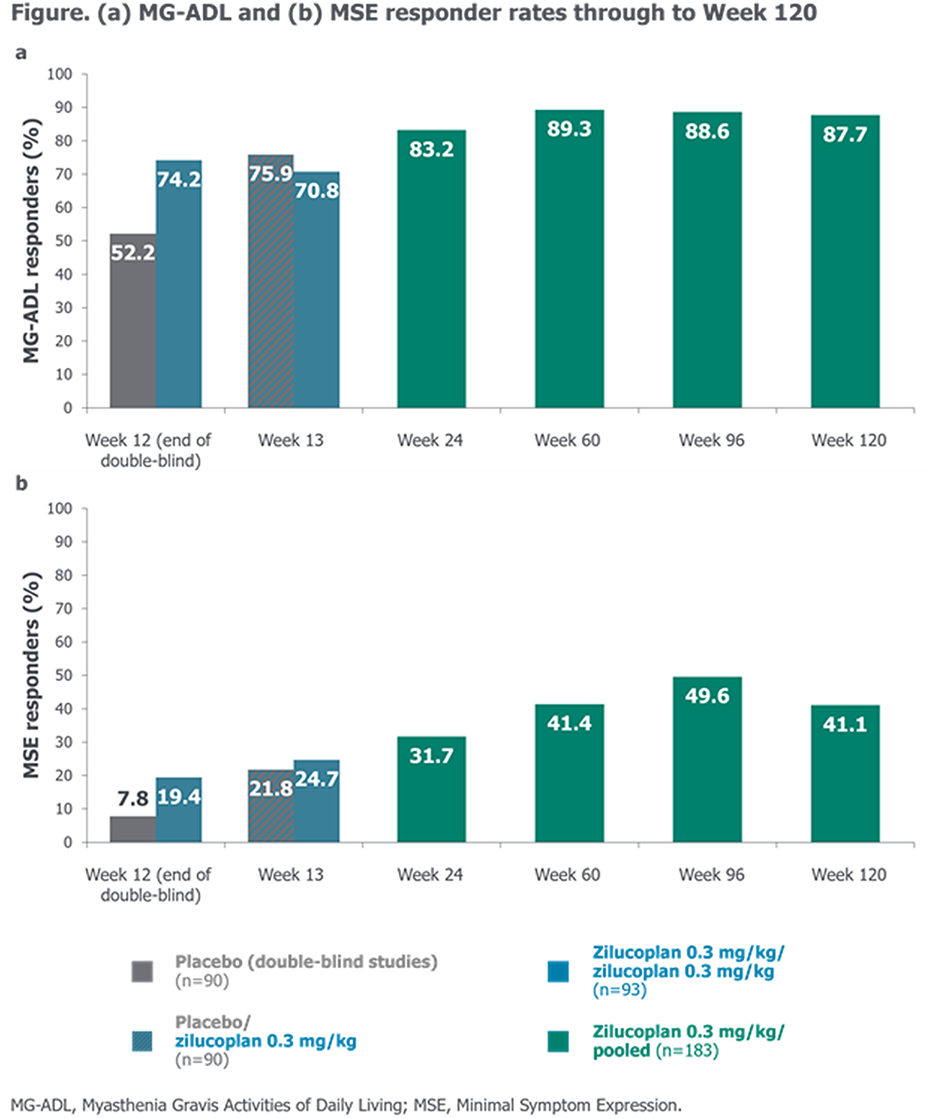
OS.03.07
OS.03.07 - Neurological irAE’s: Real-World Data on Age, Rechallenge, and Survival in a Large Melanoma Cohort
1Rambam Medical Center, Haifa, Israel
OS.03.08
OS.03.08 - Blood Nerve Permeability at Proximal Nerve Segments in Inflammatory Neuropathies
1Alfred Health, MELBOURNE, Australia, 2Monash University, Melbourne, Australia, 3Royal Melbourne Hospital, Melbourne, Australia
3T-MRI examinations of the lumbo-sacral spine were carried out on participants including T1 weighted DCE volume interpolated breath-hold (VIBE) sequences with gadolinium-based contrast agents. Ktrans, Kep, Ve and iAUC maps were generated using the Tofts Model.3-5
We calculated the Ktrans, Kep, Ve and iAUC at the motor root (MR), sensory root (SR), dorsal root ganglion (DRG) and the mixed spinal nerve (MSN) was calculated at the lumbar and sacral segments (L4, L5 and S1) bilaterally. Linear mixed model analysis was used to compare differences between the two groups. Functional measures of disease activity such as the Rasch-built overall disability scale (RODS) and the Medical Research Council (MRC) scale for muscle strength were also calculated.
1. Bäumer P, Reimann M, Decker C, Radbruch A, Bendszus M, Heiland S, et al. Peripheral nerve perfusion by dynamic contrast-enhanced magnetic resonance imaging: Demonstration of feasibility. Investigative Radiology. 2014;49(8):518-23.
2. Zochodne DW. Local blood flow in peripheral nerves and their ganglia: Resurrecting key ideas around its measurement and significance. Muscle and Nerve. 2018;57(6):884-95.
3. Larsson HB, Courivaud F, Rostrup E, Hansen AE. Measurement of brain perfusion, blood volume, and blood-brain barrier permeability, using dynamic contrast-enhanced T(1)-weighted MRI at 3 tesla. Magn Reson Med. 2009;62(5):1270-81.
4. Law M, Yang S, Babb JS, Knopp EA, Golfinos JG, Zagzag D, et al. Comparison of cerebral blood volume and vascular permeability from dynamic susceptibility contrast-enhanced perfusion MR imaging with glioma grade. AJNR Am J Neuroradiol. 2004;25(5):746-55.
5. Montagne A, Barnes SR, Sweeney MD, Halliday MR, Sagare AP, Zhao Z, et al. Blood-brain barrier breakdown in the aging human hippocampus. Neuron. 2015;85(2):296-302.
OS.04.03
OS.04.03 - A 50 Year Retrospective Study Describing Survival and Clinical Milestones in Duchenne Muscular Dystrophy
1Murdoch Children's Research Institute, Melbourne, Australia, 2Royal Children’s Hospital, Neurology Department, Melbourne, Australia, 3University of Melbourne, Department of Paediatrics, Melbourne, Australia, 4Royal Children’s Hospital, Department of Respiratory and Sleep Medicine, Melbourne, Australia, 5Victorian Respiratory Support Service, Melbourne, Australia, 6Austin Health, Cardiology Department, Melbourne, Australia, 7St Vincent’s Hospital, Cardiology Department, Melbourne, Australia, 8Royal Children’s Hospital, Cardiology Department, Melbourne, Australia

OS.04.04
OS.04.04 - Comprehensive Collation and Analysis of Variants in the Skeletal Muscle α-actin (ACTA1) Gene
1Harry Perkins Institute Of Medical Research, Perth, Australia, 2University of Western Ausralia, Perth, Australia, 3Folkhälsan Research Center, Helsinki, Finland, 4Murdoch Children's Research Institute, Melbourne, Australia, 5Department of Pediatrics, University of Melbourne, Melbourne, Australia
The ACTA1 gene encodes skeletal muscle alpha-actin, which forms the core of the sarcomeric thin filament in adult skeletal muscle. ACTA1 is one of six very highly conserved actin proteins that have all been associated with human disease. The first 15 pathogenic variants in ACTA1 were reported in the year 1999, which expanded to 177 by 2009. Such variants cause a range of congenital myopathies, including nemaline myopathy (NEM) and congenital fibre type disproportion (CFTD). Several ACTA1 variants also cause cardiomyopathy. As part of a ‘2023 mutation update’ we collated the 607 ACTA1 variants reported in the LOVD, HGMD, and/or ClinVar databases, which includes 343 reported pathogenic/likely pathogenic (P/LP) variants. We reviewed these against ACMG guidelines and provided suggestions as to how ACMG criteria should be used to classify ACTA1 variants. This included analysis of all P/LP missense changes in other human actins to assess which amino acid residues are most insensitive to change. Importantly, we found that missense changes at 345 out of 377 (91.5%) amino acid residues have been reported as disease-causing in at least one human actin. From a clinical perspective, we reviewed the breadth of phenotypes associated with ACTA1 variants, which has now grown to twenty. The vast majority (74%) cause nemaline myopathy, but there are increasing numbers that cause cardiomyopathy and novel phenotypes including distal myopathy. Overall, our findings and analyses provide a useful resource for interpretation of ACTA1 variant reports and diagnosis of neuromuscular disease patients.
OS.04.05
OS.04.05 - JOURNEY Natural History Study of Limb Girdle Muscular Dystrophies R3–R5: Study Cohort
Linda P. Lowes1, Carlos Ortez Gonzalez2, Kristl G. Claeys3, Chamindra G. Laverty4, Andrea Gangfuss5, Crystal M. Proud6, Jordi Diaz Manera7,
1Center for Gene Therapy, The Research Institute at Nationwide Children’s Hospital, Columbus, USA, 2Neuromuscular Unit, Hospital Sant Joan de Deu, Spain, 3Department of Neurology, University Hospitals Leuven and KU Leuven, Belgium, 4University of California San Diego, California, 5Department of Pediatric Neurology, Centre for Neuromuscular Disorders, Centre for Translational Neuro- and Behavioral Sciences, University Duisburg-Essen, Germany, 6Children’s Hospital of The King’s Daughter, Norfolk, USA, 7The John Walton Muscular Dystrophy Research Center, Newcastle University. Newcastle Upon Tyne Hospitals NHS, UK, 8Sarepta Therapeutics, Inc., Cambridge, USA, 9Dino Ferrari Center, Department of Pathophysiology and Transplantation, University of Milan, Italy, 10Foundation IRCCS Ca' Granda Ospedale Maggiore Policlinico, Neurology Unit, Italy
Sarcoglycanopathies affect ∼1/178,000 people worldwide, accounting for 15–20% of all limb girdle muscular dystrophy (LGMD) cases. However, data on the clinical characteristics and natural history of LGMD are limited. Here, participants’ baseline clinical characteristics from JOURNEY (NCT04475926), a global, multicenter, prospective, longitudinal study of LGMD2E/R4, 2D/R3, and 2C/R5 subtypes, are described.
Primary endpoints include North Star Assessment for Limb-Girdle Type Dystrophies (NSAD) score, performance of upper limb (PUL), pulmonary function, and other timed function tests (assessed every 6 months). Exploratory assessments include electrocardiogram (ECG).
A total of 137 participants were enrolled as of February 2024 (2E/R4: n=45; 2D/R3: n=49; and 2C/R5: n=43); 57.7% were female and 54.7% were ambulant (2E/R4: 25; 2D/R3: 36; and 2C/R5: 14) at baseline. Mean (SD) age was 18.4 (14.24) years for ambulatory and 26.2 (12.36) years for nonambulatory participants. Steroid use was reported in 15/75 (20.0%) ambulatory and 9/62 (14.5%) nonambulatory participants. Cardiac disorders by subtype and ambulatory status (ambulatory, nonambulatory) were present in 2E/R4: 2, 10; 2D/R3: 0, 1; and 2C/R5: 1, 6, respectively. Mean NSAD and PUL scores by subtype and ambulatory status (ambulatory, nonambulatory) were NSAD 2E/R4: 38.1, 4.1; 2D/R3: 35.1, 2.5; and 2C/R5: 27.3, 3.5, respectively, and PUL 2E/R4: 37.2, 16.8; 2D/R3: 34.8, 15.6; and 2C/R5: 31.2, 16.5, respectively. Mean % predicted forced expiratory volume was 86.7 in ambulatory and 63.2% in nonambulatory participants. ECG was normal in 21/45 (46.7%), 31/49 (63.3%), and 26/43 (60.5%) of 2E/R4, 2D/R3, and 2C/R5 participants, respectively. Clinically significant ECG abnormalities were only observed in 3/62 (4.8%) nonambulatory participants.
JOURNEY is a natural history study of LGMD, adding to the overall understanding of clinical characteristics and disease progression of individuals with subtypes 2E/R4, 2D/R3, and 2C/R5.
OS.04.06
OS.04.06 - Age at Loss of Ambulation in Patients with nmDMD from the STRIDE Registry: Sensitivity Analyses
1University College London Great Ormond Street Institute of Child Health, London, UK, 2Parent Project APS Italy, Rome, Italy, 3Unidad de Patología Neuromuscular, Hospital Sant Joan de Déu, Universidad de Barcelona, Barcelona, Spain, 4Department of Pediatrics, Queen Silvia Children’s Hospital, University of Gothenburg, Gothenburg, Sweden, 5Department of Neurology, Faculty of Medicine, University of São Paulo, São Paulo, Brazil, 6PTC Therapeutics Inc., South Plainfield, USA, 7PTC Therapeutics Germany GmbH, Frankfurt, Germany, 8Department of Pediatric Neurology, Catholic University, Italy
The value of real-world registries in informing long-term treatment benefits is increasingly recognized, particularly in rare diseases; however, registries often lack a standard-of-care comparator arm. Strategic Targeting of Registries and International Database of Excellence (STRIDE; NCT02369731) is an international, observational registry evaluating long-term safety and effectiveness of ataluren in individuals with nonsense mutation Duchenne muscular dystrophy (nmDMD) in real-world clinical practice. Propensity score matching (PSM) was performed to identify Cooperative International Neuromuscular Research Group (CINRG) Duchenne Natural History Study (NCT00468832) patients who were comparable to STRIDE patients for the following established predictors of disease progression: age at first symptom onset, age at first corticosteroid use and duration of corticosteroid use. Here we assess the robustness of the PSM methodology using sensitivity analyses.
Sensitivity analyses were performed to address potential sources of bias in PSM patient populations, including standard of care, severity of underlying disease, age and functional status at study entry as covariates. Sensitivity analyses also included alternative propensity score methods, using all CINRG patients, with propensity scores as weights and as covariates to detect potential bias.
As of January 2023, STRIDE had enrolled >300 boys with nmDMD. In the primary analysis, ataluren treatment in STRIDE patients was associated with a 3.5-year delay in loss of ambulation (LoA) compared with CINRG counterparts (Hazard Ratio [HR] 0.46; p<0.0001). HRs of sensitivity analyses were 0.43–0.49, demonstrating stability of the PSM algorithm of the primary matching and the robustness of the primary result, when using propensity-matched STRIDE and CINRG patients (Table 1).
Sensitivity analyses confirmed that major identified potential sources of bias of PSM were addressed, suggesting the observed delay in LoA in patients with nmDMD in STRIDE to be reliably attributed to ataluren.
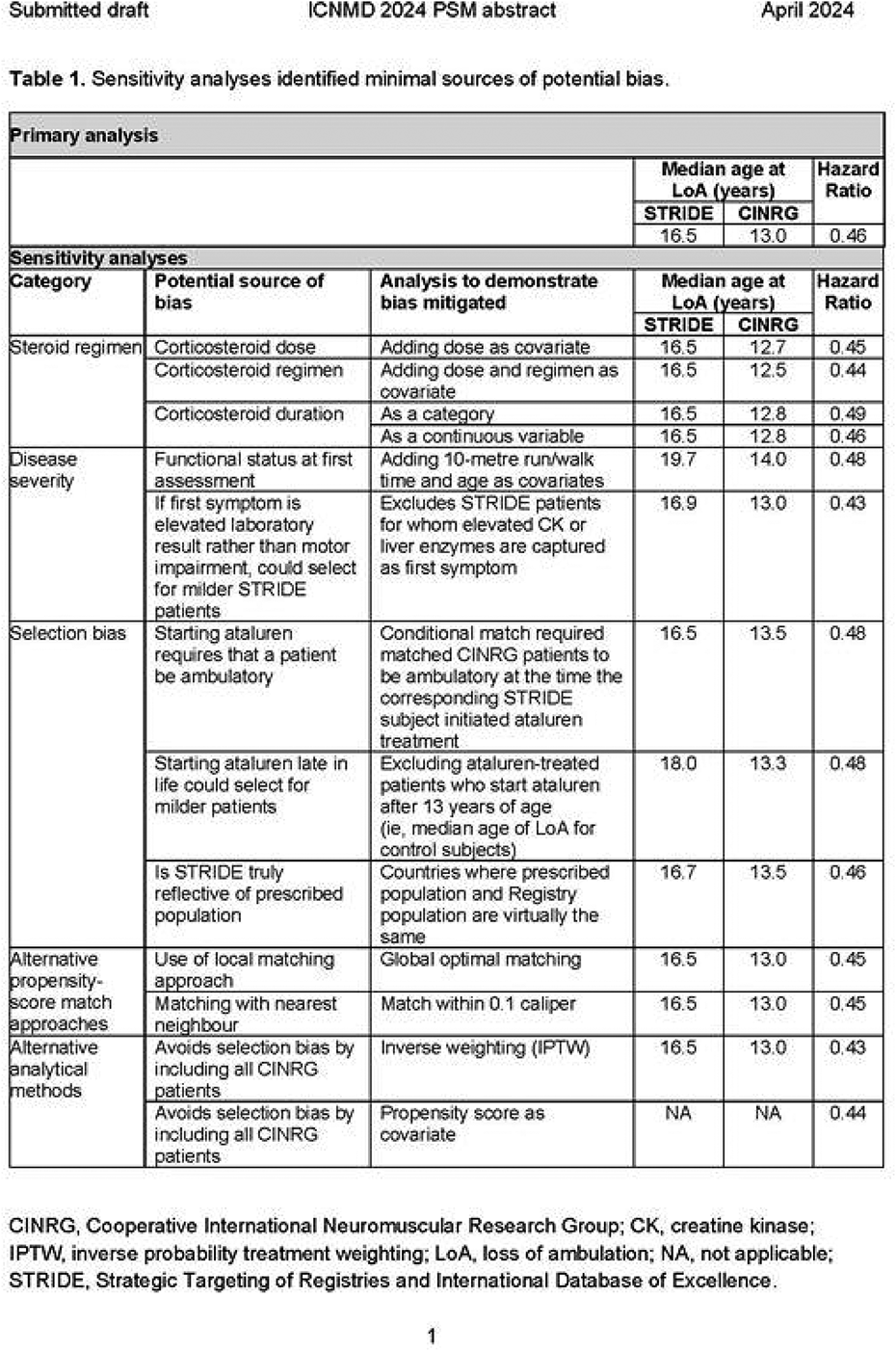
OS.04.07
OS.04.07 - Pompe Registry: Real-World Experience of LOPD Patients who Switched from Alglucosidase Alfa to Avalglucosidase Alfa
1Department of Neurology, Fiona Stanley Hospital, University of Notre Dame, Murdoch University, Perth, Australia, 2Department of Pediatrics, Division of Medical Genetics, Duke University Medical Center, Durham, USA, 3Department of Clinical and Experimental Medicine, Reference Center for Rare Neuromuscular Disorders, University of Messina, Messina, Italy, 4Sanofi, Cambridge, USA, 5Friedrich-Baur-Institute, Department of Neurology, Ludwig-Maximilians-University Klinikum München, München, Germany
Avalglucosidase alfa (AVA), a recombinant human acid α-glucosidase enzyme replacement therapy, is approved in several countries for infantile-onset and/or late-onset Pompe disease (LOPD). This analysis assesses clinical outcomes in patients with LOPD within the first year of switching from alglucosidase alfa (ALG) to AVA. Data were used from the Pompe Registry (NCT00231400), an ongoing international, observational, voluntary registry of patients with Pompe disease. To be eligible for this analysis patients needed to have ≥1 ALG record in the Registry immediately pre-switch to AVA. Pre- and post-switch respiratory, ambulatory, and biomarker data and demographics and treatment histories were collected. Increasing values for the functional outcomes and decreasing values for the biomarkers indicate improvement. As of 2 February 2024, 196 patients were identified; 98 (50%) were male. Patients were predominantly from North America (123 [63%]) and Europe (50 [26%]). Patients’ mean ± SD (range) age at diagnosis was 34.9 ± 21.4 (0.0–77.5) years; they initiated ALG at 37.4 ± 21.5 (0.0–77.7) years and were 46.3 ± 21.3 (1.0–87.1) years at the switch to AVA; most (121 [62%]) had received ALG for ≥5 years pre-switch. Last assessments pre-switch were: upright forced vital capacity % predicted (FVC%): 63.8 ± 25.1 (n=162); 6-minute walk test (6MWT): 351 ± 172 m (n=115); urinary oligosaccharides (Glc4/Hex4): 9.3 ± 15.1 mmol/mol creatinine (n=97); and serum creatine kinase (CK): 489 ± 401 U/L (n=171). For patients with both pre- and up to 1-year post-switch data, mean ± SD changes between visits were: FVC%: 1.2 ± 10.3 (n=61); 6MWT: −11 ± 116 m (n=53); GLc4/Hex4: −4.1 ± 6.6 mmol/mol creatinine (n=50); and CK: −126 ± 196 U/L (n=86), indicating stabilisation or improvement. As the Pompe Registry accrues more data for patients switching from ALG to AVA, it will further support the understanding of AVA’s effectiveness in terms of respiratory and ambulatory outcomes and biomarker levels in the real world.
OS.04.08
OS.04.08 - Gene-Based Therapies for Oculopharyngeal Muscular Dystrophy
1Montreal Neurological Institute-Hospital - McGill University, Montreal, Canada, 2Biology Department, Concordia University, Montreal, Canada
Oculopharyngeal Muscular Dystrophy (OPMD) is an autosomal dominant, late-onset muscle disorder characterized by ptosis, swallowing difficulties, and proximal limb weakness. The distinct pathological hallmark of OPMD is the presence of filamentous intranuclear inclusions in patient's skeletal muscle cells. OPMD occurs worldwide and has the highest prevalence in certain populations (for example, Quebec: 1/1000). The disease is caused by a trinucleotide repeat expansion in the polyA-binding protein nuclear 1 (PABPN1) gene, giving rise to an N-terminal expanded polyalanine tract. There is no cure for OPMD at present. Here we present two developed gene-based therapies (RNA interference and CRISPR/Cas9) that directly target the underlying mutation in OPMD. First, we demonstrate that RNA therapy combining the silencing the mutated PABPN1 gene by microRNAs and replacement it with a normal PABPN1 gene (that is microRNA resistant) is effective in treating OPMD cell models. Our results show that microRNAs are highly efficient and able to achieve ∼95% inhibition of the native PABPN1 transcript and protein levels. The optimized-codon PABPN1 enhances wild-type PABPN1 protein expression and is resistant to degradation caused by microRNAs. More importantly, co-expression of optimized-codon PABPN1 with micoRNAs protects against cell death in a stable C2C12 OPMD model. Taken together, RNA replacement therapy represents an exciting approach for OPMD treatment. Second, we describe successful CRISPR/Cas9 deletion of the expanded polyalanine-encoding tract in OPMD human patient myoblasts, and that these cells retain survival and muscle differentiation following polyalanine deletion. For the first time, this work provides a proof-of-principle that CRISPR/Cas9 gene editing could be further applied in OPMD preclinical studies in the future. Currently, we are developing and evaluating the two gene-based approaches in induced pluripotent stem cell (iPSC)-derived muscle cultures from OPMD patients, and in our OPMD transgenic mouse model. Our work represents a pre-clinical stage for a significant unmet medical need that could lead to clinical trials in the near future.
OS.05.03
OS.05.03 - Neck Triangle Nerve Enlargement in Hereditary Transthyretin Amyloidosis Correlates with the Autonomic, Cardiac, and Gastrointestinal Systems
1Department of Neurology, National Taiwan University Hospital, Taiwan, 2Department of Internal Medicine, National Taiwan University Hospital, Taiwan, 3Department of Medical Imaging, National Taiwan University Hospital, Taiwan
OS.05.04
OS.05.04 - Efficacy and Safety of Subcutaneous Efgartigimod PH20 in Chronic Inflammatory Demyelinating Polyneuropathy: ADHERE/ADHERE+ Trials
1Department of Neurology, University of Minnesota, Minneapolis, USA, 2Austin Neuromuscular Center, Austin, USA, 3Department of Neurology (Nerve-Muscle Unit), AOC National Reference Center for Neuromuscular Disorders, ALS Center, University Hospital of Bordeaux (CHU Bordeaux), Bordeaux, France, 4Department of Neurology, University of South Florida, Tampa, USA, 5Sheffield Institute for Translational Neurosciences (SITRAN), University of Sheffield, Sheffield, UK, 6Sheffield Teaching Hospitals NHS Foundation Trust, Sheffield, UK, 7Department of Neurology, Huashan Hospital, Fudan University, Shanghai, China, 8Department of Neurology, Medical University of Warsaw, Warsaw, Poland, 9European Reference Network On Rare Neuromuscular Diseases (ERN EURO-NMD), Paris, France, 10IRCCS Foundation “Carlo Besta” Neurological Institute, Milan, Italy, 11Department of Medical Biotechnology and Translational Medicine, University of Milan, Milan, Italy, 12Department of Neurology, "Victor Babes" University of Medicine and Pharmacy, Timisoara, Romania, 13Department of Neurology, Tangdu Hospital, The Fourth Military Medical University, Xi'an, China, 14Department of Neurology, Graduate School of Medicine, Chiba University, Chiba, Japan, 15argenx, Ghent, Belgium, 16School of Medicine, Duke University, Durham, USA, 17Department of Neurology, University of Copenhagen, Copenhagen, Denmark, 18Department of Neurology, Kepler University Hospital, Johannes Kepler University, Linz, Austria, 19Department of Neurology, Institute of Clinical Chemistry, Christian-Albrecht University of Kiel and University Medical Center Schleswig-Holstein, Kiel, Germany
OS.05.05
OS.05.05 - Magnitude Of Response with Subcutaneous Efgartigimod PH20 in Chronic Inflammatory Demyelinating Polyneuropathy: ADHERE/ADHERE+ Trials
Jeffrey A. Allen1, Richard A. Lewis2, Luis Querol3,4, Christian Eggers5, Satoshi Kuwabara6, Yessar M. Hussain7, Kelly Gwathmey8, Jeffrey T. Guptill9,10, Geoffrey Istas9, Benjamin Van Hoorick9, Arne De Roeck9, Ivana Basta11,
1Department of Neurology, University of Minnesota, Minneapolis, USA, 2Department of Neurology, Cedars-Sinai Medical Center, Los Angeles, USA, 3Department of Neurology, Neuromuscular Diseases Unit, Hospital de La Santa Creu I Sant Pau, Universitat Autònoma de Barcelona, Barcelona, Spain, 4Centro Para La Investigación Biomédica en Red en Enfermedades Raras (CIBERER), Madrid, Spain, 5Department of Neurology, Kepler University Hospital, Johannes Kepler University, Linz, Austria, 6Department of Neurology, Graduate School of Medicine, Chiba University, Chiba, Japan, 7Austin Neuromuscular Center, Austin, USA, 8Department of Neurology, Virginia Commonwealth University, Richmond, USA, 9argenx, Ghent, Belgium, 10School of Medicine, Duke University, Durham, USA, 11Neurology Clinic, University Clinical Center of Serbia, Faculty of Medicine, University of Belgrade, Belgrade, Serbia, 12Department of Neurology, Erasmus MC, University Medical Center, Rotterdam, the Netherlands
OS.05.06
OS.05.06 - Characterizing Monomelic Presentations of Demyelinating Neuropathy, or “Chronic Inflammatory Demyelinating Mononeuropathy” (CIDM)
1Mayo Clinic, United States, 2Sheba Medical Center, Tel Aviv, Israel, 3Mayo Clinic, United States, 4University of Alberta, Canada
Weakness was reported in most patients (n=49/54 [91%]), along with sensory symptoms (n=40/54 [74%]). Pain was uncommon (n=10 [18%]). Neurological impairment score [NIS] at presentation was significantly higher in the CIDM developing multifocal CIDP patients compared to the ones that stayed focal (p<0.001). Neurological examination findings were similar except for more than one limb involvement in multifocal CIDP patients.
Most CIDM patients did not fulfill EAN/PNS electrophysiological criteria for demyelination (definite n=5/40[13%]; possible n=10/40[25%]). CSF protein was mildly elevated (median 50 mg/dL) in most patients (n=29/33). Focal MRI nerve abnormalities (diffuse T2 hyperintensity/thickened nerve roots) of the affected limb were found in all patients which helped guide targeted sites of nerve biopsy in most cases (n=62) (dorsal lumbar rootlet n=4; targeted fascicular nerve n=45 and plexus n =13.Thirty-two CIDM patients started immunotherapy (IVIg n=25[78%]; intravenous methylprednisolone n=3[9%]; plasmapheresis n=4[13%]). CIDM patients showed significant improvement in median NIS at last follow-up (18 to 11.7, p=0.042). All CIDM and multifocal CIDP patients had abnormal neurological examinations at post-treatment follow-up however many showed marked improvement of symptoms and had reduced numbness and paresthesia.
OS.05.07
OS.05.07 - Neurologic Clinical, Electrophysiologic, and Pathologic Characteristics of Primary versus Secondary Neurolymphomatosis
1Neurological Institute Of Thailand, Bangkok, Thailand
OS.05.08
OS.05.08 - Phase 3 Trial Designs Evaluating Riliprubart, a C1s-Complement Inhibitor, in Chronic Inflammatory Demyelinating Polyneuropathy
Dr. Richard A. Lewis1, Dr. Jeffrey A. Allen2, Dr. Ingemar S.J. Merkies3,4,
1Cedars Sinai Medical Center, Los Angeles, USA, 2Department of Neurology, Division of Neuromuscular Medicine, University of Minnesota, Minneapolis, USA, 3Department of Neurology, Maastricht University Medical Center, Maastricht, The Netherlands, 4Curaçao Medical Center, Willemstad, Curaçao, 5Erasmus MC, University Medical Center, Rotterdam, The Netherlands, 6Neurologische Klinik und Poliklinik, Universitätsklinikum, Würzburg, Germany, 7Sanofi R&D, Neurology Development, Cambridge, USA, 8Sanofi R&D, Biostatistics and Programming, Bridgewater, USA, 9Neuromuscular Diseases Unit, Department of Neurology, Hospital de la Santa Creu i Sant Pau, Barcelona, Spain, 10Centro para la Investigación Biomédica en Red en Enfermedades Raras (CIBERER), Madrid, Spain
OS.06.03
OS.06.03 - Intravenous and Intrathecal Onasemnogene Abeparvovec Gene Therapy in Symptomatic and Presymptomatic SMA: Long-Term Follow-Up Study
1Department Of Neurology, Sydney Children’s Hospital Network, Sydney, Australia, 2School of Clinical Medicine, UNSW Medicine and Health, UNSW Sydney, Australia, 3Center for Gene Therapy, Nationwide Children’s Hospital, Columbus, United States, 4The Ohio State University College of Medicine, Columbus, United States, 5Department of Paediatric Neurology and Nemo Clinical Centre, Catholic University, Rome, Italy, 6Clinic for Special Children, Strasburg, United States, 7Penn Medicine-Lancaster General Hospital, Lancaster, United States, 8Department of Pediatrics and Department of Molecular, Cell & Cancer Biology, University of Massachusetts School of Medicine, Worcester, United States, 9Department of Neurology, Stanford University Medical Center, Stanford, United States, 10National Taiwan University Hospital, Taipei City, Taiwan, 11Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy, 12Novartis Global Drug Development – Neuroscience & Gene Therapy, East Hanover, United States, 13Novartis Pharmaceuticals, Basel, Switzerland, 14Novartis Gene Therapies, Inc., Bannockburn, United States, 15BioMedical Research, Novartis, Cambridge, United States, 16Department of Neurology, Boston Children’s Hospital, Harvard Medical School, Boston, United States
OS.06.04
OS.06.04 - RAINBOWFISH: 2-Year Efficacy and Safety Data of Risdiplam in Infants with Presymptomatic Spinal Muscular Atrophy
1Sydney Children’s Hospital Network And UNSW Medicine, UNSW Sydney, Sydney, Australia, 2Center for Experimental Neurotherapeutics, St Jude Children’s Research Hospital, Memphis, USA, 3MDUK Oxford Neuromuscular Centre, Department of Paediatrics, University of Oxford, Oxford, UK, 4Division of Child Neurology, Centre de Références des Maladies Neuromusculaires, Department of Pediatrics, University Hospital Liège & University of Liège, Liège, Belgium, 5Russian Children Neuromuscular Center, Veltischev Clinical Pediatrics and Pediatric Surgery Research Institute of Pirogov Russian National Research Medical University, Moscow, Russia, 6Department of Neurology, Faculdade de Medicina, Universidade de São Paulo, São Paulo, Brazil, 7Department of Neurosciences, King Faisal Specialist Hospital & Research Center-Riyadh, Riyadh, Kingdom of Saudi Arabia, 8Pediatrics Department, Faculty of Medicine, Federal University of Rio de Janeiro, Rio de Janeiro, Brazil, 9Department of Physical Therapy, University of Texas Southwestern Medical Center, Dallas, USA, 10Pharma Development, Safety, F. Hoffmann-La Roche Ltd, Basel, Switzerland, 11PDMA Neuroscience and Rare Disease, F. Hoffmann-La Roche Ltd, Basel, Switzerland, 12Roche Pharmaceutical Research and Early Development, Roche Innovation Center Basel, Basel, Switzerland, 13Roche Products Ltd, Welwyn Garden City, UK, 14Genentech, Inc., South San Francisco, USA, 15Research Unit of Neuromuscular and Neurodegenerative Disorders, Bambino Gesù Children’s Research Hospital IRCCS, Rome, Italy
Risdiplam is a centrally and peripherally distributed, oral survival of motor neuron 2 (SMN2) pre-mRNA splicing modifier that has been widely approved for the treatment of spinal muscular atrophy (SMA).
RAINBOWFISH (NCT03779334) is a global, open-label, single-arm, multicenter, Phase 2 study assessing the efficacy, safety and pharmacokinetics/pharmacodynamics of risdiplam in infants with genetically diagnosed and presymptomatic spinal muscular atrophy (SMA) from birth to 6 weeks of age (at first dose), regardless of SMN2 copy number.
The study enrolled 26 infants: eight infants had two SMN2 copies, 13 infants had three SMN2 copies and five infants had ≥4 SMN2 copies. The primary efficacy (PE) population (n=5) had two SMN2 copies and baseline compound muscle action potential (CMAP) amplitudes ≥1.5mV. Drug dosage was adjusted to achieve a target exposure of approximately 2,000 ng· hr/mL.
The primary endpoint was met after 12 months of risdiplam treatment with 4/5 (80%) infants in the PE population able to sit without support for ≥5 seconds (Item 22 of the Gross Motor Scale of the Bayley Scales of Infant and Toddler Development, third edition [BSID-III]). Irrespective of CMAP amplitude, 7/8 infants with two SMN2 copies were able to sit without support for ≥30 seconds (BSID-III Item 26), including all infants with CMAP amplitude <1.5 mV (n=3). Out of 26 infants, 24 (92%) were able to sit without support, 13 (50%) were able to stand unaided and 12 (46%) were able to walk independently at Month 12, as assessed by the Hammersmith Infant Neurological Examination, Module 2.
At Month 12, all infants were alive without permanent ventilation, maintained their swallowing and feeding abilities and no adverse events (AEs) led to withdrawal or treatment discontinuation. Most AEs were not considered treatment-related and resolved over time. One infant met the criteria for development of clinically manifested SMA.
Here we report the 2-year efficacy and safety data from RAINBOWFISH.
OS.06.05
OS.06.05 - Real-World Assessment of Onasemnogene Abeparvovec Treatment in Patients with SMA: RESTORE/Post-Marketing Surveillance in Japan
1Institute Of Medical Genetics, Tokyo Women’s Medical University, Tokyo, Japan, 2Analytics and Clinical Development Management, Novartis Pharma K.K., Tokyo, Japan, 3Novartis Gene Therapies Switzerland GmbH, Switzerland, 4Neuroscience and Gene Therapies Clinical Development, Novartis Pharma K.K., Japan, 5Analytics, Novartis Pharma K.K., Japan, 6Novartis Gene Therapies, Inc., Bannockburn, United States, 7Center for Experimental Neurotherapeutics, St. Jude Children’s Research Hospital, Memphis, United States
OS.06.06
OS.06.06 - Long-term Comparative Efficacy and Safety of Risdiplam Versus Nusinersen in Children with Type 1 SMA
Dr. Christos Kokaliaris1, Neil Hawkins2,3, Rachel Evans2, Cyrill Gasser1, Anadi Mahajan4,
1Global Access, F. Hoffmann-La Roche Ltd, Basel, Switzerland, 2Visible Analytics, Oxford, UK, 3Institute of Health & Wellbeing, University of Glasgow, Glasgow, UK, 4Bridge Medical Consulting Ltd., London, UK, 5University of Southampton, UK
The objective of this study was to compare long-term efficacy and safety of risdiplam versus nusinersen in children with Type 1 SMA treated in clinical trials for at least 4 years.
Unanchored matching-adjusted indirect comparisons were used to compare outcomes between risdiplam and nusinersen groups, adjusting for age at first dose, disease duration and Children’s Hospital of Philadelphia Infant Test of Neuromuscular Disorders (CHOP-INTEND) total score at baseline. Cox proportional-hazards models were used to compare overall survival, event-free survival and the times to Hammersmith Infant Neurological Examination, Module 2 (HINE-2) motor milestone responses, CHOP-INTEND responses and the occurrence of any serious adverse event (SAE). The median follow-up time was estimated through the reverse Kaplan–Meier method to account for censoring in the data when more than 50% of the trial population survived.
OS.06.07
OS.06.07 - Real-World Outcomes Following Onasemnogene Abeparvovec in Patients with SMA and One SMN2 Gene Copy
1Center For Experimental Neurotherapeutics, St. Jude Children’s Research Hospital, Memphis, United States, 2Department of Pediatrics and Department of Neurology, University of Iowa Carver College of Medicine, Iowa City, United States, 3University of Arizona College of Medicine, Phoenix, United States, 4University of Louisville, Norton Children’s Medical Group, Louisville, United States, 5Department of Clinical and Molecular Genetics, Hospital Vall d’Hebron, Barcelona, Spain, 6Novartis Gene Therapies, Inc., Bannockburn, United States, 7Novartis Gene Therapies Switzerland GmbH, Rotkreuz, Switzerland, 8Department of Paediatrics, MDUK Oxford Neuromuscular Centre & NIHR Oxford Biomedical Research Centre, University of Oxford, Oxford, United Kingdom, 9Department of Pediatrics, Neuromuscular Reference Center, University and University Hospital of Liège, Belgium
OS.06.08
OS.06.08 - Evaluation of Ambulatory Function with Ankle Wearable Technology in Ambulant DMD Below 4 Years Old
1University Of Neuromuscular Reference Center, Department of Pediatrics, University Hospital Liege & University of Liege, Belgium, Liege, Belgium, 2University department of neurology, CHR Citadelle, Liege, Belgium, 3SYSNAV, Vernon, France, 4University Children's Hospital, Ljubljana, Slovenia, 5Pediatric Center, Semmelweis University, Budapest, Hungary, 6F. Hoffmann-La Roche Ltd., Basel, Switzerland, 7University of Oxford, Oxford, United Kingdom
Treating Duchenne muscular dystrophy (DMD) patients at a younger age is increasingly perceived as important. However, assessing ambulatory function in patients below 4 years old is challenging. There is thus an urgent need to develop tools to evaluate the efficacy of disease-modifying therapies in young patients. Using a wearable sensor measuring accurately and precisely lower limb function in the daily life of patients, we recently obtained primary endpoint qualification of SV95C (95th centile of stride velocity, the 5% most rapid strides) by the European Medicines Agency, in ambulant DMD from the age of 4.
To determine the feasibility, robustness & sensitivity of SV95C to assess ambulatory function in young subjects, patients and controls below 4 years old were enrolled in a 3-year longitudinal multicentric study, ActiLiege-Next. Sensor is worn by patients continuously for the first 3 months and then for 1 month every 3 months, and by controls for 1 month every 6 months. SV95C reliability was assessed by comparing the 2 two-week period of the first recording month using intraclass correlation coefficient (ICC). Ability to differentiate patients from controls was assessed using a Mann Whitney U test.
27 patients (median age [min-max]: 34 months [16- 47]) and 21 healthy volunteers (32 months [12-47]) were enrolled so far. First analysis on subjects with complete baseline data showed that 12 out of 13 patients and all 7 controls recorded ≥50h of data required to derive the digital measure. SV95C reliability was excellent with ICC of 0.92 for patients and 0.98 for controls. Median SV95C at baseline was significantly different in patients and controls (p<0.001). All available baseline and 6-month data will be shared at the congress.
If these promising preliminary results and good metric properties are confirmed, SV95C could be the first functional assessment that can be used in all ambulant DMD. It could hence be a key element to enable development and approval of treatments in patients below 4 years of age.
OS.07.03
OS.07.03 - Safety of Eteplirsen in 6- to 48-Month-Old Patients With DMD: An Open-Label Extension Study
Eugenio Mercuri1,2, Andreea M. Seferian3, Nicolas Deconinck4, Larry Orogun5, Xiao Ni5, Wenfei Zhang5, Kerri Drummond5, Ihor Sehinovych5, Rachel Salazar5,
1Pediatric Neurology Unit, Università Cattolica del Sacro Cuore Roma, Italy, 2NeMO Clinical Center, Fondazione Policlinico Universitario A Gemelli IRCCS, Italy, 3Assistance Publique Hôpitaux de Paris, Sorbonne Université, Institut de Myologie, AFM-Téléthon, Essais Cliniques I-Motion Enfants, Hôpital Armand Trousseau, France, 4Centre de Référence Neuromusculaire and Paediatric Neurology Department, Hôpital Universitaire des Enfants Reine Fabiola, Université Libre de Bruxelles, Belgium, 5Sarepta Therapeutics, Inc., Cambridge, USA, 6Dubowitz Neuromuscular Centre, University College London, Great Ormond Street Institute of Child Health, UK, 7National Institute for Health Research Great Ormond Street Hospital Biomedical Research Centre, UK
Eteplirsen is indicated for treatment of exon 51 skip-amenable patients with Duchenne muscular dystrophy (DMD). Study 4658-102 (NCT03218995), a phase 2, open-label, dose-escalation trial, showed that eteplirsen (30 mg/kg IV once weekly) was well tolerated in patients aged 24 to 48 months (cohort 1, n=9) and 6 to <24 months (cohort 2, n=6) up to 96 weeks. Results from the open-label extension (OLE), which assessed the ongoing safety and tolerability in patients who were treated for up to ∼5 years (258 weeks) are reported. Safety endpoints included incidence and frequency of treatment-emergent adverse events (TEAEs) and clinically significant laboratory abnormalities. Fourteen (93.3%) of 15 patients were enrolled in the OLE when the study was terminated by the sponsor to reduce trial burden (ensuring continued treatment if desired). At OLE baseline, 4/15 (26.7%, cohort 1) were receiving corticosteroids, with 11 patients receiving corticosteroids during the OLE. From the start of the OLE, patients received a mean (SD) of 108.5 (37.1) eteplirsen infusions and were on eteplirsen for a mean (SD) of 120.1 (35.79) weeks. All patients experienced ≥1 TEAE; most were mild, consistent with those commonly seen in pediatric populations (cough, pyrexia, rhinorrhea, nasopharyngitis), and reduced in frequency and severity compared with TEAEs in the parent study. Three patients experienced mild treatment-related TEAEs (catheter-site swelling, chromaturia, abnormal urine albumin/creatinine ratio); there were no port-related infections. One serious TEAE was reported (influenza; unrelated to treatment). Eteplirsen was well tolerated, with no treatment-related discontinuations, deaths, or evidence of kidney toxicity. The safety profile of eteplirsen in the OLE was consistent with the known safety profile, with no new safety signals up to 162 weeks of treatment and no discernible difference between cohort 1 and 2. These data support the safety and tolerability of eteplirsen in patients as young as 6 months old.
OS.07.04
OS.07.04 - Ocular Symptoms in Patients with Generalised Myasthenia Gravis Receiving Rozanolixizumab: Post hoc Analysis of MycarinG
1MDA ALS and Neuromuscular Center, University of California, Irvine, Irvine, USA, 2Neurology Department, Indiana University School of Medicine, Indiana University Health, Indianapolis, USA, 3Department of Neurology, Rigshospitalet, University of Copenhagen, Copenhagen, Denmark, 4Department of Neurology, University of South Florida Morsani College of Medicine, Tampa, USA, 5UCB Pharma, Madrid, Spain, 6UCB Pharma, Slough, UK, 7University Health Network, Toronto, Canada
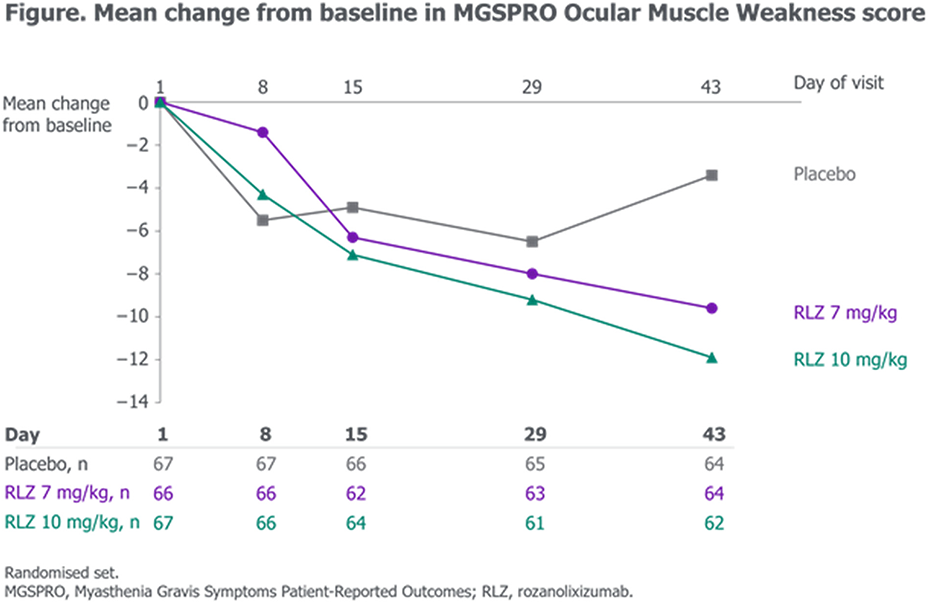
OS.07.05
OS.07.05 - Cost-Effectiveness Analysis of Efgartigimod Versus IVIg for Treatment of AChR-Ab+ Generalized Myasthenia Gravis in Canada
1Department of Neurology and Neurosurgery, McGill University, Montreal, Canada, 2argenx, Vaughan, Canada, 3University of Alberta, Edmonton, Canada, 4argenx, Ghent, Belgium, 5CRG-EVERSANA Canada Inc., Burlington, Canada
OS.07.06
OS.07.06 - Safety and Efficacy of Subcutaneous Efgartigimod PH20 in Generalized Myasthenia Gravis in the ADAPT-SC+ Study
Heinz Wiendl1, Yuebing Li2,
1Department of Neurology, University of Münster, Münster, Germany, 2Cleveland Clinic, Cleveland, USA, 3Department of Neurology, The University of North Carolina, Chapel Hill, USA, 4Department of Neurology, University of South Florida Morsani College of Medicine, Tampa, USA, 5State Budgetary Healthcare Institution of Novosibirsk Region "State Novosibirsk Regional Clinical Hospital", Novosibirsk, Russia, 6argenx, Ghent, Belgium, 7Department of Neurology, Hanamaki General Hospital, Hanamaki, Japan, 8NRSO Department, Federico II University of Naples, Naples, Italy, 9Ghent University Hospital, Ghent, Belgium, 10Department of Neuroimmunology and Neuromuscular Diseases, Fondazione Istituto Neurologico Carlo Besta, Milan, Italy
OS.07.07
OS.07.07 - Safety and Efficacy of Subcutaneous Immunoglobulin in Patients with Generalized Myasthenia Gravis
1University Of Toronto, Toronto, Canada, 2University Health Network, Toronto, Canada
OS.07.08
OS.07.08 - Efficacy and Safety of Nipocalimab in Patients with Generalized Myasthenia Gravis: Topline Results from VIVACITY-MG
Dr. Tuan Vu1, Dr. Carlo Antozzi2, Dr. Sindhu Ramchandren3, Dr. Richard J. Nowak4, Dr. Constantine Farmakidis5, Dr. Vera Bril6, Dr. Jan De Bleecker7, Dr. Huan Yang8, Dr. Eduard Minks9,
1University of South Florida, Tampa, USA, 2Neurology IV - Neuroimmunology and Neuromuscular Diseases Unit, Fondazione I.R.C.C.S. Istituto; Neurologico Carlo Besta, Italy, 3Janssen Research & Development, LLC, Titusville, NJ, USA, 4Yale University School of Medicine, New Haven, CT, USA, 5University of Kansas Medical Center, Kansas City, USA, 6University of Toronto, University Health Network, Toronto, Canada, 7Ghent University Hospital, Belgium, 8Xiangya Hospital, Central South University, China, 9First Department of Neurology, Faculty of Medicine, Masaryk University and St. Anne’s Hospital, Brno, Czech Republic, 10Department of Neurology, School of Medicine, Kyungpook National University, Kyungpook National University Chilgok Hospital, Daegu, South Korea, 11Centrum Medyczne Neuro-Protect Ul, Poland, 12Silesian Neurology Medical Center, Poland, 13Hospital Universitari i Politècnic and IIS La Fe/University of Valencia, Spain, 14Charité - Universitätsmedizin, Germany, 15The Neuromuscular Research Center and Neuromuscular Clinic of Arizona, Phoenix, USA, 16Janssen Research & Development, LLC, Cambridge, USA
OS.08.03
OS.08.03 - FSHD Global Registry Project: Whole-body Muscle-level Analysis for Advancing Research and Empowering Patients
Silvia Blemker1,
1 FSHD, Australia
The FSHD Global registry project aims to perform whole-body MRI in Australian patients with FSHD to advance research, develop a clinical trial readiness passport for each patient, and assess the utility of providing patients with their own MRI-derived muscle metrics. An initial analysis of the first 11 patients is presented here.
Leveraging accelerated AI-based muscle segmentation, fat fraction was quantified within 59 unique skeletal muscles bilaterally, totaling 118 muscles per patient. The analysis including patients with a range of disease states (total muscle fat fraction ranged from 11% to 72% across patients). Despite this range, all patients had at least 4 individual muscles with fat fraction above 30%, demonstrating the potential for detecting early signs of disease progression. While the fat fraction of some muscles correlated highly with total muscle fat fraction (e.g., rectus abdominus), other muscles had no correlation with total muscle fat fraction (e.g., soleus). Comparing total muscle fat fraction in the upper body versus the lower body, some patients demonstrated higher average fat fraction in upper body muscles (n=7) while other patients demonstrated higher average fat fraction in the lower body muscles (n=4). These results motivate the need for muscle-level analysis across the whole body to capture individual patient disease state. Collected feedback from patients revealed strong support for providing patients with MRI-derived muscle reports
OS.08.04
OS.08.04 - The Screen4Care Project: Genetic Newborn Screening and Early Whole Genome Sequencing Impact on Neuromuscular Diseases
Prof. Alessandra Ferlini1, Dr Marianna Farnè1, Dr. Fernanda Fortunato1, Dr. Alice Margutti1, Prof. Rita Selvatici2, Dr Emanuele Agolini3, Dr. Silvia Ottombrino3, Dr. Antonio Novelli3, Prof. Enrico Bertini3, dr Aldona Zygmunt4, Dr. Michela Zuccolo5, Dr Markos Mihalatos5, Dr Matt Rodesch5, Dr Moshe Einhorn6, Dr Sergi Beltran7, Dr Leslie Matalonga7, Prof Andreas Clemens8, Dr Stefaan Sansen9, Dr Christina Saier10, Prof Janbernd Kirschner10,
1Medical Genetics Unit, University Of Ferrara, Ferrara, Italy, 2Consorzio Futuro in Ricerca, Ferrara, Italy, 3Bambino Gesù Pediatric Hospital, Medical Genetics, Rome, Italy, 4Pfizer Inc., Collegeville, Collegiville, Pennsylvania, United States, 5F. Hoffmann-La Roche AG, Basel, Switzerland, 6GENOOX LTD, NA, Israel, 7Centro Nacional de Análisis Genómico - CNAG, Centro Nacional de Análisis Genómico, Barcelona, Spain, 8Novartis Pharma AG Basel-Stadt, Basel, Suisse, 9Sanofi, Diegem, Machelen, Machelen, Belgium, 10Gottingen University, Germany, 10University of Freiburg, Department of Neuropediatric and Muscle Disorders, Freiburg, Germany
The Screen4Care is a research project funded by EU-IHI with the aim of shortening the path to diagnosis for rare diseases through genetic newborn screening (gNBS) and digital technologies. It is composed of six work packages dedicated to different activities and tasks. In work package three (3) dedicated to gNBS, we have designed a pipeline we plan to apply in Italy, Germany, France, and Greece. This pipeline will be used for early identification of rare diseases (RDs) using a two-tier approach based on next generation sequencing (NGS) -based genetic newborn screening (gNBS), and early whole genome sequencing (WGS).
We have designed a NGS-based gene panel (TREAT panel) for running gNBS in up to 25.000 neonates for early identification of rare diseases (RDs) with approved treatments (from which the definition TREAT panel since detecting treatable diseases only). We adopted a mandatory criterion which is treatability, and other specific criteria as mendelian inheritance, natural history knowledge, clinical utility, and amenability of NGS. Both small and copy number variants will be identified using dedicated algorithms.
We selected by cut off-narrowed scores 245 genes which are associated with the following phenomics: 106 metabolic, 33 blood and coagulation, 29 endocrine, 26 immunological, 25 neurological and neuromuscular, 9 renal, 6 mendelian syndromes, 4 cardiological, and 7 other phenotypes (see Figure 1). Among them, 168 are autosomal recessive, 24 autosomal dominant, 20 X-linked, and 33 with both AD and AR inheritance. Infants, whose gNBS using the TREAT panel yields a negative result, but who become symptomatic during the first year of life, will be screened using whole genome sequencing (WGS) in a two-tier, post-NBS enrolment approach. This will allow us to identify the targeted 245 gene-related diseases and many other RDs that can be picked at the neonatal-perinatal age by WGS. This will also provide some insights into the frequency of early symptomatic RDs escaping the TREAT panel detection. All 24 European Reference Networks (ERNs) will be involved in the diagnostic work-up, treatment referral, and follow-up. Impact on neuromuscular diseases will be high in the list of priorities to allow an early diagnosis in 25 neuromuscular conditions for which treatments are available and approved, and of which 24 are currently completely disregarded in newborn screening programmes. In addition to these 25 diseases, infants with negative gNBS results using the TREAT panel but with early symptoms will be tested with WGS. At the end, we expect to gain not only data from the gNBS, but also from the early diagnosis of all neuromuscular and potentially other treatable conditions with onset before 2 years of age via the WGS approach. We believe that S4C will design and validate a gNBS and post-gNBS WGS approach to be possibly utilized by diverse health systems as a sustainable, ethical, and cost-effective two-tier gNBS approach. Grants: This work has been funded by the EU-IHI Screen4Care project, grant number No 101034427
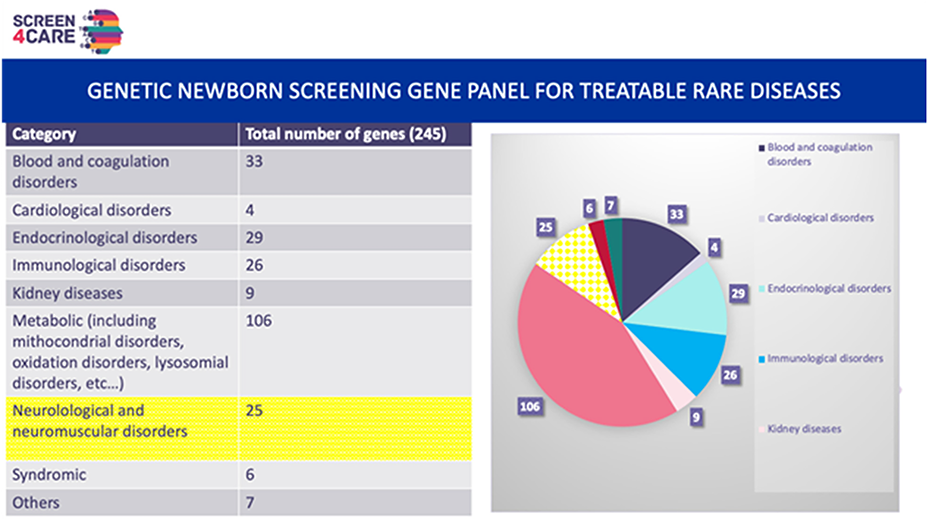
OS.08.05
OS.08.05 - Evaluating the Delivery of Genomic Medicine for Neuromuscular Patients in Latin America (Latin SEQ+)
1Newcastle Hospitals NHS Foundation Trust, Newcastle upon Tyne, United Kingdom, 2Newcastle University, Newcastle upon Tyne, United Kingdom
This paper discusses the outcomes of engaging with patients and the public around genetic testing for neuromuscular diseases in Latin America and reports on early collaborative encounters with clinicians in the Latin SEQ+ study.
The Latin SEQ+ study is a collaborative international project funded by the UK Medical Research Council to evaluate the delivery of Whole Exome Sequencing (WES) to 1000 patients with undiagnosed neuromuscular disease across 15 countries in Latin America. Unprecedented access to WES is available to Latin American patients via a pharma-funded project, the Latin SEQ study (LSS). However, in most Latin American countries, genetic counselling is not a well-resourced or registered profession and patients having WES will not have prior access to genetic counselling. WES in the LSS will be delivered by their neuromuscular specialist clinicians. Delivering genomics without genetic counselling is concerning for patient outcomes and the concern is shared by clinicians in the UK where WES is also increasingly delivered by non-genetics specialist clinicians.
Genetic counsellors holistically consider how a genetic diagnosis impacts patients and families. Genetic testing can affect individuals psychologically and socially and it can change family relationships. Genetic counselling is important to help individuals reach an informed contextually specific decision that is right for them at the point of testing. It prepares patients for the potential and actual outcomes of a genetic test including those that are not directly looked for which is a real possibility in genomic medicine
Across Latin America, some of the benefits of a genetic diagnosis such as tailored treatment, better supportive care, and prenatal testing options may not be realised. Gene therapy may be unachievable given limited resources. Cultural and religious beliefs may influence acceptability/accessibility of prenatal testing.
The Latin SEQ + evaluation uses mixed methods (surveys, qualitative interviews, and ethnography) to explore how genetic counselling is delivered in contexts without dedicated genetic counselling resources. Indicative aims and scope are outlined below, which will be iteratively developed and adjusted following extensive stakeholder engagement.
The evaluation aims to:
1) highlight the experiences of clinicians giving a diagnosis and patients’ and families' experiences of receiving a genetic diagnosis.
2) understand how, and if, any changes are made to patients’ care depending on the outcome of WES (explanatory variant found, no variant found, variant of uncertain significance found)
3) identify, describe, and understand how genetic information is communicated within families.
4) understand culturally specific influences on delivering and receiving diagnoses via WES.
Early Stakeholder Engagement
The paper discusses survey findings from patients and public involved in neuromuscular disease charities in Latin America and initial responses and concerns from collaborators gleaned from meeting observations and survey.
Ultimately, evaluation outputs will develop educational input and resource; share findings with partners to grow their genetics services and improve patient outcomes; highlight areas of good practice and share findings with Health Education England and Genomics England to inform UK service development. The project will generate new knowledge about delivering effective genetic counselling to improve patient outcomes in Latin America and the UK.
OS.08.06
OS.08.06 - Rasch Evaluation of North Star Ambulatory Assessment and NSAD in Becker Muscular Dystrophy
1John Walton Muscular Dystrophy Research Centre,Newcastle upon Tyne Hospitals NHS Foundation Trust+Newcastle University, Newcastle Upon Tyne, United Kingdom, 2Nationwide Children's Hospital, Columbus, USA, 3ATOM International, United Kingdom, 4University of California Davis, Sacramento, USA, 5Rigshospitalet, University of Copenhagen, Copenhagen, Denmark, 6Rare Disease Research, Atlanta, USA, 7Edgewise Therapeutics, Boulder, USA
The North Star Ambulatory Assessment (NSAA) and North Star Assessment for Limb-Girdle type muscular dystrophies (NSAD) are motor performance scales specifically developed for Duchenne muscular dystrophy (DMD) and Limb Girdle muscular dystrophy (LGMD), respectively. Both have been used in natural history studies of Becker muscular dystrophy (BMD), a dystrophinopathy sharing clinical characteristics of DMD and LGMD. We examined the psychometric performance of the NSAA and NSAD across three clinical trials of sevasemten (EDG-5506) enrolling ambulant subjects with BMD aged between 12 and 65 years.
Data were examined using Rasch Unidimensional Measurement Model (RUMM) as an innovative strategy for understanding the suitability of the NSAA and NSAD to measure the motor function of individuals with BMD. Psychometric evaluation was completed examining NSAA and NSAD performance in seven areas: targeting, response categories, fit, reliability, dependency, stability and unidimensionality using RUMM2030 software. A total of 182 assessments included screening and/or baseline visits for participants in the EDG-5506-002 (NCT05160415), EDG-5506-201 (NCT05291091) and EDG-5506-202 studies were utilized.
NSAA and NSAD both demonstrated unidimensional construct of functional motor performance and high reliability with a Person Separation Index (PSI) of 0.96. Although a ceiling effect still existed for the strongest symptomatic subjects, the motor performance of ambulant subjects was targeted successfully by the items of the NSAA and NSAD. 28/29 NSAD items and 14/17 NSAA items demonstrated ordered response categories, meaning the scoring categories for each item are logical and appropriate for BMD. The items fit well together to make use of the total score appropriate.
The NSAA and NSAD perform well in BMD, with the NSAD providing additional items to test both non-ambulant and those very able individuals with BMD. This analysis supports use of NSAA and NSAD as clinically meaningful outcomes in clinical trials of BMD.
OS.08.07
OS.08.07 - The Phase 3 Myasthenia Gravis Inebilizumab Trial (MINT): Topline Efficacy and Safety Results
1University of North Carolina, Chapel Hill, United States, 2Hanamaki General Hospital, Hanamaki, Japan, 3University of Miami, Miller School of Medicine, Miami, United States, 4University of Rochester, Rochester, United States, 5University of Oxford, Oxford, United Kingdom, 6University of Copenhagen, Copenhagen, Denmark, 7Amgen Inc., Rockville, United States, 8Yale University, New Haven, United States
1. Howard JF, Jr. M Ann N Y Acad Sci. 2018;1412(1):113-128.
2. Dresser L, et al. J Clin Med. 2021;10(11).
OS.08.08
OS.08.08 - Digital Phenotyping and Lifestyle Intervention in Patients with Myasthenia Gravis: The DIG-MG RCT
1Uppsala University, Uppsala, Sweden, 2Kinǵs College London, London, United Kingdom
Myasthenia Gravis (MG) is a chronic autoimmune disorder characterized by autoantibodies targeting the acetylcholine receptor or muscle-specific tyrosine kinase, leading to fatigable skeletal muscle weakness and general fatigue. Previous short-term supervised studies have shown that physical exercise is safe and beneficial for MG patients, but longer, unsupervised studies are needed to establish guidelines for physical activity. Additionally, the impact of sleep on MG-related fatigue remains underexplored.
The objective of the DIG-MG trial was to evaluate the effects of a digital lifestyle intervention on fatigue in MG patients, utilizing remote digital supervision via the smart OURA ring. A randomized controlled trial (ClinicalTrials.gov ID NCT05992025) was conducted with 83 MG patients from all over Sweden. Patients wore the OURA ring for 7 months, with an initial 6-week acclimatization period followed by a 3-month intervention and a subsequent 6-week observation phase.
Participants were randomized into three groups: 1) physical activity counseling (n=26), 2) sleep hygiene counseling (n=28), and 3) observation only (n=29). The primary outcome measure was the MG-Activities-of-Daily-Living (MG-ADL) scale, while secondary outcomes included the Fatigue Severity Scale and Chalder Fatigue Scale. Exploratory outcomes were based on OURA ring data, including heart rate variability, temperature fluctuations, and sleep and activity parameters.
This study demonstrated that digital group counseling and remote monitoring via the OURA ring provided valuable insights into the effects of physical activity and sleep hygiene on MG-related and general fatigue in a home-based setting.
The DIG-MG trial highlights the potential of digital lifestyle interventions in managing fatigue in MG patients. It will pave the way for refined physical activity guidelines and a deeper understanding of the role of sleep in mitigating MG-associated fatigue.
OS.09.03
OS.09.03 - Clinical Features are Distinctive in Biopsy-Confirmed Nerve Large-arteriole Vasculitis and Nerve Microvasculitis Cases
1Department of Neurology, Mayo Clinic, Rochester, USA, 2Division of Neurology, Department of Medicine, Siriraj Hospital, Mahidol University, Bangkoknoi, Thailand, 3Division of Neurology, Department of Medicine, Dalhousie University, Halifax, Canada, 4Department of Neurology, Mayo Clinic, Phoenix, USA, 5Department of Neurology, Mayo Clinic, Jacksonville, USA, 6Division of Rheumatology, Department of Internal Medicine, Mayo Clinic, Rochester, USA
Nerve microvasculitis was significantly related to non-systemic vasculitis (71% vs 21%, odd ratio, 9.1; (95%CI), 5.2-15.9; p<0.0001). Nerve microvasculitis had more autonomic involvement (24.2% vs 7.2%, odd ratio, 4.1; (95% CI), 1.9-8.9; p = 0.0002) and higher disability score at the last follow-up NIS sum (31.0 vs 20.3; p = 0.02), mRS (1.7 vs 2.3; p = 0.006).
OS.09.04
OS.09.04 - Compensatory Effect of Dnm2 Gain-of-Function and Loss-of-Function Mutations in Centronuclear Myopathy and Charcot-Marie-Tooth-Neuropathy in Vivo
1IGBMC (Institut de Génétique et de Biologie Moléculaire et Cellulaire), Inserm U1258, CNRS UMR7104, Université de Strasbourg, 67404, Illkirch-Graffenstaden, France
Different dominant mutations in DNM2, encoding dynamin, a GTPase implicated in membrane trafficking, result in two distinct neuromuscular diseases: Centronuclear myopathy (CNM) is characterized by myofiber structural anomalies and Charcot-Marie-Tooth neuropathy (CMT) by peripheral nerve defects and sensory and coordination alterations. Ultimately, both diseases are associated with muscle atrophy and weakness. It is unknown how different mutations in the same gene cause two distinct diseases. The pathological impact of these heterozygous mutations is also unclear. In addition, no cure is approved for any of the diseases.
Here we showed that CNM and CMT mutations have an opposite impact on the GTPase activity of dynamin, with gain-of-function effect in CNM and loss-of-function in CMT. Dynamin oligomerizes to achieve its cellular functions. Thus, to investigate the opposite impact of CNM and CMT mutations on dynamin functions in vivo and their potential compensatory effect, we crossed Dnm2S619L/+ CNM mice with Dnm2K562E/+ CMT mice, and evaluated the Dnm2S619L/K562E offspring through behavioral, functional, morphological and biochemical investigations at 8 weeks. Both Dnm2S619L/+ and Dnm2K562E/+ mice have decreased motor function and body weight while the Dnm2S619L/K562E littermates were larger with increased general muscle force. Moreover, Dnm2S619L/K562E mice did not display the coordination defects observed in Dnm2K562E/+ mice on the treadmill, and the in situ muscle force was significantly higher compared with Dnm2S619L/+ mice. Compared with Dnm2S619L/+ and Dnm2K562E/+ mice, histological analyses of Dnm2S619L/K562E muscle sections showed an increase of myofiber diameter. The CNM histopathological hallmarks of Dnm2S619L/+ mice, mis-localization of nuclei and mitochondria, were improved in Dnm2S619L/K562E muscle. Sciatic nerve analysis of both individual disease models revealed a decreased g-ratio, attributed to axonal hypotrophy, while Dnm2S619L/K562E nerve fibers presented a normalized g-ratio due to axonal hypertrophy and myelin thickening. qRT-PCR data also indicated a downregulation of acetylcholine receptor levels, suggesting an improved muscle innervation.
Taken together, these results show an improvement of the neuromuscular functions, muscle structure and innervation in Dnm2S619L/K562E mice, compared with Dnm2S619L/+ and Dnm2K562E/+ mice. Overall, this study is the first to report a compensatory effect of two different mutations in the same gene, causing two different disorders. This study confirms in vivo the loss-of-function mechanism in DNM2-CMT, and supports the increase of DNM2 activity as potential therapeutic strategy.
OS.09.05
OS.09.05 - Clinical Predictors of RFC1 Expansion Positivity in a Cohort of Patients with Sensory Predominant Neuropathy
Dr Anthony Garvey Garvey, Ms Zay Melville5, Dr Rachael Taylor5, Dr Carolin Scriba6, Ms Miriam Rodrigures1,2, Dr Vivien Yong, Dr Justin Kao2, Dr Melanie Doerflinger1, Dr Luciana Pelosi1, Dr Shilpan Patel2, Dr Thomas Chang2, Prof. Nigel Laing7, Associate Professor Gianina Ravenscroft6,
1Centre for Brain Research, Neurogenetics Clinic, University of Auckland, Auckland, New Zealand, 2Te Whatu Ora - Health New Zealand - Te Toka Tumai (Auckland City Hospital), Auckland, New Zealand, 3Te Whatu Ora - Health New Zealand - Southern, Dunedin, New Zealand, 4Te Whatu Ora - Health New Zealand - Hauora a Toi - Bay of Plenty, Tauranga, New Zealand, 5Dept of Physiology, Universtiy of Auckland, Auckland, New Zealand, 6Rare Genetic Diseases and Functional Genomics Group, Centre for Medical Research, University of Western Australia, Harry Perkins Institute of Medical Research, QEII Medical Centre, Nedlands, Australia, 718 Preventive Genetics Group, Centre for Medical Research, University of Western Australia, Harry Perkins Institute of Medical Research, QEII Medical Centre, Nedlands, Australia
While the gene was originally described in patients who had the full spectrum of the syndrome, once the gene was found, patients with more restricted clinical manifestations have been identified. The most widely reported restricted presentation has been the identification of patients with sensory neuropathy. However, to date, the studies which have investigated this have not thoroughly interrogated the patients to look for evidence of vestibulopathy, in particular. Also, there is some debate as to whether the nerve involvement in RFC1 expansion disease is restricted to a dorsal root ganglionopathy, or whether RFC1 expansion can cause a sensory axonal neuropathy.
We deep-phenotyped a cohort of patients with sensory-predominant neuropathy, to identify the clinical features which predict which patients were positive for RFC1 expansion.
Participants were invited to undergo five assessments which were performed blind to the other assessments.
1. Neurological exam with close attention to clinical features of CANVAS including cough, formal autonomic testing.
2. Repeat nerve conduction studies including bilateral sensory assessments.
3. Nerve ultrasound.
4. Video vestibular testing – VVOR, three plane VORs, VEMPs.
5. Blood tests for common causes of acquired neuropathy.
Genetic testing for RFC1 expansion used flanking and repeat-primed PCR (RP-PCR) to detect known pathogenic expansions.
Five of the remaining 44 patients tested positive for a pathological AAGGG expansion.
Features favouring a positive test included chronic cough, sensory neuronopathy and small nerves on ultrasound. All five had either a neuronopathy picture on NCS, or small average upper limb nerves – all had chronic cough.
Only one of the five patients had formal vestibular failure; one had a length dependent sensory neuropathy but her nerves were small.
• Though the final numbers of patients who were RFC1 expansion positive in our cohort were small we have confirmed that not all patients with RFC1 repeat expansions have vestibular involvement and that a small proportion have nerve conductions indistinguishable from axonal neuropathy - these patients may have small nerves on ultrasound.
• Nerve ultrasound is a valuable adjunct in investigating genetic neuropathy.
• The finding of a pure sensory neuropathy in a patient with chronic cough should trigger testing for RFC1 genetic testing.

OS.09.06
OS.09.06 - The Effect of efgartigimod on Pain, Mental Health and Fatigue in CIDP Patients
Ms. Febe Brackx1,
1SHE BV, Brussels, Belgium, 2Argenx, Ghent, Belgium, 3Department of Neurology, University of California, Irvine, Irvine, USA
Pain was measured using the Brief Pain Inventory – Short Form (BPI-SF), with both the Pain Severity and Pain Interference score ranging from 0 (no pain/interference) to 10 (severe pain/interference). Mental health was measured using the Hospital Anxiety and Depression Scale (HADS), with both the Depression and Anxiety scores ranging from 0 (no anxiety/depression) to 21 (severe anxiety/depression). Fatigue was measured using the Rasch-transformed Fatigue Severity Scale (RT-FSS), ranging from 0 (no fatigue) to 21 (severe fatigue).
Mean changes from baseline after max 12 weeks of treatment with efgartigimod (stage A) and after max 48 weeks treatment with efgartigimod or placebo (stage B) are reported and tested for significance using a one-sample t-test, separately by stage and treatment arm.
Furthermore, a reduction in the HADS Anxiety and HADS Depression scores were noted among efgartigimod-treated patients in stage A (Anxiety: -1.3 (SD=3.1), p<0.0001; Depression: -0.8 (SD=3.1), p<0.0001). Data from stage B confirmed the favorable stabilization of HADS scores in the efgartigimod group (Anxiety: -0.1 (SD=2.6), p=0.612; Depression: -0.2 (SD=3.0), p=0.426); whereas a worsening was found in the placebo group (Anxiety: 0.6 (SD=3.1), p=0.003; Depression: 0.7 (SD=3.4), p=0.064).
Similarly, in the efgartigimod-administered stage A, a reduction in the RT-FSS Fatigue score was found (-2.6 (SD=5.0), p<0.0001). In stage B, the RT-FSS scores remained at a similar level in the efgartigimod group (0.0 (SD=5.6), p=0.971), and an increase was observed in the placebo group (0.5 (SD=5.0), p=0.299).
OS.09.07
OS.09.07 - Creating Disease Models for Charcot-Marie-Tooth with CRISPR-Edited Stem Cells
Camille Loret1, Amandine Pauset4,5, Dr Pierre-Antoine Faye1,2, Dr Valérie Prouzet-Mauléon4,5, Dr Ioanna Pyromali1, Angélique Nizou1, Dr Federica Miressi1, Pr Franck Sturtz1,2, Dr Amandine Rovini1, Pr Frédéric Favreau1,2, Dr Béatrice Turcq4,5,
1Limoges University - UR20218 - NeurIT, Limoges, France, 2Limoges Hospital - Biochemistry and Molecular Genetics Dept, Limoges, France, 3Limoges Hospital - Bioinformatics Dept, Limoges, France, 4CRISP'edit, TBMCore UAR CNRS 3427, US Inserm 005, Bordeaux, France, 5Modeling transformation and resistance in leukemia, BRIC Inserm U1312, Univ. of Bordeaux, Bordeaux, France
Charcot-Marie-Tooth disease (CMT) stands out as the most prevalent inherited peripheral neuropathy, associated with over 100 identified genes. Most of them, such as SH3TC2, the most frequently mutated gene for the demyelinated autosomal recessive CMT form (AR-CMT), are expressed predominantly in neuronal cells. Studies of disease pathophysiology and the exploration of novel therapeutic approaches require suitable cellular models, particularly when accessing relevant cells, as in the case of CMT patients, is challenging. In this regard, human induced pluripotent stem cells (hiPSCs) are a powerful tool, but their reprogramming is time-intensive, and generated clones with diverse genetic backgrounds. Recent advancements in CRISPR base editing, now allow for the precise conversion of a base pair in the targeted DNA sequence, enhancing the generation of hiPSC isogenic cellular models for disorders arising from single nucleotide alterations.
Our objective was to use the CRISPR-Cas9 base editing technology to create the first two CMT hiPSC models harboring SH3TC2 alterations. Starting with a healthy individual's hiPSCs, we introduced two variations previously identified in patients : c.2860C>T (resulting in p.Arg954*) and c.211C>T (resulting in p.Gln71*). Due to the time and cost associated with growing and engineering hiPSCs, we optimized the generation of SH3TC2 variations in cultivable and transfectable HEK-293T cells. This allowed for a quick and easy evaluation of different CRISPR base editing approaches, subsequently, applied to hiPSCs. This approach resulted in 90.5% and 93% On-Target activity for c.211C>T, p.Gln71* and c.2860C>T, p.Arg954*, respectively, ensuring correct editing in at least one allele. As SH3TC2 is predominantly expressed in Schwann cells, we initiated the differentiation of both CMT clones and studied the disparity in SH3TC2 expression between our patient models and control ones throughout the differentiation process.
This study successfully establishes the first two Schwann cell models derived from hiPSCs harboring pathogenic CMT variations in SH3TC2. These cellular models serve as valuable tools for gaining a deeper understanding of the pathophysiology associated with this CMT type and for exploring novel therapeutic strategies.
OS.09.08
OS.09.08 - Clinical, Electrophysiological &MR Neurography Profile of Paraproteinemic Neuropathy – A Single Center Experience from South India
1NIMHANS, Bangalore, India, 2NIMHANS, Bangalore, India, 3NIMHANS, Bangalore, India, 4NIMHANS, Bangalore, India, 5NIMHANS, Bangalore, India, 6NIMHANS, Bangalore, India, 7NIMHANS, Bangalore, India, 8NIMHANS, Bangalore, India
The conductions parameters were demyelinating in 20 (64.5%), 11 (35.5% ) had axonal changes, 28 (90.3%) had motor and 27 (87.1%) had sensory involvement . The median and ulnar terminal latency index (TLI) were 0.55 and 0.65 respectively; none had abnormal TLI.
The most common was IgG lambda 9/21 (42.8%) followed by Ig A lambda 5/21 (23.8%). The haematological classification were 16 (51.6% ) MGUS, 7 (22.6%) with POEMS, 6 (19.4%) had multiple myeloma and one each had AL amyloidosis (3.2%) and solitary plasmacytoma(3.2%) . 2 patients had Anti MAG positivity and 9 patient had undergone antiganglioside antibody testing which were negative . The mean CSF protein levels were 143.2 (22/31) .
The mean brachial cross sectional area (CSA), measured as average of CSA from right C5-T1 is 0.24±0.07 cm2 (17/31) and mean lumbosacral CSA, average from L1-L5 is 0.35 ±0.17 cm2 .MR neurography showed diffuse symmetric thickening of roots (16/17), 29.4% (5/17 ) had thickening of only lower brachial trunk and 26.6% (4/15) had lower lumbar plexus thickening .The fascicular architecture and perineural fat integrity were maintained in majority, disrupted in 17.6% and 11.8% respectively
There were no significant associations between haematological syndrome, paraprotein elevation with clinical presentation, electrophysiology or radiological findings in the current study.
PP.001
PP.001 - Assessing Toxicity of Splice Switching Antisense Oligonucleotides on Different Backbone Chemistries using Zebrafish Larvae
1ZeBRLab, Department Of Biochemistry, Faculty Of Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand, 2Department of Chemistry and Biochemistry, University of Colorado at Boulder, Boulder, Colorado 80309, United States, Boulder, US, 3Centre for Molecular Medicine and Innovative Therapeutics, Health Futures Institute, Murdoch University, 90 South Street, Murdoch, WA, 6150, Australia., Perth, Australia, 4Perron Institute for Neurological and Translational Science, Perth, WA, 6009, Australia, Perth, Australia
Splice switching antisense oligonucleotide analogues have become a class of molecules that modify gene expression to ameliorate disease severity in neuromuscular conditions, i.e. Duchenne muscular dystrophy and spinal muscular atrophy. Phosphorodiamidate morpholino oligomer (PMO) has a great target binding affinity with resistance to nucleases, and this backbone chemistry has great attributes suitable for AO-induced splice switching strategy. Four PMO drugs, i.e. Exondys51, Amondys45, Vyondys 53 and Viltepso have been approved for treatment for patients with DMD. The great potential of this backbone chemistry put forth the requirement for developing efficient synthesis of PMO and PMO analogues. Thiophosphoramidate morpholino oligomer (TMO), a PMO analogue, was demonstrated as a potential AO candidate for splice switching AO in a cell model of DMD.
We synthesized oligos with the same sequence (zebrafish Gata1) on five different backbones, i.e. TMO, PMO, 2OMe, MOE, DNA-PS and investigated their toxicity in zebrafish larvae. Five concentrations of two-fold dilution of all five oligos were injected into newly fertilized zebrafish embryos. Two nanolitres of oligos were microinjected into yolk of each embryo. Survival of zebrafish larvae was determined at 24 hours post fertilization (hpf), while abnormal morphology was micrographed at 48, and 72 hpf. Survival of zebrafish embryos injected with PMOs at five concentrations, i.e. 100, 50, 25, 12.5 and 6.25 uM, were between 39 to 80%, while survival of those injected with TMOs at the similar concentrations were between 5 to 45% in a concentration dependent manner. Survival of zebrafish larvae injected with other oligos at similar concentrations were between 1 to 16%. The abnormal morphologies, i.e. reduced yolk absorption, kyphosis and pericardial edema, were observed between 70 to 5% at different concentrations in TMO and PS-DNA injected zebrafish embryos.
Our results suggested that PMO is the least toxic backbone, while PS-DNA, 2’OMe and 2’MOE injection led to substantially more toxicity in our zebrafish embryo toxicity test. TMO yielded less the embryo toxicity compared to PS-DNA, 2’OMe and 2’MOE injected zebrafish. Testing for splice switching efficiency of each oligotide should provide an insight on how TMOs compare to PMOs.
Figure demonstrates the chemical structures of splice switching antisense oligonucleotides on with different backbone chemistries.
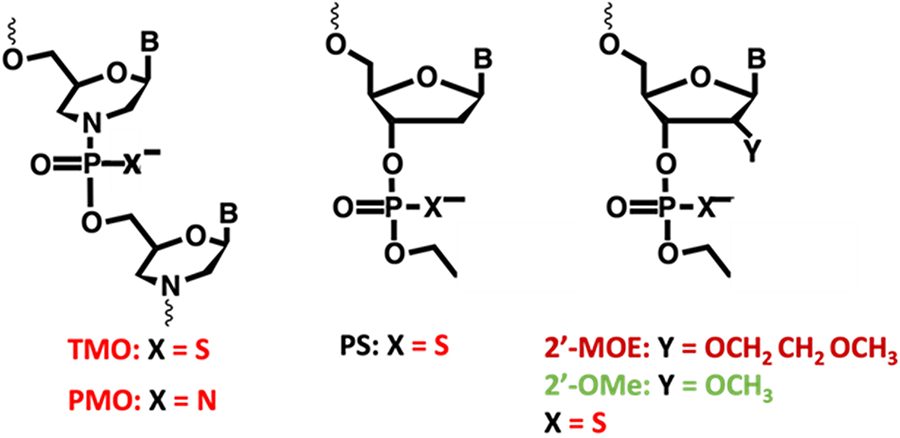
PP.002
PP.002 - Six-Year Long-Term Safety and Efficacy of Golodirsen in Patients With DMD vs Mutation-Matched External Controls
Francesco Muntoni1,2, Andreea M. Seferian3, Volker Straub4, Michela Guglieri4, Laurent Servais5,6, Ewa Wilk-Durakiewicz7, Xiao Ni7, Ping Gao7, Menghan Hu7, Joel Iff7, Lorna Hill7, Ihor Sehinovych7, Larry Orogun7,
1Dubowitz Neuromuscular Centre, University College London, Great Ormond Street Institute of Child Health, United Kingdom, 2National Institute for Health Research Great Ormond Street Hospital Biomedical Research Centre, United Kingdom, 3Assistance Publique Hôpitaux de Paris, Sorbonne Université, Institut de Myologie, AFM-Téléthon, Essais Cliniques I-Motion Enfants, Hôpital Armand Trousseau, France, 4John Walton Muscular Dystrophy Research Center, Translational and Clinical Research Institute, Newcastle University and Newcastle Hospitals NHS Foundation Trust, United Kingdom, 5Division of Child Neurology, Centre de Références des Maladies Neuromusculaires, Department of Pediatrics, University Hospital Liège & University of Liège, Belgium, 6MDUK Oxford Neuromuscular Centre & NIHR Oxford Biomedical Research Centre, University of Oxford, United Kingdom, 7Sarepta Therapeutics, Inc., Cambridge, MA, Italy, 8Pediatric Neurology Unit, Università Cattolica del Sacro Cuore Roma, Italy, 9Nemo Clinical Centre, Fondazione Policlinico Universitario A Gemelli IRCCS, Italy
Golodirsen is FDA approved for the treatment of Duchenne muscular dystrophy (DMD) in patients with exon 53 skip-amenable mutations. Results from Study 4053-101 (NCT02310906) and the open-label extension (OLE) 4045-302 (NCT03532542) evaluating the safety and efficacy of golodirsen treatment up to ∼6 years in patients with progressive disease deterioration are described. Post hoc analyses comparing ambulatory and pulmonary function of golodirsen-treated patients with matched (including age, mutation, steroid use) external controls (ECs) were performed. Twenty-five patients received treatment at mean age 8.8 years; 18/25 (72%) completed the OLE up to 6.49 years. Overall, golodirsen was well tolerated: treatment-emergent adverse events were generally mild, nonserious, and unrelated to treatment; there were no treatment-related discontinuations, kidney abnormalities, or port-related infections in the OLE. At year 3, loss of ambulation (LOA) occurred in 4/25 (16%) of golodirsen-treated patients compared with 12/54 (22.2%) age- and mutation-matched ECs, representing a 91.1% risk reduction (HR 0.089; P=0.0224). Over 6 years, 15 golodirsen-treated patients experienced LOA (10.7–19.5 years), with 7 patients still ambulant at OLE completion (12.4–20.3 years). Compared with age- and mutation-matched ECs (n=16), golodirsen-treated patients experienced a median delay in time to LOA of ∼2.4 years, representing a 47.4% risk reduction (HR 0.526; P=0.149). A separate post hoc analysis suggested that golodirsen-treated patients (≥10 years) experienced a statistically significant and clinically meaningful attenuation in the annual rate of percent predicted forced vital capacity decline compared with mutation-matched ECs (2.9% vs 6.67%; P<0.01). Overall, golodirsen treatment up to ∼6 years demonstrates a favorable, consistent safety profile and supports its long-term efficacy vs mutation-matched ECs. This is the longest follow-up of safety and functional benefit of golodirsen in a declining DMD population.
PP.003
PP.003 - Deep Phenotype Digitalization in Neuromuscular Diseases: Challenges and Prospectives from InGene 2.0
1University of Pisa, Pisa, Italy, 2IFC-CNR, Pisa, Italy, 3IRCCS Stella Maris Foundation, Calambrone-Pisa, Italy
Here, we present an integrated, InGene-based, and multiparametric approach, converging on a single cloud-based, GDPR-compliant platform, named Health360, featuring a user-friendly interface, created with the aim to perform a comprehensive phenotype digitalization of the neuromuscular patient as an innovative technological tool for care optimization and sharing data between different centers.
PP.004
PP.004 - FORTIFY: A Phase 3 Study to Evaluate Safety, Tolerability, and Efficacy of BBP-418 in LGMD2I/R9
Dr. Divya Reddy1, Tricia Blankenbiller1, Amy Rainey1, Lindsay Reklis1, M.D., M.Sc. Douglas Sproule1,
1ML Bio Solutions, A BridgeBio Company, Palo Alto, United States
BBP-418 is an investigational oral substrate supplementation intended to saturate the partially functional FKRP enzyme, drive increased glycosylation of αDG, and potentially stabilize or improve muscle integrity.
In an ongoing open-label Phase 2 study in 14 individuals with LGMD2I/R9, BBP-418 was well-tolerated. Following 21+ months of dosing, stabilization in measures of ambulation (10MWT, 100mTT) and motor performance (NSAD) was observed. Marked increases in glycosylated αDG and reductions in serum CK were initially noted at 3 months post-dosing and sustained over 21 months. Based on these data, a Phase 3 study was designed to further assess the safety, tolerability, and efficacy of BBP-418 in LGMD2I/R9.
The primary endpoint will be change in NSAD from baseline in BBP-418-treated individuals relative to placebo at 36 months. Secondary endpoints evaluated will include FVC, PUL 2.0, and ambulatory measures (100mTT and 10mWT). Troponin I, Electrocardiograms and Echocardiograms will be obtained to assess progression of cardiomyopathy. An interim analysis of at least 42 participants will evaluate change in glycosylated αDG levels, and selected clinical measures, at 12 months.
PP.005
PP.005 - Sevasemten, a Fast Myosin Inhibitor, in Becker Adults Reduced Muscle Damage Biomarkers and Stabilized Function
H Phan1, A Russell2, B Barthel2, L Thaler2, N Kilburn2, M Amato2,
1Edgewise Therapeutics, Boulder, USA, 2Rare Disease Research, Atlanta, USA
Fast (Type II) muscle fibers are affected early and disproportionately in dystrophinopathies. Sevasemten (EDG-5506) is an orally administered, once daily, investigational product that modulates fast skeletal muscle myosin and, in DMD disease models, decreased muscle damage biomarkers and fibrosis while increasing muscle strength and activity.
ARCH is a 24-month Phase 1b open-label study of sevasemten, assessing safety, pharmacokinetics, biomarkers of muscle damage and functional measures in adults with Becker. Twelve ambulatory adults with Becker received daily oral doses of sevasemten. At baseline, North Star Ambulatory Assessment (NSAA) ranged from 5 to 31 (mean 15.5). Decreased muscle mass was evident by low serum creatinine and DXA lean muscle mass. After 24 months, sevasemten was well tolerated without serious adverse events, withdrawals due to AEs, or dose modifications. Rapid (within 1-2 months) reductions in biomarkers of muscle damage were sustained to 24 months, including creatine kinase, myoglobin and fast skeletal muscle troponin I. Alongside acute and chronic biomarker changes, trends toward functional improvement were noted with NSAA with a mean change of -0.2 versus an expected decline of -2.4 or more as predicted from natural history (Bello 2016, Van de Velde 2021, De Wel 2024). The 100-meter timed test and grip strength were unchanged over 24 months.
In summary, sevasemten was well tolerated with rapid and sustained reductions in biomarkers of muscle damage. Treatment resulted in acute and sustained reductions in muscle damage biomarkers. Functional improvements compared to the expected natural history decline in the NSAA score were observed. Phase 2 trials in BMD and DMD are ongoing (NCT05291091 and NCT05540860), including a pivotal cohort in Becker.
PP.006
PP.006 - Biallelic Vvariants in JAG2 in Additional Patients with Limb-Girdle Muscular Dystrophy: A Case Report
1Harry Perkins Institute of Medical Research, Centre for Medical Research, University of Western Australia, Perth, Australia, 2Department of Anatomical Pathology, Royal Brisbane and Women's Hospital, Brisbane, Australia, 3Department of Medical Imaging and Nuclear Medicine, Queensland Childrens Hospital, Brisbane, Australia, 4Greg Marzolf Jr. Muscular Dystrophy Center and Department of Neurology, University of Minnesota Medical School, Minneapolis, Minnesota; Institute for Translational Neuroscience, University of Minnesota, Minneapolis, USA, 5Neurosciences Department, Queensland Children's Hospital, Brisbane, Australia, 6Diagnostic Genomics, PathWest, Perth, Australia
Limb-girdle muscular dystrophy recessive 27 (LGMD R27) is a progressive disorder associated with biallelic pathogenic variants in JAG2, encoding the JAG2 Notch ligand. To date, 24 affected individuals from multiple unrelated families have been described in two reports. Here, we present two additional families and three novel missense variants in JAG2 identified by disease gene panel sequencing and exome sequencing. This includes a homozygous variant (c.1021G>T, p.(Gly341Cys)) in two siblings of Pakistani origin, and compound heterozygous variants (c.703T>C, p.(Trp235Arg); c.2350C>T, p.(Arg784Cys)) in an Australian proband of European ancestry. The patients’ phenotypes resemble the relatively severe LGMD R27 cases with early-onset progressive myopathy and limb weakness leading to walking difficulties or loss of ambulation. Muscle MRI of two patients revealed widespread fatty infiltration of the torso and limb muscles and showed characteristic patterns previously described in LGMD R27. This includes the outward to inward progression of fatty infiltration of the quadriceps muscles, with central areas of less affected muscle. Muscle pathology revealed myopathic changes including fatty infiltration and type 1 fibre predominance, with evidence of muscle fibre degeneration and regeneration in one patient. The p.(Gly341Cys) and p.(Arg784Cys) substitutions introduce a cysteine within the epidermal growth factor-like repeats (EGF3 and EGF14, respectively) of JAG2 and are predicted to disturb native disulphide bonding and EGF folding, a mechanism reported in LGMD R27 and other Notch-related disorders. The p.(Trp235Arg) substitution located within the JAG2 Delta/Serrate/Lag-2 domain affects a conserved residue which has been associated with JAG1-related disease and is predicted by FoldX to be destabilising (ΔΔG 2.75 ± 0.16 kcal/mol). The mechanisms of these variants, like other JAG2 substitutions identified to date, remain to be functionally elucidated. This report highlights the need for JAG2-centric guidelines for variant characterisation and interpretation to help rapidly and appropriately classify and report variants. Muscle MRI may help narrow the differential diagnosis among other limb-girdle muscular dystrophies and provide guidance towards attaining a genetic diagnosis. Given the severe, rapidly progressive nature of JAG2-related myopathy in some patients and the 1 in 4 risk of disease reoccurrence, a diagnosis for families is essential to enabling appropriate genetic counselling for subsequent pregnancies and family planning.
PP.007
PP.007 - Unraveling Dysferlin-Deficient Mice: Comprehensive Analysis of Disease Pathology in the BLAJ Mouse Model
1Murdoch Childrens Research Institute, Parkville, Australia
Dysferlinopathies represent a heterogeneous group of limb girdle muscular dystrophies characterized by the loss of dysferlin protein due to mutations in the dysferlin (Dys) gene. Despite being clinically variable, dysferlinopathies uniformly manifest as progressive muscle weakness, for which there are limited treatment options, underscoring the urgent need for effective therapeutic interventions.
The development of targeted treatments has been hindered by a limited understanding of disease mechanisms and suitable pre-clinical laboratory models. This study aims to elucidate the pathophysiology of dysferlin deficiency using dysferlin-deficient (dysf-/-) BLAJ mice.
A cohort of 13-month-old dysf-/- BLAJ and age-matched wild-type C57BL/6J healthy control mice were utilized. Animal ethics approval was obtained (AEC approved A946), and mice were donated by the Jain Foundation. Our experimental approach involved a comprehensive analysis comprising functional muscle assessments and histological examination, coupled with immune cell profiling.
Functional analyses encompassed grip strength measurement and ex vivo muscle physiology evaluation of the extensor digitorum longus (EDL) and soleus muscles. Grip strength assessments revealed a significant decrease (14%) in dysf-/- BLAJ mice compared to controls. Interestingly, EDL muscle physiology showed no discernible differences between genotypes, suggesting a differential impact of dysferlin deficiency on muscle function. Notably, dysf-/- BLAJ mice exhibited enhanced fatigue resistance and recovery in soleus muscles compared to controls, highlighting potential compensatory mechanisms in the posterior hindlimb.
Furthermore, we conducted histological analyses to assess muscle pathology, including perilipin deposition and fibrosis. Histological examination of quadriceps (QUAD) muscles revealed elevated perilipin deposition and increased fibrosis in dysf-/- BLAJ mice, indicative of progressive pathological changes associated with dysferlin deficiency.
Immunohistochemical profiling of immune cell infiltration in QUAD muscles unveiled a notable increase in F4/80 and CD8 expression in dysf-/- BLAJ mice, suggesting heightened immune response in affected muscles. Additionally, muscle mass quantification demonstrated significant decreases in gastrocnemius (20.3%), quadriceps (27.4%), and psoas (31%) muscles, contrasted with an increase in soleus mass (18%) in dysf-/- BLAJ mice.
In conclusion, our comprehensive analysis provides valuable insights into dysferlin-deficient mice, shedding light on critical aspects of disease pathophysiology and highlighting the potential use of this model in the development of future therapeutic targets. Further investigations into muscle function phenotypes are warranted with ongoing research currently assessing the in vivo muscle function parameters in this mouse model. By improving our understanding of this pre-clinical model we aim to elucidate novel therapeutic avenues for dysferlinopathies, ultimately improving patient outcomes.
PP.008
PP.008 - Severe Neonatal Hypotonia – X Lynked Myotubular Myopathy CaseREPORT
1NATIONAL UNIVERSITY HOSPITAL FOR CHILDREN NEUROREHABILITATION "DR NICOLAE ROBANESCU' ", BUCHAREST, Romania, 2University of Medicine and Farmacy “Carol Davila”, BUCHAREST, Romania, 3Emergency Hospital "Bagdasar Arseni", BUCHAREST, Romania
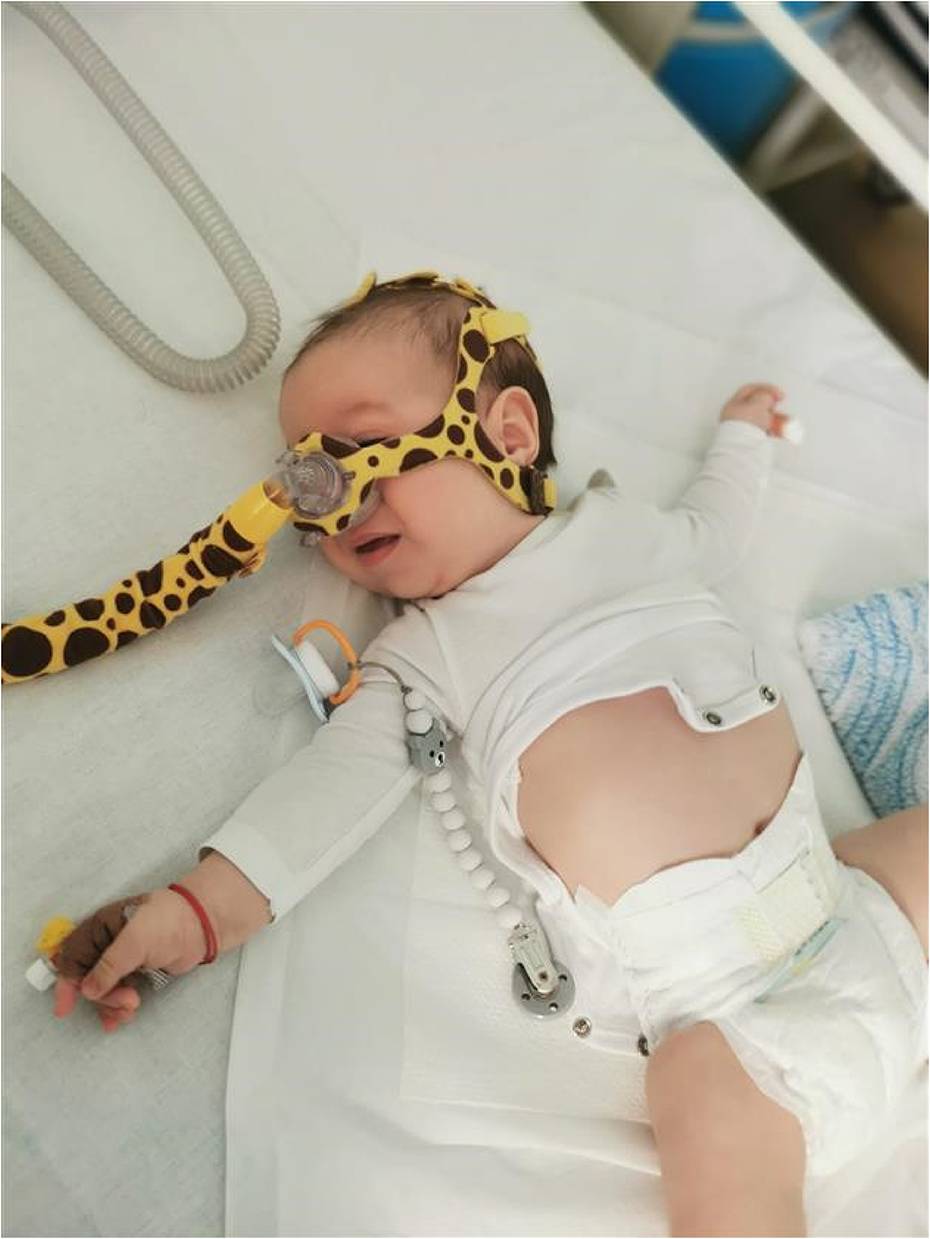
PP.009
PP.009 - Exploring Energy Expenditure in Duchenne Muscular Dystrophy
1Monash University, Department Nutrition, Dietetics and Food, Melbourne, Australia, 2Murdoch Children's Research Institute, Melbourne, Australia, 3Royal Children's Hospital, Department of Neurology, Melbourne, Australia, 4University of Melbourne, Department of Paediatrics, Melbourne, Australia, 5University of Queensland, Brisbane, Australia
Considering participants within a healthy weight range, energy expenditure was similar between boys with DMD and control participants across all measures: REE DMD: 4797 (752) kJ vs control: 4884 (1324) kJ (p 0.895), TEE 7041 (970) kJ vs 7397 (2320) kJ (p 0.709), PAL 1.58 (0.37) vs 1.52 (0.27) (p 0.921) and AEE 2589 (1349) kJ vs 2513 (1361) kJ (p 0.708).
Considering participants above a healthy weight range, there was a considerable (but not statistically significant) difference in TEE (7676 (930) kJ vs 9927 (2657) kJ, p 0.188) largely driven by a significant difference in AEE between boys with DMD and control participants, respectively (2457 (1037) kJ vs 4278 (2153) kJ, p 0.041). There were no statistically significant differences in REE (5133 (513) kJ vs 5649 (1814) kJ, p 0.612), or PAL (1.49 (0.24) vs 1.83 (0.59), p 0.344) between boys with DMD and control participants, respectively; however, we note the differences in REE and PAL may be clinically important.
PP.010
PP.010 - Awareness and Adherence to Guidelines for Allied Health Assessment in Duchenne Muscular Dystrophy
1Murdoch Children’s Research Institute, Melbourne, Australia, 2Royal Children’s Hospital, Department of Neurology, Melbourne, Australia, 3Monash University, Department Nutrition, Dietetics and Food, Melbourne, Australia, 4University of Sydney School of Health Sciences, Faculty of Medicine, Sydney, Australia, 5Sydney Children’s Hospitals Network, Sydney, Australia, 6University of Melbourne, Department of Paediatrics, Melbourne, Australia, 7University of Melbourne, School of Health Sciences, Faculty of Medicine, Dentistry and Health Sciences, Melbourne, Australia
Of the 68 survey respondents, 52 (76%) completed questions specific to allied health assessments. Table 1 describes the use and barriers to use of selected assessment recommendations from the allied health guideline. Over half of respondents reported using goniometry, the North Star Ambulatory Assessment, and height and weight measures. Barriers to use varied by assessments; however, lack of access to equipment/treatment space, insufficient time and knowledge of the assessment were barriers identified across all assessments.
PP.011
PP.011 - Oxidised Albumin in Plasma as Biomarker of Myonecrosis and Oxidation in Duchenne Muscular Dystrophy
1School of Human Sciences, The University of Western Australia, Perth, Australia, 2School of Molecular Sciences, The University of Western Australia, Perth, Australia, 3Proteomics International Pty Ltd, Harry Perkins Insitute of Medica Research, Perth, Australia
There is an urgent need for robust blood biomarkers in blood to rapidly and reliably measure increased inflammation and associated oxidative stress in damaged tissues, of humans for various conditions, especially for use in clinical trials and in experimental animal models for various neuromuscular diseases. Our data strongly support the use of plasma albumin thiol oxidation (AlbOx), for cysteine 34 (Cys34), as a validated simple blood biomarker to track increased levels of inflammation and oxidative stress closely associated with intrinsic ongoing necrosis of skeletal muscles (myonecrosis), the key feature that occurs in most dystrophic skeletal muscles in boys with Duchenne muscular dystrophy (DMD).
In the mdx mouse model of DMD, we have visualised high levels of various oxidised molecules localised specifically within foci of myonecrosis, with associated high levels of neutrophils and macrophages that generate highly reactive oxygen species (Iwasaki et al, 2022. PMID: 36270048). In intimate contact with myofibres, albumin in the interstitial fluid that circulates throughout all muscles (from the blood, and then returns to the blood) is exposed to these oxidants, resulting in high levels of AlbOx in plasma. Such high levels of plasma AlbOx are demonstrated in dystrophic animal models for DMD, including mdx mice, dogs and rats (Terrill et al, 2016 & 2013. PMID: 27679740 & PMID: 36837851): with associated oxidised proteins in urine also elevated in dystrophic mice and dogs (Terrill et al, 2018. PMID: 33031394).
Recent studies comparing redox modifications of oxidised albumin Cys34 in the plasma and muscles of mdx mice, confirm that plasma AlbOx is an accurate blood biomarker of myonecrosis and disease progression, and tracks with experiments that reduce or increase myonecrosis (Terrill et al, 2024. PMID: 38929159). To facilitate convenient clinical testing for AlbOx, we have developed a simple, cost effective and widely accessible dried blood card (DBC) collection method using small fingerprick (ear or heel prick) blood sample, that can be easily used at home (repeatedly), stored at room temperature and mailed for rapid protein analysis of AlbOx (Grounds et al, 2020. PMID: 32224496). We have successfully trialled DBCs at home with normal human volunteers and demonstrated that plasma AlbOx is responsive to changes in inflammation and oxidative stress within 2 days of exercise-induced muscle damage (James et al, 2024 PMID: 38627299). These combined studies strongly support the wider use of plasma AlbOx as a blood biomarker for animal and human studies; specifically to rapidly measure the capacity of therapeutic clinical trials that aim to protect, and hence reduce the severity of myonecrosis in DMD.
PP.012
PP.012 - Patient Demographics, Clinical Characteristics and Genetic Mutations of DMD and BMD Patients in Qatar
Dr. Mahmoud Osman1,
1Sidra Medicine, Doha, Qatar
are rare X-linked neuromuscular childhood disorders that result in functional decline, loss of ambulation and early death due to cardiac or respiratory failure.
PP.013
PP.013 - Epidemiology and Clinical Features of Dystrophinopathies in Australia - A Registry Based Study
1Murdoch Children's Research Institute, Melbourne, Australia, 2The Royal Children's Hospital, Melbourne, Australia, 3Queensland Children's Hospital, Brisbane, Australia, 4Women's and Children's Hospital, Adelaide, Australia, 5Sydney Children's Hospital, Randwick, Australia, 6School of Clinical Medicine, UNSW Sydney, Sydney, Australia, 7Discipline of Child and Adolescent Health, University of Sydney, Sydney, Australia, 8Western Australian Department of Health, East Perth, Australia, 9Princess Alexandra Hospital, Brisbane, Australia, 10Perth Children's Hospital, Perth, Australia
PP.015
PP.015 - Muscle MRI Outcomes in Delandistrogene Moxeparvovec-Treated Patients with Duchenne Muscular Dystrophy from EMBARK Part 1
Dr. Krista Vandenborne1, Dr. Glenn Walter2, Prof. Volker Straub3, PhD Rebecca Willcocks1, PhD Sean Forbes1, Sravya Ennamuri4,
1Department of Physical Therapy, University of Florida, Gainesville, USA, 2Department of Physiology and Aging, University of Florida, Gainesville, USA, 3Newcastle University, Newcastle Upon Tyne, John Walton Muscular Dystrophy Research Centre, UK, 4Sarepta Therapeutics, Inc., Cambridge, USA, 5Roche Products Ltd, Welwyn Garden City, UK, 6F. Hoffmann-La Roche Ltd, Basel, Switzerland
Delandistrogene moxeparvovec, an rAAVrh74-based gene transfer therapy designed to address absent functional dystrophin by delivering a transgene encoding an engineered micro-dystrophin, is approved in the US, UAE, Qatar, Kuwait, Bahrain, and Oman (March 2024) for ambulatory pediatric patients aged 4 through 5 years with Duchenne muscular dystrophy (DMD) with a confirmed DMD mutation. Delandistrogene moxeparvovec showed stabilization of disease trajectory and manageable safety in patients aged ≥4–<8 years in a Phase 3 trial (EMBARK; NCT05096221). We report prespecified, exploratory analyses of muscle magnetic resonance (MR) data from EMBARK Part 1 (Week 52) to test the effect of delandistrogene moxeparvovec on muscle MR measures of disease progression.
A subset of patients who received delandistrogene moxeparvovec (1.33×1014 vg/kg; n=19) or placebo (n=20) underwent quantitative muscle MR, including 8-point Dixon (MR imaging [MRI]) and proton MR spectroscopy (MRS) to measure muscle fat fraction (FF) and multi-slice spin echo imaging to measure transverse relaxation time (T2). MRS FF was measured in the soleus and vastus lateralis, while MRI T2 and FF were measured in 5 preselected leg muscles/groups with an important role in ambulation. A global statistical test (GST) combining all muscles and modalities assessed overall treatment effect.
Overall, treated patients showed less disease progression vs. placebo. In all muscles, MRI FF and MRS FF magnitude of changes favored the treated group compared with placebo. The placebo group also showed increases (worsening) in T2 (all muscles) compared with reductions (improvement) in treated patients (4/5 muscles; baseline to Week 52 change ranged from −1.05 to 0.05 msec [delandistrogene moxeparvovec] and 1.11 to 3.02 msec [placebo]). GST supports overall treatment benefit (P=0.0328).
Muscle MRI changes were consistent with previously reported timed function tests, suggesting disease stabilization with delandistrogene moxeparvovec.
PP.016
PP.016 - Biomarker Panel for Duchenne Muscular Dystrophy Clinical Trials
1The University of Western Australia, Perth, Australia
Duchenne Muscular Dystrophy (DMD) is a fatal X-linked muscle wasting disease, which results in premature death from cardiac or respiratory complications. DMD is characterised by severe skeletal muscle loss, due to chronic bouts of inflammation and oxidative stress, resulting in ongoing myonecrosis. The current gold standard treatments of DMD, corticosteroids, have extended the lifespan of DMD patients, but are not curative, with patients still dying around 30 years of age. Creating new treatments for DMD is challenging and time consuming as the current clinical outcomes depend on changes in muscle function which can take over a year to become evident.
Molecular biomarkers in blood which can respond very rapidly to effective treatments for DMD have the potential to substantially improve timelines. These could be used, for example as a go/no-go checkpoint, so that no-go clinical trials could cease early, which would benefit families and free resources for testing alternative therapies. Short-term testing using blood biomarkers would also be highly desirable for clinicians to more efficiently adjust regimens (e.g. dosage) of current treatments to minimise side effects without compromising treatment efficacy.
We propose biomarkers in blood linked to myonecrosis, as well as the associated oxidative stress and inflammation, will be early indicators of likely treatment efficacy. We have identified a panel of plasma biomarkers that have previously been shown to be elevated in dystrophy, and are associated with key aspects of the dystropathology. The panel includes proteins that respond to inflammation, oxidative stress, fibrosis and muscle degradation. We will present findings outlining the response of the panel to exercise-induced skeletal muscle damage and corticosteroid treatment in dystrophic mice.
PP.017
PP.017 - Effect of Ataluren on Upper Limb Function in nmDMD Patients from the STRIDE Registry
Prof Francesco Muntoni1, Filippo Buccella2, Dr Andrés Nascimento Osorio3, Prof Már Tulinius4, Dr Maria Bernadete Dutra de Resende5, Shelley Johnson6,
1University College London Great Ormond Street Institute of Child Health, London, UK, 2Parent Project APS Italy, Rome, Italy, 3Unidad de Patología Neuromuscular, Hospital Sant Joan de Déu, Universidad de Barcelona, Barcelona, Spain, 4Department of Pediatrics, Queen Silvia Children’s Hospital, University of Gothenburg, Gothenburg, Sweden, 5Department of Neurology, Faculty of Medicine, University of São Paulo, São Paulo, Brazil, 6PTC Therapeutics Inc., South Plainfield, USA, 7PTC Therapeutics Germany GmbH, Frankfurt, Germany, 8Department of Pediatric Neurology, Catholic University, Rome, Italy
Strategic Targeting of Registries and International Database of Excellence (STRIDE; NCT02369731) is an international, observational registry evaluating the long-term safety and effectiveness of ataluren in individuals with nonsense mutation Duchenne muscular dystrophy (nmDMD) in real-world clinical practice. This study investigated whether patients in the STRIDE Registry experienced a slower decline in upper limb function versus patients with DMD receiving standard of care (SoC) alone in the Cooperative International Neuromuscular Research Group (CINRG) Duchenne Natural History Study (NCT00468832).
Data were extracted on 31 January 2023. Upper limb function was assessed using the PUL module in the STRIDE Registry and the Brooke Scale in the CINRG cohort. Kaplan–Meier analyses estimated the age at loss of upper limb function (loss of overhead reach, loss of hand-to-mouth function and loss of distal hand function).
The mean age at the last assessment was 14.0 years and 15.2 years in propensity score-matched STRIDE and CINRG cohorts (N=277 per cohort), respectively. The median (95% CI) ages at loss of overhead reach (STRIDE vs CINRG) were 15.8 (12.1, 19.2) and 12.7 (10.3, 14.3) years, respectively (HR [95% CI], 0.412 [0.220, 0.771]; p=0.0095). In CINRG patients, the median (95% CI) age at loss of hand-to-mouth function was 20.5 years (17.1, NC) and the median age at loss of distal hand function was 30.1 years (27.6, NC). These outcomes were not yet estimable for STRIDE Registry patients.
These results indicate that treatment with ataluren plus SoC in routine clinical practice may help preserve upper limb function in patients with advanced nmDMD.
PP.018
PP.018 - Novel Inhibitor PK007 Reduces Muscle Inflammation and Myonecrosis in Mdx Mouse Models
1School of Biodemedical Sciences, The University of Queensland, Brisbane, Australia, 2Institute for Molecular Bioscience, The University of Queensland, Brisbane, Australia, 3School of Anatomy and Human Biology, University of Western Australia, Perth, Australia
PP.019
PP.019 - Integrated Machine Learning-Based Platform Model to Study FSHD: From Deep Phenotyping to Predictive Biomarkers
Dr. Giulia Ricci1, Dr. Giulio Gadaleta2, Dr. Alessandro Tonacci3, Dr. Francesco Sansone3, Dr. Raffaele Conte3, Prof. Tiziana Mongini2, Prof. Massimiliano Filosto4,
1Department of Clinical and Experimental Medicine, University of Pisa, Pisa, Italy, 2Neuromuscular Unit, Department of Neurosciences "Rita Levi Montalcini", University of Turin, Turin, Italy, 3Institute of Clinical Physiology, National Research Council of Italy (IFC-CNR), Pisa, Italy, 4NeMO-Brescia Clinical Center for Neuromuscular Diseases, Brescia, Italy
FSHD is a complex disease with high clinical variability and a still unsolved genetic landscape. Although detection of the molecular signature is the main determinant of diagnostic criteria, this paradigm does not completely fulfill the understanding of disease natural history and often results on pitfalls in familial genetic counseling.
Several molecules run both in pre-clinical and clinical trial studies, further prompting the need for prognostic indicators and biomarkers describing disease progression. A diagnostic flow-chart based only on genetic signature may not highlight the differences among patients and possibly bias the interpretation of response to a certain treatment. On the other hand, the clinical complexity hides further molecular mechanisms to be investigated.
Based on these considerations, we wanted using FSHD as a disease model and develop a dedicated digital platform that can comprehensively collect all the different variables involved, clinical, laboratory molecular and investigational, enabling a deep phenotyping towards a personalized plan of diagnosis and care. This is be achieved through an integrated and multiparametric approach, converging on a single support cloud-based, GDPR-compliant platform.
The study includes a multicenter collaboration between neurologists and IT-professionals to develop a platform starting from the structure of the Clinical Comprehensive Evaluation Form, a tool shared by the Italian Clinical Network for FSHD, and as at this stage comprises modules for phenotype and pedigree, genetics and MRI.
A machine-learning approach will inform automatic algorithms for recognition of clinical, genetic and imaging patterns and provide clinicians with correlation analysis among variables.
PP.020
PP.020 - Motor Outcomes to Validate Evaluations in Pediatric FSHD (MOVE Peds) – Study Outline
1The Royal Children's Hospital, Parkville, Australia, 2The Murdoch Children's Reearch Institute, Parkville, Australia, 3Duke University, Durham, USA, 4Seattle Children's Hospital, Seattle, USA, 5University of Rochester, Rochester, USA, 6University of Kansas Medical Centre, Kansas City, USA, 7Kennedy Krieger Institute, Baltimore, USA, 8UC Irvine Medical School, Orange, USA
PP.021
PP.021 - Early-Onset FSHD Natural History and Pre-Clinical Research Pipeline at Melbourne Children’s Campus
1The Royal Children's Hospital, Parkville, Australia, 2Murdoch Children's Research Institute, Parkville, Australia
FSHD is a genetically complex neuromuscular disorder that typically presents in the second or third decades of life. The manifestation of symptoms in early childhood is less common and thought to correlate with greater symptom severity and more rapid disease progression. Early-onset FSHD is rare and less is known about disease natural history or factors that may impact disease severity and progression.
The Royal Children’s Hospital (RCH) and Murdoch Children’s Research Institute (MCRI) are in the third year of a childhood onset FSHD longitudinal outcome study (iFSHD-LOS) involving the collection of biological samples (blood and saliva), functional and patient reported outcome measures and whole-body MRI. This project involves three pillars of work 1) the clinical / MRI assessment and longitudinal analysis of children with early-onset FSHD, 2) biobanking of patient material (saliva, blood and induced pluripotent stem cell (iPSC) lines) for current and future research initiatives and 3) analysis of the role that other biological or environmental factors play in FSH disease mechanism and outcomes.
The ultimate goal of this project is to understand the disease trajectory and the impact of development and childhood growth, validate existing outcome measures, and develop a framework for laboratory-based pre-clinical drug testing to facilitate wholistic patient management and research into childhood onset FSHD.
PP.022
PP.022 - Longitudinal Remote Monitoring Of Motor Function In Inclusion Body Myositis Using Wearable Sensors
Ram Kinker Mishra1, Adonay Nunes1, Rylee Cole1, Andrew Heim2, Samantha Colgan2, Ali Russo2, Abby Davis2, Ilia Parish2, Sandhya Sasidharan2, Matthew Varon2, Constantine Farmakidis2, Mamatha Pasnoor2, H. Jeffrey Wilkins3, Ashkan Vaziri1,
1BioSensics, LLC, Newton, United States, 2University of Kansas Medical Center, Kansas City, United States, 3Aburco, Inc., Newton, United States
Sporadic inclusion body myositis (IBM) causes gradual weakness in the hands and proximal legs, eventually affecting all muscles and leading to physical disability and loss of independence. Current assessment methods involve time-consuming in-clinic muscle strength examinations by experts, which are insensitive to functional changes, exhibit poor inter-rater reliability, and only offer a snapshot of disease progression. Our study aims to develop wearable-based digital health technologies to monitor upper and lower limb function in IBM, allowing for frequent at-home monitoring of motor function, which could be integrated into future clinical trials and routine care.
Ten individuals diagnosed with IBM (69.7 ± 7.6-year-old, 4 female) participated in our study and are being followed for up to 24 months. An additional 10 individuals are being recruited. The 10-point IBM Functional Rating Scale (IBMFRS) is assessed during each clinical visit, at baseline, 3-month intervals twice and then every 6 months along with strength measures and patient-related outcomes. Participants wear at home a PAMSys™ pendant and two PAMSys ULM™ wrist sensors (one on each wrist) for a week after each visit to measure daily physical activity and hand goal-directed movements (GDMs).
Our findings based on analyzing sensor-derived measures at the baseline indicate significant associations between sensor-derived physical activity metrics and the lower extremity subdomain score of IBMFRS (sum of items 7-10) with r = -0.558 to -0.846, p < 0.05. Additionally, GDM-related metrics showed moderate to high correlations with the upper extremity subdomain scale (sum of items 2-6) with r = 0.649 to 0.775.
These pilot results demonstrate the potential of at-home wearable-based monitoring for tracking motor dysfunction in IBM. Digitized assessments offer objective measurements, improved compliance, convenience, scalability, and cost-effectiveness, thereby enhancing clinical care and research in IBM.
PP.023
PP.023 - Inclusion Body Myositis in a Neurological Referral Center in Thailand
1Neurological Institute of Thailand, Department of Medical Services, Ministry of Public Health, Bangkok, Thailand, 2Department of Pathology, Faculty of Medicine Siriraj Hospital, Mahidol University, Bangkok, Thailand
Inclusion body myositis (IBM) was most common acquired myopathy in people over 40 years of age. The disease was slowly progressed. It had unique clinical presentation such as weakness in several muscle groups especially long finger flexors and quadriceps. Prevalence of IBM varied among the ethnic groups. IBM was thought to be rare in Asian countries, however the number of the patients had increased overtime and several case series had been reported in the recent years. We reported here the clinical characteristic of 6 IBM patients seen at Neurological Institute of Thailand which was the neurological referral center.
The first patient was seen 8 years ago and, to our knowledge, he was the first Thai patient diagnosed with IBM. The others were 2 male and 3 female patients, therefore our male to female ratio was 1:1. Age at onset of male patients was slightly younger than those of female, which is 50, 56 and 67 years old VS 58, 67 and 70 years old respectively. The most common presentation was proximal leg weakness (5/6 patients) while one female patient presented with dysphagia. All patients had typical clinical presentation of IBM such as asymmetrical forearm atrophy, long finger flexors weakness and quadricep muscles weakness and atrophy. Their CK levels were mildly elevated around 400-900 U/L. The anti-cN1A antibody was tested in only 4 patients as it become available in Thailand just few years ago. The result showed positivity in 2 out of 4 patients. None of our IBM had associated autoimmune diseases. HLA-DRB1*0301 allele was not found in 2 patients being sent for HLA study.
In our patients, muscle biopsies revealed marked inflammatory infiltrate in one patient while rimmed vacuoles were presents is most patients. In 2 patients, special stain with p62 was done on their muscle specimen, p62 Positivity was evident in many fibers in dot-like/patchy pattern.
In terms of treatment, only one patient received treatment other than physiotherapy. Intravenous immunoglobulin (IVIg) was prescribed in a lady whose major problem was dysphagia. Unfortunately, her swallowing problem was not improved after three monthly IVIg injection.
To conclude our findings, even though the number of patients was small, we observe 1:1 male to female ratio which is different form the published data from the bigger cohort especially in Western countries. Male patients tended to be affected earlier than female. Anti-cN1A antibody presented in about half of the patients. Data collection and analysis in larger Thai IBM patient cohort would provide more understanding of the disease in our setting.

PP.024
PP.024 - Neuromyotonia in Idiopathic Inflammatory Myopathy Associated with Anti-Mi2 Positivity and Immune Myopathy with Perimysial Pathology
1Alfred Health, MELBOURNE, Australia
She had distal and proximal weakness in the upper limbs including deep finger flexors, facial and neck flexion weakness. In the lower limbs she had pyramidal weakness, brisk tendon reflexes and positive left Babinski. There were no nailbed/skin changes.
EMG revealed myopathic motor unit potentials with fibrillation potentials in weak muscles. NMTs were seen in the tongue/tibialis anterior.(4) Laboratory investigations demonstrated CK level of 3579U/L, anti-nuclear antibody(ANA) of 1:2560 with positive Mi-2a/Mi- 2b antibodies. Video fluoroscopy revealed oropharyngeal dysphagia.
MRI thigh showed bilateral increased STIR signal in the anteromedial compartment of the thigh and subcutaneous oedema. MRI brain/spine was unremarkable. FDG-PET was negative for malignancy.
Muscle biopsy (left vastus lateralis) demonstrated changes consistent with immune mediated perimysial myopathy(IMPP). A Pathwest(5) neuromuscular genetic panel was negative. C9orf72/ DM1 testing was negative and whole exome sequencing did not reveal any pathogenic mutations.
She was treated with intravenous methylprednisolone and immunoglobulin, with monthly maintenance doses with poor response. Plasma exchange was then undertaken with some clinical and biochemical improvement. She commenced rituximab and has continued stable weakness.
Anti-Mi2 is strongly associated with dermatomyositis characterized by muscle and cutaneous involvement, immunomodulatory responsiveness and occasionally interstitial lung disease and malignancy.(6) IMPP is usually a multisystemic disorder, sometimes associated with antibodies including Anti-Mi-2 but the pathogenicity of these antibodies remains unclear.(7) Our patient did not have typical extramuscular manifestations except for NMT and upper motor neuron features. There have been three reported cases of NMT with dermatomyositis, two of which were Anti-Mi-2 seropositive.(8-10)
At this stage, our patient does not meet diagnostic criteria for motor neuron disease or have any other features of PNH. We hypothesize an autoimmune cause for her neuromyotonia.
1. Clinical Neurophysiology. Rubin DI, editor:Oxford University Press; 2021 01-Oct-2022.
2. Maddison P. Neuromyotonia. Clin Neurophysiol. 2006;117(10):2118-27.
3. Spagni G et al. Clinical, neurophysiological and serological clues for the diagnosis of neuromyotonia and distinction from cramp-fasciculation syndrome. Neuromuscul Disord. 2023;33(8):636-42.
4. Rodrigues E. Neuromyotonia[Video]. YouTube.https://youtu.be/wanPXyw4IU8?si=Wa2Y_mSC39SFR0QU2023.
5. Department of Health WA. Clinician Resources [Available from: https://pathwest.health.wa.gov.au/∼/media/PathWest/Documents/Our-Services/Clinical-Services/Diagnostic-Genomics/Neuromuscular-gene-panel-Neuro-v6-gene-list-boxed-180821.pdf.
6. Liang L et al. Anti-Mi-2 antibodies characterize a distinct clinical subset of dermatomyositis with favourable prognosis. Eur J Dermatol. 2020.
7. Bucelli RC, Pestronk A. Immune myopathies with perimysial pathology: Clinical and laboratory features. Neurol Neuroimmunol Neuroinflamm. 2018;5(2):e434.
8. Lertnawapan R, Kulkantrakorn K. Isaacs' syndrome in a patient with dermatomyositis: case report and review of the literature. Int J Rheum Dis. 2017;20(8):1039-45.
9. Oh SJ et al. Myokymia, neuromyotonia, dermatomyositis, and voltage-gated K+ channel antibodies. Muscle Nerve. 2003;27(6):757-60.
10. Bailey G, Trivedi JR. An Unusual Presentation of Dermatomyositis With Muscle Hypertrophy. Cureus. 2023;15(6):e41005.
PP.025
PP.025 - Adult Onset NXP2-Dermatomyositis Related Gastrointestinal Vasculopathy: A Case Series
1Neurology Department, St John Of God Hospital Midland, Perth, Australia, 2Neurology Department, Royal Perth Hospital, Perth, Australia, 3Neuropathology, Royal Perth Hospital, Perth, Australia, 4Perron Institute for Neurological and Translational Science, Perth, Australia
Case 1 had preceding ischemic colitis, with abdominal pain and bloody diarrhoea three months before her myositis presentation. There were endoscopic and histological features of ischaemic colitis. Subsequent to this, she presented with rhabdomyolysis with a peak CK of 20153. She developed a second episode of ischaemic colitis, soon after commencement of steroid treatment. Following diagnosis and management of dermatomyositis, with high dose oral steroids, methotrexate, and intravenous immunoglobulin (IVIG), her symptoms of myositis resolved. She has had no further clinical episodes of ischaemic colitis, her histological changes of colitis resolved, and she has not developed malignancy.
Case 2 initially presented with subacute onset proximal muscle pain and weakness, with erythema over his cheeks and nose. He was positive for both NXP2 and anti-isoleucyl tRNA synthetase (OJ) antibodies. His gastrointestinal symptoms occurred at 4 years and 8 years following the diagnosis: both episodes occurred in the context of dermatomyositis flare due to treatment cessation by the patient. Endoscopy demonstrated extensive stomach and colon ulceration, and histopathology demonstrated ischaemic changes due to small vessel vasculitis or vasculopathy. He was treated with steroids, methotrexate, and IVIG, and continues to do well on maintenance methotrexate and prednisolone alone.
PP.026
PP.026 - Magnetic Resonance Imaging in Idiopathic Inflammatory Myopathies
1Nimhans, Bangalore, Bangalore, India, 2Nimhans, Bangalore, India, 3NIMHANS, Bangalore, India
In DM, 53.12% showed moderate to severe edema in adductor group. In the leg, 46.8% showed moderate to severe edema in the peronei, and 65.6% had significant fasciitis. Fasciitis and subcutaneous edema were significant in 65.6% (21 patients). Almost all muscles demonstrated grade 1 atrophy, except for tensor fascia lata, which showed grade 1.5 atrophy, and there was no specific pattern of atrophy observed.
In IMNM, 52.1% displayed severe edema in rectus femoris & semimembranosus. Interestingly, in the leg, 78.2% of IMNM patients lacked edema in peroneii. Regarding muscle atrophy grading, minimal atrophy (grade 2) was seen in gluteal muscles and posterior thigh muscles, while no atrophy was observed in the rest of the muscles.
For OM subgroup, there was no specific pattern of involvement. In seronegative subset, vastus lateralis showed severe edema, while in the leg, deep posterior compartment and peroneal muscle group exhibited subtle to no edema.
Muscle edema and atrophy displayed symmetricity on both sides in 94.4% and 97.6% patients respectively. In this study, the correlation between the MMT-8 score and muscle edema exhibited a trend but did not reach significance, with a p-value of 0.077. The MDAAT score did not exhibit correlations with either CK values or muscle edema in thigh muscles. In leg muscles, the muscle atrophy of Tibialis anterior, Extensor digitorum, and Extensor hallucis longus demonstrated significant correlations with MDAAT, MMT-8, and CK values, with a p-value <0.05.
The involvement of peroneus muscles in leg emerged as a characteristic feature of DM, they were conspicuously spared in other subgroups. Mild muscle atrophy was observed in all groups except for IMNM, where it was more pronounced.
PP.027
PP.027 - Disease Progression of Inflammatory Myositis and Treatment Response
1Nimhans, Bangalore, India, 2NIMHANS, Bangalore, India, 3AIIMS, Mangalagiri, Vijayawada, India, 4NIMHANS, Bangalore, India, 5NIMHANS, Bangalore, India, 6NIMHANS, Bangalore, India, 7NIMHANS, Bangalore, India
Muscle pains (76%), muscle tenderness (38%), muscle contractures (28%), wasting(53%), respiratory symptoms (32%), cardiac abnormality (3.3%), skin (67%), joint involvement (42%) and malignancy (4.3%) .
Dermatomyositis(DM) and overlap myositis (OM) were 37% each, necrotising myositis (NAM)19% and anti-synthetase syndrome (ASS)7% . Positive ANA (39%), myositis specific antibody (MSA) 66% and myositis associated antibody (MAA) 58%, paraneoplastic antibody -6.5% .
Muscle biopsy done in 36% cases showed muscle necrosis(28%) perimysial infiltrates(20%), endomysial infiltrates(22%), perifasicular atrophy(11%), fibrosis(23%), rimmed vacuoles (3.3%) and neurogenic atrophy(2.2%) .
Pelvis and adductors muscle MRI showed fatty replacement (FR) in 83.6% (41/49), edema 85%(42/49) .Anterior thigh (AT) FR-81.6%, edema 85.7%, Posterior thigh(PT) FR-89.7% and edema 81.6% .Ant distal lower limb (ADLL) FR-63.2% and edema -69.3% .In PDLL 67.3% FR and edema 63.2% . ILD was observed in 13% .
FDG uptake also showed good correlation with histopathological features (severity of muscle inflammation, interstitial edema) and muscle MRI (muscle edema in T2-STIR). SUV mean, SUV max ratio were higher among non responders.
Profile of treatment -IVMP pulse (97%), oral steroids (60%), azathioprine(29%), MMF(22%), methotrexate(30%), Inj cyclophosphamide(40%), PLEX(13%), Rituximab (29%), HCQ(9.2%), IvIg (4.5%) and cyclosporine(1.1%) . Mean duration of IVMP pulses -6 months, oral steroids 8 months, 96% required addition of new immunomodulatory agent, mean duration of maintenance-14 months .
Outcomes showed best treatment responders(36%), satisfactory responders(39%), unsatisfactory responders(13%) and non responders (11%) . Severe muscle weakness (55%), rapidly progressive course(56%), monocyclic illness(57%), chronic continuous(27%), relapse 30.5%(on maintenance(16%), high dose steroids(3.4%), low dose steroids (5.7%) and off treatment(5.4%) . Complications in 17.5% of which infections were most common . 7.6% had expired.
Non responders had rapidly progression of weakness, higher relapses, higher MSA positivity, higher edema and fatty replacement on MR, higher SUV mean and max ratio on PET, more florid necrosis & inflammation in biopsy and they required higher doses, duration of steroids and addition of more number of maintenance agents for longer duration and had higher complications .
PP.028
PP.028 - Diagnostic Accuracy in Idiopathic Inflammatory Myopathies (ADAPT-Study): A Prospective, Over-Complete Design, Diagnostic Study
1Amsterdam University Medical Centre, Amsterdam, Netherlands, 2Division of Rheumatology and Clinical Immunology, Department of Medicine, University of Pittsburgh Medical Center, Pittsburgh, USA, 3Department of Internal Medicine and Clinical Immunology, Pitié-Salpêtrière University Hospital, Sorbonne Université, AP-HP, Paris, France, 4Department of Radiology and Nuclear Medicine, Amsterdam University Medical Centre, Amsterdam Movement Sciences, Amsterdam, The Netherlands, 5Department of (Neuro)Pathology, Amsterdam University Medical Centre, Amsterdam Neuroscience, Amsterdam, The Netherlands, 6Department of Experimental Immunology, Amsterdam University Medical Centre, Amsterdam Institute for Infection & Immunity, Amsterdam, The Netherlands, 7Sanquin, Amsterdam, The Netherlands, 8Department of Epidemiology and Data Science, Amsterdam UMC, University of Amsterdam, Amsterdam, the Netherlands, Amsterdam, The Netherlands
Therefore, we aim to determine the most optimal diagnostic strategy, with the lowest burden in patients with clinically suspected IIM.
PP.029
PP.029 - Allogeneic CD19-Targeted CAR-T Therapy for Severe Refractry Immune-Mediated Necrotizing Myopathy
1BRL Medicine Inc., Shanghai, China, 2Department of Rheumatology and Immunology, Changzheng Hospital, Shanghai, China, 3National key laboratory for immunity and inflammation, China, 4Shanghai Frontiers Science Center of Genome Editing and Cell Therapy, Shanghai Key Laboratory of Regulatory Biology and School of Life Sciences, East China Normal University, Shanghai, China, 5Department of Rheumatology, the Second Affiliated Hospital Zhejiang University 20 School of Medicine, Hangzhou, China
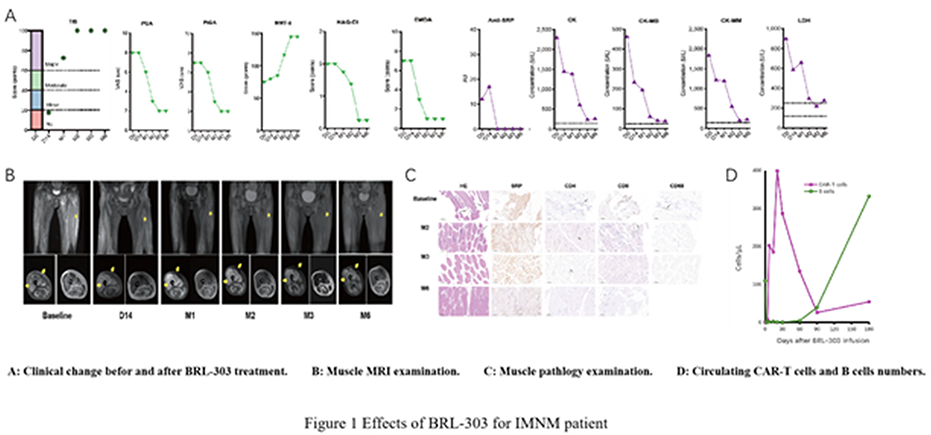
PP.030
PP.030 - TIMMDC1-Related Multisystemic Disease in Adulthood: Mismatch of Biochemical and Clinical Phenotype
1University Hospital of Bonn, Department of Neuromuscular Disorders, Center for Neurology, Bonn, Germany, 2University Hospital of Bonn, Department of Epileptology, Bonn, Germany, 3Medical Genetics Center MGZ, Munich, Germany, 4Goethe University, Functional Proteomics, Frankfurt am Main, Germany
TIMMDC1 is a protein located within the inner mitochondrial membrane, functions as a linker of two sub-membrane arms of complex I and contributes to complex I and respirasome assembly. Loss of TIMMDC1 leads to “mitochondrial complex I deficiency, nuclear type 31” (AR OMIM 618251). To our knowledge, TIMMDC1 defects have been reported in 7 children to date, each presenting with an early-onset, severe and fatal neurodegenerative phenotype. We here report on a 25-years-old male complaining on exercise intolerance at first presentation to our neuromuscular outpatient department at the age of 18 years. His past medical history was remarkable for a hypertrophic cardiomyopathy diagnosed shortly after birth with spontaneous remission in adolescence. In early childhood, he suffered from recurrent lactic acidosis, episodes of hypoglycemia, hypotonia and generalized muscular weakness resulting in transient loss of ambulation at the age of 8 years. He was further diagnosed with a submucous cleft palate, migraine, mild cognitive impairment and bilateral optic atrophy leading to poor vision. Over time, muscular weakness massively improved, and the patient was able to participate in running events. However, mild weakness reoccurred in early adulthood. At the age of 18 years, the patient presented with visual impairment, mild hip girdle and intrinsic hand muscle weakness, minor ideomotor apraxia and spasticity of the legs. Brain MRI showed optic nerve atrophy, callosal dysgenesis and hippocampal malrotation. Skeletal muscle histology including enzyme histochemistry at the ages of 3 months and 19 years was unremarkable. Respiratory chain enzyme analyses from muscle and fibroblasts showed a severe complex I deficiency with a residual activity less than 10% of controls. Subsequent immunohistochemistry detected a structural complex I defect in skeletal muscle. Molecular genetic testing in childhood (mitochondrial nuclear gene panel, mitochondrial DNA analyses) was normal. Re-testing at the age of 19 years identified two compound heterozygeous TIMMDC1 sequence variants: c.596del p.(Gly199Alafs*4), likely pathogenic; c.751G>T p.(Glu251*), variant of unknown significance. Western blot analyses in muscle tissue demonstrated a massively reduced amount of TIMMDC1, and sequencing of skeletal muscle cDNA identified the frame shift mutation in 10% and the stop codon mutation in 90% of the TIMMDC1 transcripts. Mitochondrial complexome profiling in fibroblasts showed a severe complex I assembly defect and mitochondrial complex I intermediate assembly complex accumulation. In detail, we found a significant decrease in complex I and assembled supercomplexes, including complexes I, III and IV. In addition, intermediates of complex I assembly were identified along with the assembly factors NDUFAF1, ACAD9, ECSIT, TMEM126A, TMEM126B and TMEM70 suggesting a stalled assembly at this step. We characterize for the first time an adult patient with a TIMMDC1-related disease presenting with a severe phenotype in childhood, improvement in adolescence and a secondary, however moderate deterioration in early adulthood. Nevertheless, the biochemical consequences of the TIMMDC1 deficiency are severe. The unusual clinical course is not fully understood and highlights the risk of a decades-long diagnostic gap during the transition of childhood to adult neurology.
PP.031
PP.031 - Three Heterozygous GAA Cases Mimicking Late-Onset Pompe Disease
11Center for Cardiovascular Genetics and Gene Diagnostics, Swiss Foundation for People with Rare Diseases, Schlieren-Zurich, Switzerland, 2Motol University, Neuromuscular Centre, Prague, Czech Republic, 3Seegarten Klinik AG, Kilchberg, Switzerland, 4University Hospital Brno, Department of Neurology, Brno, Czech Republic
PP.032
PP.032 - Innovations for Orphan Public Health: A Digital Based Evidence and Novel Test in Muscle Glycogenosis
1Vall D'hebron Research Institute, Barcelona, Spain, 2Universidad Europea de Madrid, Villaviciosa de Odón, Spain, 3Universidad Pablo de Olavide, Sevilla, Spain, 4Universaldoctor/UhDa Health, Barcelona, Spain
Glycogen Storage Disorders (GSD) are a group of rare diseases characterized by a defect in gene expression of specific proteins involved in glycogen metabolism, frequently resulting in the increase of the glycogen stores in different tissues. From the 14 different GSD currently described in ORPHANET, 11 present with skeletal muscle involvement (types 0, II, III, IV, V, VII, IX, X, XII, XIII and XV; globally referred to as GSD-SM). Exercise intolerance, fatigue, muscle cramps and/or contractures, and muscle weakness are common clinical manifestations in GSD-SM. Their incidence varies from 1:45.000 to <1:1000.000 individuals. Although no curative treatment is currently available, neurologists have found that maintaining an active lifestyle and engaging in progressive patterns of physical training significantly improve the quality of life of these patients. Unfortunately, a lack of efficacious support for this patient population remains a concerning barrier for improved disease management. These patients have not been follow-up systematically and consequently it has not been possible to distinguish the best management strategy. This is a reality confronted by hundreds of minority diseases. In the present work we have used technology to deliver a medical and social breakthrough, improving the equity disease management to these orphan populations. The usage of a customized and customisable technological platform by the final users facilitates the prediction and the dynamic evolution of the disease and at the same time influence their daily behavior with a real patient-centric focus. In these rare pathologies the experience of “patients experts” will be dynamically updated in the platform with easy elements of customization and transformation by these experts. This social innovative platform based already in a platform known for targeting social determinants of health, will proportionate an innovative building block mechanism of self-customisation by the patients and supporting health care workers with an initial focus on the collection of the physiological data, with the consequence capacity to research in these pathologies, and at the same time motivate the user to motivate other orphan patients experts to implement this customizable platform in their unattended diseases. In the present work we summarize the results obtained in the remote monitorization of GSD III and V patients during the daily life routines and exercise training programmes using the UhDa Health digital platform.
PP.033
PP.033 - Enzyme Replacement Therapy for Pompe Disease in Taiwan
1Department of Neurology, Chang Bing Show Chwan Memorial Hospital, Changhua, Taiwan, 2Management office for Health Data, China Medical University Hospital, Taichung, Taiwan, 3Department of Exercise and Health Promotion, College of Kinesiology and Health, Chinese Culture University, Taipei, Taiwan
PP.034
PP.034 - Myotonia Congenita, a Case Series of CLCN1 Possible Founder Mutation
1King Faisal Specialist Hospital And Research Centre, Jeddah, Saudi Arabia
MC is characterized by episodic muscle stiffness, myotonia, warm up phenomena and muscle hypertrophy. Until now more than 200 pathogenic disease causing variants were described in the literature. The exact mechanism by which mutations in the CLCN1 gene cause both dominant and recessive diseases remains unclear. Most disease causing CLCN1 mutations lead to loss-of-function phenotypes in the ClC-1 channel and thus increase membrane excitability in the muscles.
We are describing a founder mutation from Saudi Arabia due to a novel CLCN1 variant identified in five individuals from four unrelated families with classical childhood onset autosomal recessive myotonia congenita.
Involving the previously described case by Monies et al., we are describing the phenotype of a total of five individuals from four different families known to the author with clinical phenotype of MC and a confirmed genetic variant for the hypothesized founder homozygous variant (CLCN1; NM_000083.2: c.2365-1G>T) [Table1]. Despite the limited number of cases in our cohort, the reported variant lead to an autosomal recessive (Becker disease).
The characterization of the phenotype and the confirmation of the (CLCN1:c.2365-1G>T) mutations provided in this report adds to the previously reported CLCN1 disease causing variant and will aid in the diagnosis and counseling for patients with this rare mutation. We are aiming to expand our observation through a national collaboration.
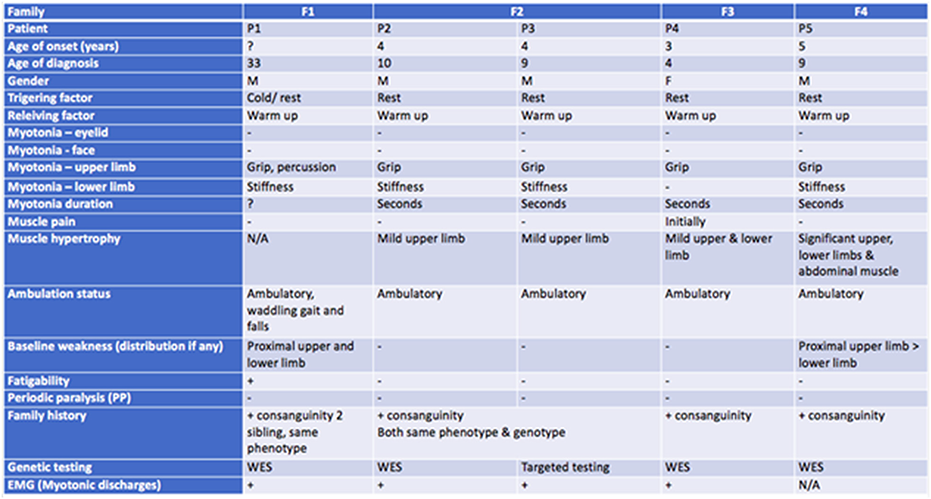
PP.035
Exploring PP.035 - Tremor Phenotypes in Myopathies: Insights from Sarcomeric Gene Mutations
1Synergy Superspecial Hospital, Rajkot, JAMNAGAR, India, 2 H J Doshi Trust Hospital, Rajkot, India
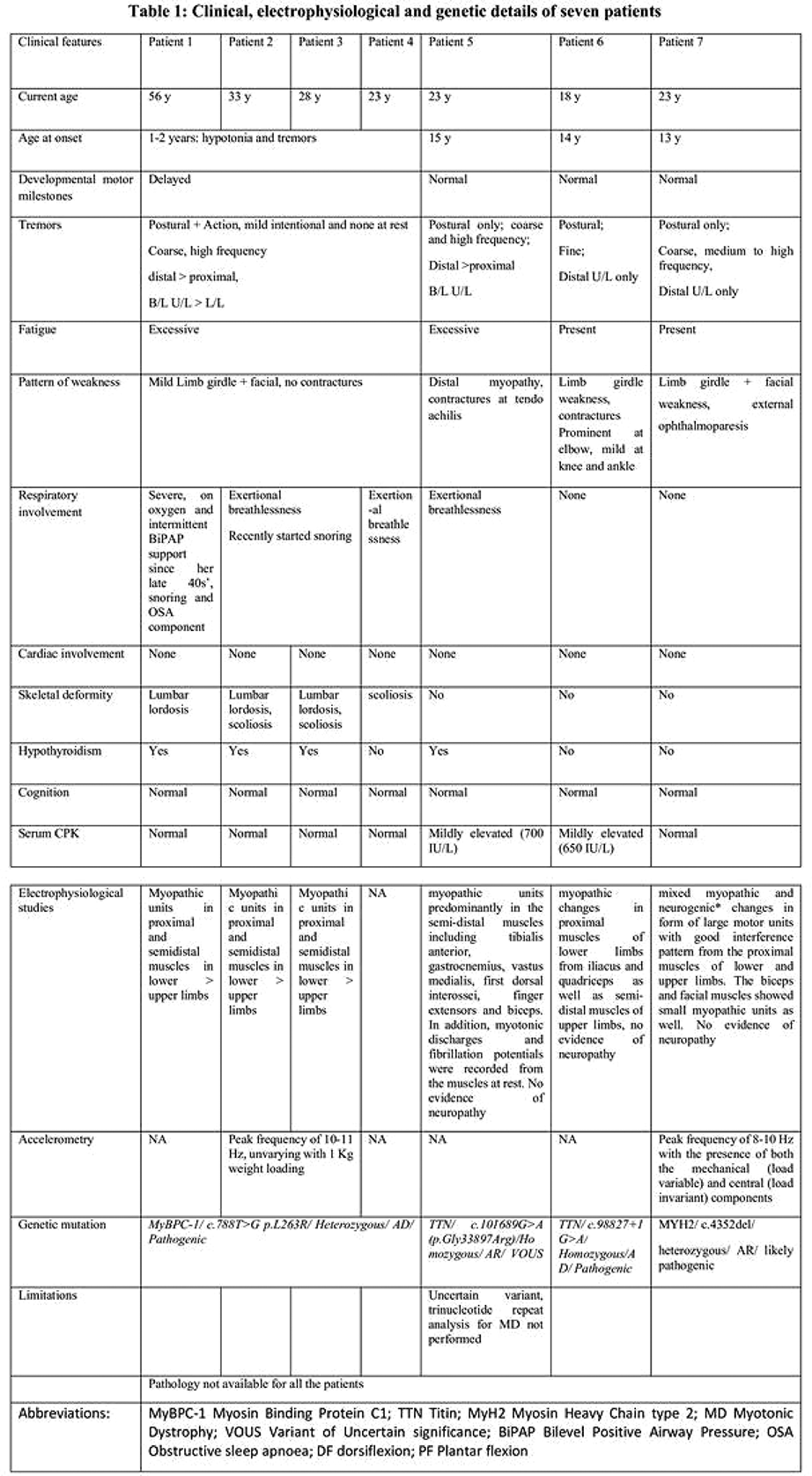
PP.036
PP.036 - Predominance of the c.2120A>G Variant in CAPN3 Among Korean Patients Diagnosed with Calpainopathy
1Department Of Neurology, Gangnam Severance Hospital, Yonsei University College Of Medicine, Seoul, Republic of Korea
PP.037
PP.037 - Dilated Cardiomyopathy in B3GALNT2-Related Congenital Muscular Dystrophy: Case Report and Single Centre Paediatric Cohort Review
1Dubowitz Neuromuscular Centre, UCL Great Ormond Street Institute of Child Health & Great Ormond Street Hospital, London, United Kingdom, 2Paediatric Cardiology Department, Great Ormond Street Hospital, London, United Kingdom, 3NIHR Great Ormond Street Hospital Biomedical Research Centre, Great Ormond Street Institute of Child Health, UCL & Great Ormond Street Hospital Trust, London, United Kingdom
Biennial screening echocardiography remained normal until age 14 years, when mild systolic impairment was noted (left ventricular fractional shortening (LVFS) 25%). Dilated cardiomyopathy progressed rapidly over the next 4 months, with a globular and dilated left ventricle (LVFS 15%). There was mild mitral regurgitation, and right ventricular function was normal. He was commenced on captopril. Over the following year, his echocardiogram was largely unchanged, and his N-terminal pro-Brain Natriuretic Peptide increased from 117 to 323 pg/ml despite the addition of carvedilol. He remained asymptomatic from a cardiac perspective throughout, with a normal ECG.
We then reviewed internal cohorts of paediatric alpha-dystroglycanopathy patients to identify occurrence of cardiac involvement. Among 49 individuals, 8 were identified with dilated cardiomyopathy. Most patients harboured variants in genes previously associated with cardiac complications, including FKRP, GMPPB, POMT2, and fukutin. Age at onset of cardiomyopathy ranged between 9-16 years, with four FKRP patients developing cardiomyopathy at ages 11, 12, and 16 (two patients), one GMPPB patient at 13, one POMT2 patient at 14, and one patient with a fukutin variant at 9 years of age. One patient with FKRP-related LGMD underwent successful cardiac transplantation at age 16 years.
PP.038
PP.038 - IPSCs Generation, Characterization and Differentiation for LGMDR4 Patients
1Board of Directors of Family Group of Beta-sarcoglycanopathy (GFB ETS), Talamona, Italy, 2Ph.D., Centro cardiologico Monzino, Milan, Italy, 3Ph.D., Centre de recherche CHU de Quebec, Université Laval, Canada, 4Chairman of Family Group of Beta-sarcoglycanopathy (GFB ETS), Talamona, Italy, 5Ph.D., Laboratory of Stem Cells of the Department of Medical-surgical Pathophysiology and Transplantation, University of Milan, Foundation IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy
LGMDR3, LGMDR4, LGMDR5 are recessive autosomal devastating diseases with no specific treatments. In 2013 the Foundation named Family Group of Beta-sarcoglycanopathy (GFB ETS; www.lgmd2e.org) was established to stimulate and support both basic and clinical research on these diseases.
In 2023, GFB promoted and funded the IPSCs GENERATION, CHARACTERIZATION AND DIFFERENTIATION FOR LGMDR4 PATIENTS Project, to create a new tool to study this disease and to evaluate the impact of new therapies on these cells.
Three cell lines of patients presenting cardiomyopathy were produced by Dr. F. Gros-Louis, Ph.D. at the Centre de Recherche CHU de Quebec, Université Laval, Canada. Two lines involve patients with mutation c.377_384dup in homozygosity on exon 3 and the third line with mutation c.341C>T and c.906insATGTTTG, in heterozygosity on exons 3 and 6 respectively.
The lines produced will now be differentiated into cardiomyocytes by Dr. G. Pompilio, Ph.D., at Centro cardiologico Monzino, Milan, Italy. Subsequently, they will be differentiated into muscle tissue by Dr. Massimiliano Cerletti, Ph.D., at the UCL University College London in the UK and by Dr. Carles Sanchez Riera, Ph.D., at La Sapienza University, Rome, Italy. Dr. Yvan Torrente, Ph.D., at University of Milan, Foundation IRCCS Ca' Granda Ospedale Maggiore Policlinico in Milan, Italy. will transform them into neuromuscular cells.
PP.039
PP.039 - Observational Study: The Quality of Life in Patients with Beta-Sarcoglycan, Alpha-Sarcoglycan, and Gamma-Sarcoglycan Gene Mutation
1Board of Directors of Family Group of Beta-sarcoglycanopathy (GFB ETS), Talamona, Italy, 2Chairman of Family Group of Beta-sarcoglycanopathy (GFB ETS), Talamona, Italy, 3Ph.D., UCL Research Department for Surgical Biotechnology, UCL Institute for Immunity and Transplantation, Centre for Surgical Innovation, Organ Repair and Transplantation (CSIORT), University College London, London, UK, 4Ph.D., Histology and embryology unit DAHFMO, La Sapienza University, Rome, Italy, 5Chairman of the Board. NGO "Ukrainian Association of Neuromuscular Diseases and Diseases of the Peripheral Nervous System", Ukraine, 6Professor of Neurology, Head of Child Neurology Department, Hedi Chaker hospital, Director of research-Sfax medical school, Sfax, Tunisia, 7Ph.D., Laboratory of Stem Cells of the Department of Medical-surgical Pathophysiology and Transplantation, University of Milan, Foundation IRCCS Ca' Granda Ospedale Maggiore Policlinico, Milan, Italy
LGMDR3, LGMDR4, LGMDR5 are recessive autosomal diseases, there are no specific treatments.
In 2013 the Foundation named Family Group of Beta-sarcoglycanopathy (GFB ETS; www.lgmd2e.org) was established to stimulate and support both basic and clinical research on these diseases.
Since 2012, families of the GFB ETS funded a gene therapy research project for LGMDR4, under the supervision of Prof. J. Mendell, at the Ohio State University (Columbus, Ohio, USA). The first clinical trial, conducted by Sarepta Therapeutics, is currently underway.
In 2018 GFB ETS promoted a natural history study on Italian patients affected by LGMDR4, published on 1st July 2021 in the journal “Frontiers in Neurology”, with the title "Clinical Determinants of Disease Progression in Patients with Beta-sarcoglycan Gene Mutations", led by Prof. Yvan Torrente, of the University of Milan and Ospedale Maggiore Policlinico in Milan, Italy.
Since 2021 GFB ETS has started an ongoing observational study on the quality of life in patients with Beta-sarcoglycan, Alpha-sarcoglycan, and Gamma-sarcoglycan gene mutation, again in collaboration with Prof. Yvan Torrente.
The study is aimed at international GFB patients. At this time 191 patients from 44 countries have joined the Project: in particular, 76 with LGMDR4, 53 with LGMDR5, 62 with LGMDR3. 606 questionnaires have already been filled out.
The ACTIVLIM, ABILHAND, EK, PEDSQL and PROMIS scales are used in the study, both in paper and electronic format. The study will run for 3 years and the scales will be given to patients every 6 months. Patient recruitment is still open at segreteria@beta-sarcoglicanopatie.it.
PP.040
PP.040 - Double Whammy! TTN and FLNC Related MFM- Presenting as ALS Mimic
1Medanta The Medicity, Gurugram, India, 2All India Institute of Medical Sciences, New Delhi, India
On examination he had atrophy of pectoralis major, supraspinatus, thenar, hypothenar, first dorsal interossei (FDI) and quadriceps bilaterally (right > left ). Fasciculations were seen in quadriceps, calf muscles, deltoid and polyminimyoclonus of the hands was noted. Power is depicted in table 1. Mild neck flexor and truncal weakness was observed. Beevors was absent. All reflexes were brisk with bilateral flexor plantar. Sensory system examination was normal. He had a waddling gait along with bilateral asymmetrical foot drop (right> left). Cranial nerve examination was normal. Rest of systemic examination was normal.
Clinically he was suspected to have clinically probable amyotrophic lateral sclerosis (ALS) with upper motor neuron (UMN) plus lower motor neuron (LMN) signs in cervical and lumbar region.
In view of young onset ALS diagnosis, we performed whole exome sequencing which revealed two heterozygous gene mutation, TTN [(c.82022G>A, p. Arg27341Gln), Exon 326] and FLNC [c.2560G>A, p.Ala854ThrHeterozygous, Exon 17]. Both were variants of unknown significance. For genotype and phenotype correlation, he underwent muscle biopsy which confirmed the diagnosis of MFM. Biopsy revealed disarray of fascicular architecture, marked variation in fibre size, regenerating and degenerating fibres with rimmed vacuoles along with normal fibre type distribution. Electron microscopy revealed destruction of Z disc and myofibrillary disorganisation. Immunohistochemistry showed desmin positive aggregates in degenerating fibres.
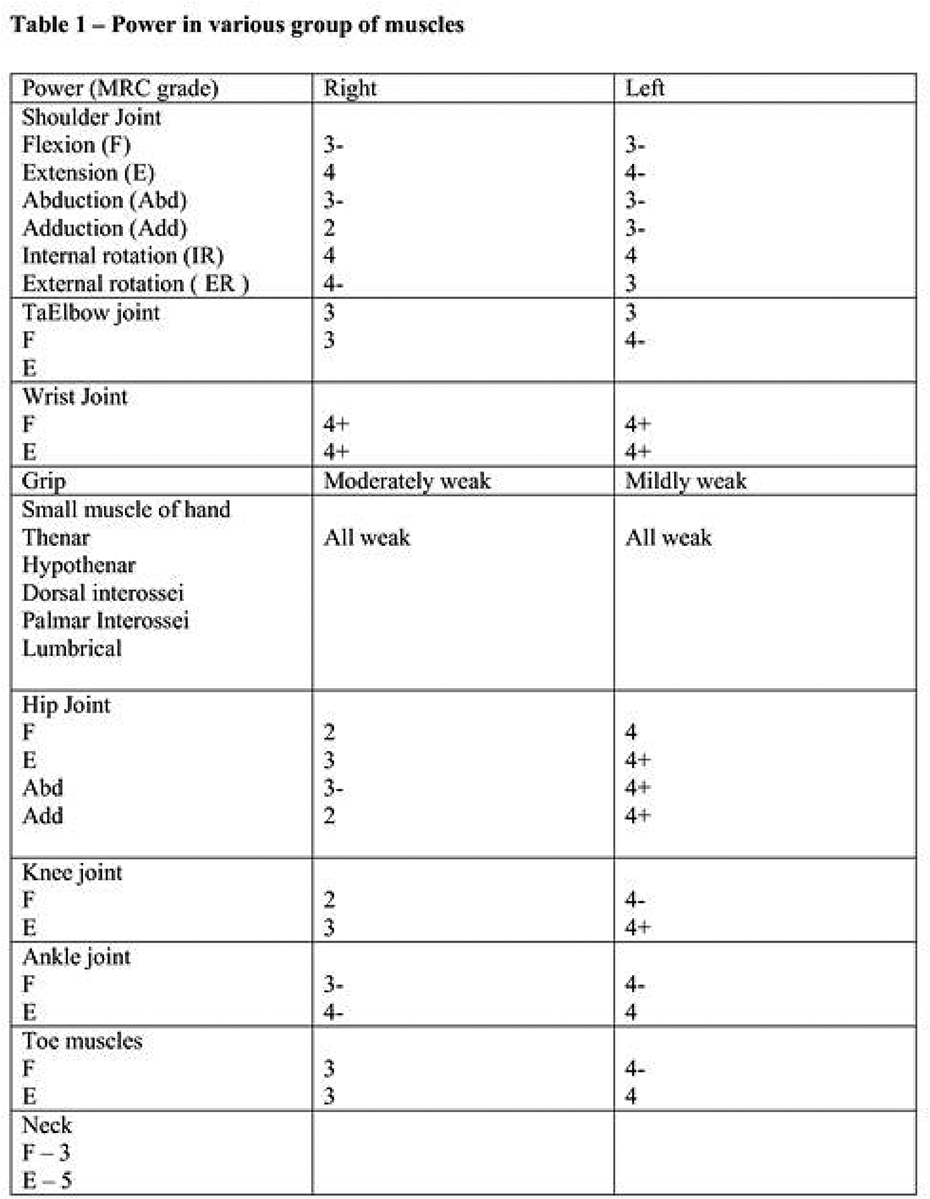
PP.041
PP.041 - Desminopathy with Novel Muscle MRI Findings
1National Institute of Mental Health and Neurosciences, Bengaluru,Karnataka, India
PP.042
PP.042 - Small Molecule Inhibition Reverses miRNA Expression and Disease Hallmarks in Myotonic Dystrophy
1ETH Zurich, Zurich, Switzerland, 2Freiderich Baur Institute, LMU Munich, Munich, Germany, 3University of Geneva, Geneva, Switzerland
Sequestration of MBNL1 by expanded CUG or CCUG repeats causes myotonic dystrophy (DM). Besides its activity as a splicing factor, MBNL1 binds to a UGC motif located within the terminal loop region of pre-miR-1 and prevents access to the RNA from LIN28, an RNA binding protein which mediates uridylation and subsequent degradation of miRNA precursors by TUT4. Several groups are investigating peptides or small molecules as possible inhibitors of the MBNL1.CUGexp interaction as potential therapeutic approaches for treatment of DM. However, targeting of MBNL1 might also interfere with non-pathological MBNL1-RNA interactions. Therefore, we are investigating an alternative high risk approach by inhibition of LIN28. Cardiac defects and myotonic dystrophies in DM patients are directly associated with dysregulation of a small number of ion channels that are targets of miRNAs, e.g. miR-1, miR-9, miR-30, miR-107 and miR-181. We have shown that precursors of these miRNAs bind to MBNL1 and LIN28 in in vitro assays, and furthermore are lowly expressed in myotubes from DM1 patients compared to those from healthy volunteers. Two prominent ion channels in cardiac defects are CACNA1C and KCNJ2, both of which have conserved predicted binding sites for some of these miRNAs in their 3'UTRs. Treatment of cells with a small molecule NTPA, increased levels of several of these miRNAs in DM1 myotubes, and reduced levels of KCNJ2 and other ion channels, possibly via altered biogenesis of miR-1 and miR-9, both of which are predicted to regulate KCNJ2. We are currently testing other small molecule as a RNA therapeutics strategy towards pathophysiology of myotonic dystrophies or cardiac defects.
PP.043
PP.043 - Pulmonary Embolism as a Rarely Presenting Manifestation of Myotonic Dystrophy Type 1
1Taoyuan General Hospital, Ministry of Health and Welfare, Taiwan, 2School of Medicine,National Yang Ming Chiao Tung University, Taiwan, 3the Neurological Institute, Taipei Veterans General Hospital, Taiwan
Myotonic dystrophy type 1 (DM1), which is the most frequent hereditary muscle disorder caused by pathogenic expansion of CTG repeats in 3’-UTR of DMPK gene, has a wide spectrum of clinical manifestations. Muscle weakness involving cranial, trunk and distal limbs, myotonia and early cataract are core features of DM1. We report a female patient obtaining the diagnosis of DM-1 resulted from a rarely presenting manifestation of pulmonary embolism.
However, a neurological consultation was referred for persistent weakness in her trunk and limbs. She only ambulated with wheelchair and had difficulty of standing with hyperextension of her knees. She experienced distal limb weakness which disturbed writing and using chopsticks. The patient reported that she had bilateral cataract surgery at her age 30. Her family history was relevant for her father and sister who had early cataract surgery and distal limb weakness. The neurological examination was significant for temporal muscle atrophy, bilateral facial weakness, and symmetric proximal and distal limb weakness. Muscle strength in MRC score disclosed 4/5 for neck flexor, 4/5 for bilateral arm abduction, 3/5 for wrist extension, 3/5 for finger flexion, 2/5 for bilateral hip flexion, 4/5 for knee extension, and 2/5 for ankle extension. Generalized hyporeflexia, grip myotonia and percussion myotonia were observed at distal hands. The neurophysiology study revealed sensory-motor polyneuropathy, axonal type. The needle electromyography disclosed spontaneous activity with myotonic discharges and typical myopathic change.
Repeat primed-PCR analysis of DMPK gene showed a heterozygous mutation with pathogenic CTG expansion (>300 bp), which confirmed the genetic diagnosis of myotonic dystrophy type 1.
Investigation of multi-system involvement in patients with DM-1 expand spectrum of clinical manifestations and provide a more comprehensive management for clinical care.
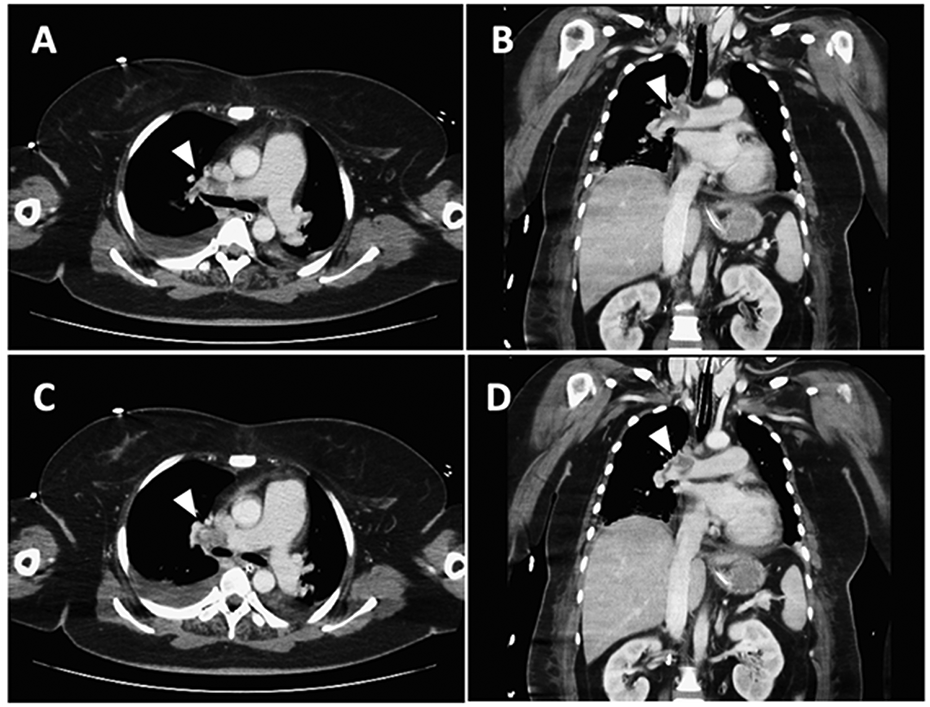
PP.044
PP.044 - Spectrum of Skeletal Muscle Channelopathies in Czechia and Novel Mutation in CLCN1 Gene
1University Hospital Brno, Brno, Czech Republic
Skeletal muscle channelopathies are a group of rare inherited diseases caused by mutations in muscle ion channels that result in increased or decreased muscle membrane excitability leading to a spectrum of related clinical disorders, most often the nondystrophic myotonias and the periodic paralyses.
Until March 2024 our records indicate that 125 patients with skeletal muscle channelopathies were observed by the Czech national registry (REaDY, Registry of Muscular Dystrophies). 84 patients had mutations in the chloride channel (CLCN1) gene (50 patients with Myotonia congenita Becker and 34 patients with Myotonia congenita Thomsen). Most of CLCN1 mutations were recessive and occur as compound heterozygous mutations. The most frequently found CLCN1 mutation was c.2680C>T, (p.Arg894*), it was found 39 times in both dominant and recessive forms, and the most frequent recessive mutation c.1437_1450del, (p.Pro480Hisfs*24) was found 15 times. Fourty one patients had sodium channel (SCN4A) gene mutations (38 patients with congenital paramyotonia and 3 patients with periodic paralyses). The most frequently found SCN4A mutations were c.3938C>T (p.Thr1313Met) and c.3917G>T (p.Gly1306Val), both found 9 times. The prevalence of congenital myotonias and congenital paramyotonias in Czechia is in the line with estimated worldwide prevalence and with the fact that CLCN1 mutations are more common in European populations than SCN4A mutations.
Although genetic testing is the gold standard by making a definitive diagnosis, a mutation might not be identified in many patients with a well-supported clinical diagnosis. We present a case of a patient with typical clinical symptoms of myotonia congenita from childhood, who at 82 years old underwent neurological investigation because of finding myotonic discharges on electromyography. We found the novel mutation c.958_964delinsTGTG (p.Ala320_Trp322delinsCysGly) in CLCN1 gene in heterozygous form in this patient. This new undescribed variant replaces three amino acids with two others. In correlation with the patient's phenotype and EMG findings, it is very likely a causal variant. His both sons with no clinical presentations of myotonias, but clear myotonic discharges on electromyography, have the same mutation in CLCN1 gene. It confirms that most of the channelopathies can show clinical variability for a given mutation even within a one family through generations.
PP.045
PP.045 - Late Presentation of Oculopharyngeal Muscular Dystrophy (OPMD) Associated With Axonal Sensorimotor Peripheral Neuropathy
1Neurology and Stroke Department, Joondalup Health Campus, Joondalup, Australia
WE was a 78 year old Caucasian male who was referred by his General Practitioner for leg weakness on a background of 30 years of progressive dysarthria and dysphagia. WE reported 5 years of progressive difficulty in climbing stairs, particularly with the right leg, as well as 5 years of progressive symmetric numbness and paresthesia in the feet.
Medical background included hypertension, gastroesophageal reflux, osteoporosis, ex-smoker, and osteoarthritis. Patient had prior surgery to correct bilateral ptosis "a long time ago".
Medications included Alendronate, Rabeprazole, Amitriptyline (for neuropathic pain), Paracetamol, and Oxycodone/Naloxone.

PP.046
PP.046 - Rare yet Reversible Presentation of Muscle Contracture in Addison’s Disease -A Clinical Enigma
1Neurology Department Manipal Hospitals Bengaluru, Bengaluru, India
At the time of presentation vitals were stable, general physical examination showed hyper-pigmentation of knuckles, palms and feet. Flexion contractures at bilateral knee joints noted, other neurological examination was unremarkable .
Investigation showed mild hyponatremia (128mol/L), serum 8 am cortisol was low -(9 nmol/L, range :138-635),ACTH levels was high (382pg/ml, range : 7-60).Needle EMG studies from right biceps femoris short head showed reduced activity, right biceps femoris long head and right flexor digitorum profundus showed no activity .PET scan showed FDG avid nodular lesions involving the bilateral adrenal glands likely favors infective etiology, Koch’s and no metabolically active lesion is seen elsewhere.
Treatment initially comprised Hydrocortisone, Fludrocortisone for 4 weeks . Following which hyperpigmentation, upper limb contracture improved and lower limb flexion contracture improved significantly as mentioned in Figure 1(b) and he can walk with minimal support now.
Anti tubercular rifampicin based regimen was initiated 2 weeks after improvement of cortisol levels which patient tolerated well.
The underlying mechanisms of muscle contractures in Addison's disease involve electrolyte imbalances, particularly sodium dysregulation, leading to impaired muscle function and contractility. Till date only two case reports were published on Addison disease presenting as muscle contractures, however contributing EMG findings of the same have not been reported in literature till date our case had findings demonstrated as in figure 1(c)
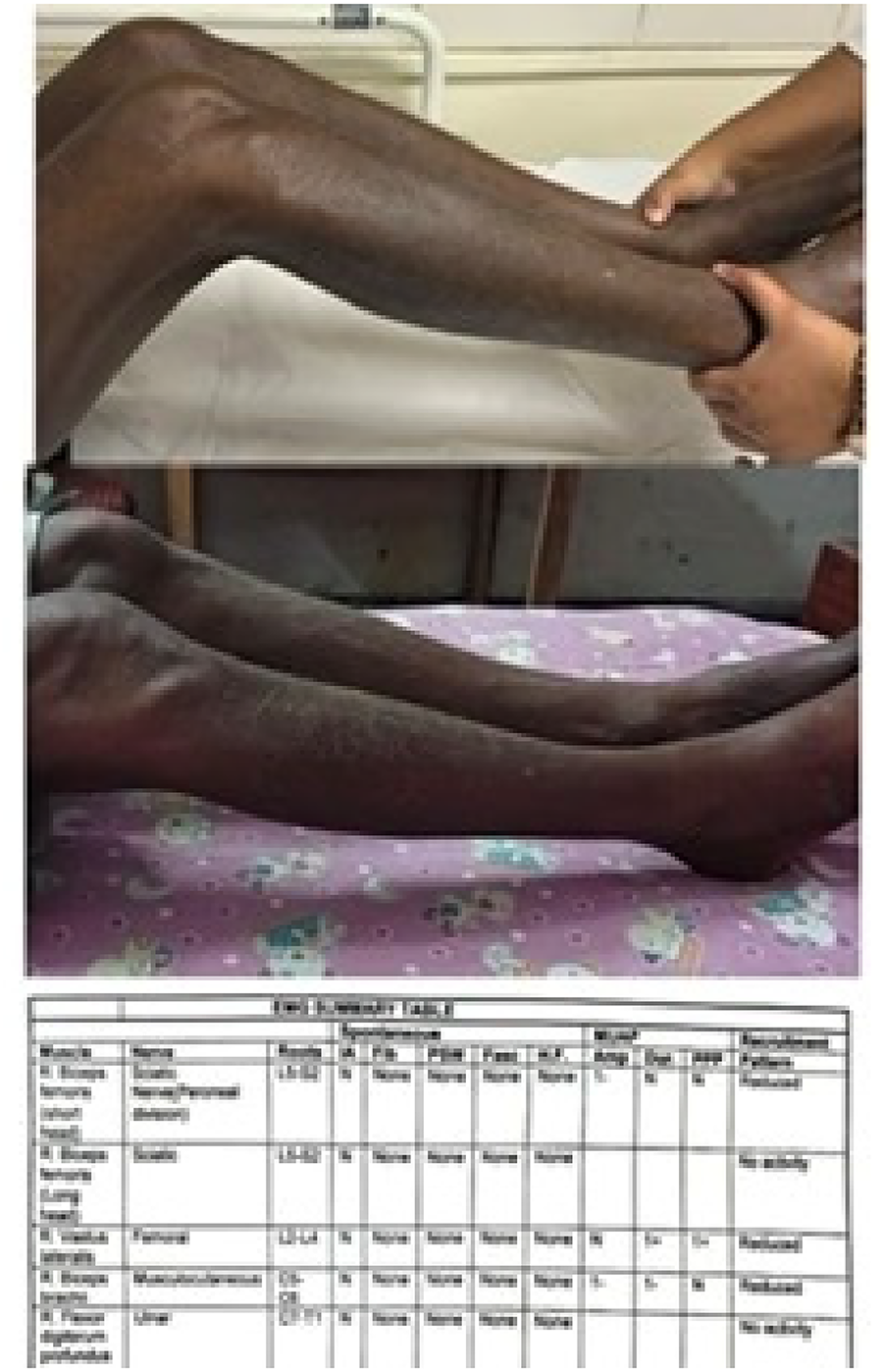
PP.047
PP.047 - Neuromuscular Junction Deficits in PURA Syndrome
1Department of Neurology, University Hospital of Basel, University of Basel, Basel, Switzerland, 2Department of Child Neurology, University Children Hospital, Lublin, Poland, 31st Paediatric Department, Developmental Centre A. Fokas, Aristotle University of Thessaloniki, Hippokration General Hospital, Thessaloniki, Greece, 4Biozentrum, University of Basel, Basel, Switzerland, 5Department of Pediatric Neurology, Centre for Neuromuscular Disorders, Centre for Translational Neuro- and Behavioral Sciences,University Duisburg-Essen, Essen, Germany, 6Johns Hopkins All Childrens Hospital, Division of Neurology, 501 6th Ave S, United States
PP.048
PP.048 - Efgartigimod in Generalized Myasthenia Ggravis with Anti-AChR Antibodies: Real-Life Experience in a Portuguese Nation-Wide Study
1Neurology Department, Hospital De Santo Antonio, Porto, Portugal, 2Unit for Multidisciplinary Research in Biomedicine, ICBAS - School of Medicine and Biomedical Sciences, University of Porto, Porto, Portugal, 3ERN EURO-NMD European Reference Network for Neuromuscular Disorders, 4Neurology Department, ULS Coimbra, Hospital Universitário de Coimbra, Coimbra, Portugal, 5Neurology Department, Centro Hospitalar Universitário do Algarve, Faro, Portugal, 6Neurology Department, ULS Tras-os-Montes e Alto Douro, Hospital de Vila Real, Vila Real, Portugal, 7Neurology Department, Hospital de São Joao, ULS São Joao, Porto, Portugal, 8Neurology Department, Centro Hospitalar Universitário Lisboa Central - Hospital de São José, Lisboa, Portugal, 9Neurology Department, Hospital Amadora Sintra, Sintra, Portugal, 10Neurology Department, Serviço de Neurologia Hospital S. Teotónio, ULS Dão-Lafões, Viseu, Portugal
In the whole group average MG ADL before treatment was 8 (4-15) and after the first treatment was 5. Twelve (80%) patients improved their MG ADL score, 2 remained unchanged and in 1 the result was not available. In those that improved the average improvement was 4 points in MG ADL score.
12 completed two cycles of treatment: 9 improved, 2 unchanged score and 1 worsened MG ADL score. The average interval between the first and second cycle was 12 weeks. Although in two patients the MG ADL was unchanged, the levels of anti-AChR antibodies and IG G were reduced. The patient that worsened stopped MMF during the first efgartigimod treatment. Nine patients completed a third treatment. Of the two that MD AGL was unchanged after the second treatment one improved from 6 to 1 and the other remained unchanged.
None of the patients needed rescue therapy with IV IG or PLEX during the follow-up period. Side effects were headache in 3, urticaria in 2 patients and nausea/diarrhoea in one.
PP.049
PP.049 - ARGX-119 in Participants With DOK7 Congenital Myasthenic Syndromes: A Phase 1b Study Design
Hanns Lochmuller1, Steven J Burden2, Javier Avendaño3, Tonke Van Bragt4,
1Children's Hospital of Eastern Ontario Research Institute; Division of Neurology, Department of Medicine, The Ottawa Hospital; and Brain and Mind Research Institute, University of Ottawa, Ottawa, Canada, 2Massachusetts General Hospital, Boston, USA, 3argenx, Ghent, Belgium, 4Curare Consulting B.V., Liempde, the Netherlands
PP.050
PP.050 - Refractory Myasthenia Gravis: Case Series
1Deparment of Neurosciences,Azienda Ospedaliera Di Padova, Padova, Italy, 2Neurophysiology,Azienda Ospedaliera Di Padova, Padova, Italy
Myasthenia gravis (MG) is an acquired autoimmune disorder of the neuromuscular junction, caused by antibodies that target the post-synaptic membrane. The predominant manifestations are muscle weakness and fatigue. Patients usually present antibodies against the acetylcholine receptor (AChR), but in a smaller proportion of cases, there are antibodies to muscle-specific tyrosine kinase (MuSK) or lipoprotein receptor-related protein 4 (LRP4). Around 10% of patients do not have these antibodies and are considered seronegative. Standard treatment of MG consists of acetylcholinesterase inhibitors, steroids immunosuppressive drugs, and/or thymectomy, but it is individualized according to the patient's characteristics. In severe weakness cases or exacerbations, IV immunoglobulin (IVIg), subcutaneous Ig (SCIg), and plasma exchange (PLEX) are used as rescue therapy. Around 10-15% of all MG patients have a disease that is refractory to standard treatment.
Refractory MG can be defined as cases that are not clinically controlled on an immunotherapy regimen, aren't able to lower immunotherapy without clinical relapse, need rescue therapy frequently, or complain of severe side effects from immunotherapy (1). Refractory cases of MG are difficult to treat and present often depression.
We report four rather severe cases: the first was refractory to most traditional MG treatments, and had some benefit from Eculizumab but still might need steroids and has residual lipo-thymic tissue, the second was a 72 years woman, with diabetes, kidney infection, AF, treated with prednisone, azathioprine, and SCIg, the third a woman with psoriatic arthritis and negative MG, the fourth an 83-year-old physician with seropositive MG had over a fortnight of permanence in the ICU for respiratory distress, AF after COVID-19 infection (Figure). He never developed pneumonia but was suspected to have bacterial and Aspergillus pulmonary/respiratory tract infection, markedly weak, although conscious throughout his intensive ICU stay. Myasthenia gravis (MG), patients have been predicted to have high rates of coronavirus disease-2019 (COVID-19) complications due to frequent involvement of respiratory muscles in MG and frequent use of immunosuppressive therapies. The risk of a severe course of COVID-19 is increased in patients suffering from Neuromuscular disorders due to the following comorbidities: muscular weakness of the chest and diaphragm, use of ventilator supports, and/or presence of tracheostomy, weak airway clearance, cardiac involvement, comorbidities such as other autoimmune disorders, steroid, and immunosuppressant treatments. Refractory myasthenia gravis refers to cases where the disease does not respond adequately to standard therapies or where these treatments are not possible, due to contraindications or significant side effects. This can pose a challenge for managing the condition effectively and may require more specialized approaches. The considerations for refractory myasthenia gravis: patients with refractory myasthenia gravis should be referred to a neurologist or a neuromuscular specialist with experience in managing complex cases of MG. In cases where conventional treatments such as pyridostigmine, corticosteroids, or immunosuppressive drugs are ineffective or not well-tolerated, alternative immunomodulatory therapies may be considered. This could include drugs like rituximab, mycophenolate mofetil, and eculizumab.
1. Mantegazza R, Antozzi C. When myasthenia gravis is deemed refractory: clinical signposts and treatment strategies. Ther Adv Neurol Disord. 2018 Jan 18;11:1756285617749134.

PP.051
PP.051 - Treatment Preferences in Myasthenia Gravis: A Discrete Choice Experiment
1University Of Toronto, Toronto, Canada
PP.052
PP.052 - Corticosteroid Use in Ravulizumab-Treated Adults With Anti-Acetylcholine Receptor Antibody-Positive Generalized Myasthenia Gravis: CHAMPION-MG Open-label Extension
Prof. Michael Nicolle1, Prof Djillali Annane2, Prof Andreas Meisel3, Prof Tuan Vu4, Prof Renato Mantegazza5, Prof Masahisa Katsuno6, Prof Vera Bril7, Dr. Rasha Aguzzi8, Dr. Glen Frick8, Prof James Howard Jr9,
1London Health Sciences Centre, London, Canada, 22Hôpital Raymond Poincaré, Poincaré, France, 33Charité Universitätsmedizin Berlin, Berlin, Germany, 44University of South Florida Morsani College of Medicine, Tampa, USA, 5Fondazione IRCCS Istituto Neurologico Carlo Besta, Carlo Besta, Italy, 66Nagoya University Graduate School of Medicine, Nagoya, Japan, 77Ellen & Martin Prosserman Centre for Neuromuscular Diseases, University Health Network, University of Toronto, Toronto, Canada, 88Alexion, AstraZeneca Rare Disease, Boston, USA, 99The University of North Carolina, Chapel Hill, USA
PP.053
PP.053 - Fixed Cycle and Q2W Dosing of Intravenous Efgartigimod for gMG: Part A of ADAPT NXT
Vera Bril1,2, Ali A. Habib3, Kristl G. Claeys4,5, Yessar Hussain6, Kelly Gwathmey7, Gregory Sahagian8, Elena Cortés-Vicente9,10, Edward Brauer11, Jeffrey Guptill11,
1Ellen & Martin Prosserman Centre for Neuromuscular Diseases, University Health Network, Toronto, Canada, 2University of Toronto, Toronto, Canada, 3Department of Neurology, University of California, Irvine, Irvine, USA, 4Department of Neurology, University Hospitals Leuven, Leuven, Belgium, 5Laboratory for Muscle Diseases and Neuropathies, KU Leuven, Leuven, Belgium, 6Austin Neuromuscular Center, Austin, USA, 7Department of Neurology, Virginia Commonwealth University, Richmond, USA, 8The Neurology Center of Southern California, Carlsbad, USA, 9Neuromuscular Diseases Unit, Department of Neurology, Hospital de la Santa Creu i Sant Pau, Barcelona, Spain, 10Biomedical Research Institute Sant Pau, Barcelona, Spain, 11argenx, Ghent, Belgium, 12Department of Neuroimmunology and Neuromuscular Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy, 13Department of Neurology and NeuroScience Clinical Research Center, Charité – Universitätsmedizin Berlin, Berlin, Germany, 14Reference Center for Neuromuscular Disorders and ALS Timone Hospital University, Marseille, France
PP.054
PP.054 - Prognosis Of Thymoma-Associated Myasthenia Gravis and Ectopic Germinal Centers
1Yonsei University College of Medicine, Seoul, Republic of Korea, 2Yonsei University College of Medicine, Yongin Severance Hospital, Yonsei University Health System, Republic of Korea
Knowledge concerning the risk factors for poor prognosis is needed for adequate clinical care and to plan for future treatment. Additional prospective studies regarding may establish a better understanding of the pathogenesis of TAMG.
PP.055
PP.055 - DNTH103, a Potentially Safer and More Convenient Novel Therapy for Generalized Myasthenia Gravis
Prof. John Vissing1, Dr Stojan Peric2, Dr Lisa Lewis3, Dr Jeffrey Stavenhagen4,
1University of Copenhagen, Copenhagen, Denmark, 2University of Belgrade, Belgrade, Serbia, 3University of Massachusetts Chan Medical School, Worcester, USA, 4Dianthus Therapeutics, Inc., New York, USA
PP.056
PP.056 - Phase 3 Trial Investigating Intravenous Efgartigimod in Anti-Acetylcholine Receptor Antibody Negative Generalized Myasthenia Gravis
1argenx, Ghent, Belgium, 2Department of Neurology, The University of North Carolina, Chapel Hill, United States
PP.057
PP.057 - Switching to Subcutaneous Zilucoplan from IV C5 Inhibitors in Myasthenia Gravis: A Phase 3b Study
1Department of Neurology, University Of North Carolina at Chapel Hill, Chapel Hill, USA, 2Atrium Health, Wake Forest University, Charlotte, USA, 3HSHS St. Elizabeth’s Hospital, O’Fallon, USA, 4Department of Neurology, University of California, San Francisco, San Francisco, USA, 5Department of Neurology, UT Southwestern Medical Center, Dallas, USA, 6The Regional MS Center and Center for Neurological Disorders, Milwaukee, USA, 7Honor Health Neurology, Bob Bové Neuroscience Institute, Scottsdale, USA, 8Department of Neurology, Eisenhower Health, Rancho Mirage, USA, 9Department of Neurology and Pediatrics, University of California Los Angeles, Los Angeles, USA, 10Department of Neurology, University of Washington Medical Center, Seattle, USA, 11UCB Pharma, Monheim, Germany, 12UCB Pharma, Braine-l’Alleud, Belgium, 13UCB Pharma, Tampa, USA, 14UCB Pharma, Slough, UK, 15Department of Neurology, The Ohio State University Wexner Medical Center, Colombus, OH, USA, Columbus, USA
PP.058
PP.058 - Long-term Efficacy and Safety of Ravulizumab, in Anti-Acetylcholine-Receptor Antibody-Positive Generalized Myasthenia Gravis: CHAMPION-MG Open-label Extension
Prof. Tuan Vu1, Prof Renato Mantegazza2, Prof. Djillali Annane3, Prof Masahisa Katsuno4, Prof. Andreas Meisel5, Prof. Michael Nicolle6, Prof. Vera Bril7, Dr. Rasha Aguzzi8, Dr. Glen Frick8, Dr. James F. Howard Jr9,
1University of South Florida Morsani College of Medicine, Tampa, USA, 2Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy, 3Hôpital Raymond Poincaré, Garches, France, 4Nagoya University Graduate School of Medicine, Nagoya, Japan, 5Charité Universitätsmedizin Berlin, Berlin, Germany, 6London Health Sciences Centre, London, Canada, 7Ellen & Martin Prosserman Centre for Neuromuscular Diseases, University Health Network, University of Toronto, Toronto, Canada, 8Alexion, AstraZeneca Rare Disease, Boston, USA, 9The University of North Carolina, Chapel Hill, USA
PP.059
PP.059 - Oral Cladribine for Generalized Myasthenia Gravis: Design of the Actively-Recruiting Phase 3 MyClad Study
1Department of Neurology & Rehabilitation Medicine, School of Medicine & Health Sciences, George Washington University, Washington DC, USA, 2Department of Neurology, University of North Carolina at Chapel Hill, Chapel Hill, USA, 3School of Public Health – Biostatistics, University of Alabama at Birmingham, Birmingham, USA, 4Departments of Neurology and Immunobiology, Yale School of Medicine, Yale University, New Haven, Connecticut, USA, 5Department of Neurology, University of Kansas Medical Center, Kansas City, Kansas, USA, 6Nuffield Department of Clinical Neurosciences, University of Oxford, Oxford, UK, 7Department of Neurology with Experimental Neurology, Charité – Universitätsmedizin Berlin, Berlin, Germany, 8Neuroimmunology and Neuromuscular Diseases, IRCCS Carlo Besta, Milan, Italy, 9Merck Healthcare KGaA, Darmstadt, Germany, 10Merck Santé S.A.S., Lyon, France, an affiliate of Merck KGaA, Darmstadt, Germany, 11Ares Trading SA, Eysins, Switzerland, an affiliate of Merck KGaA, Darmstadt, Germany, 12Merck Serono Ltd., Middlesex, UK, an affiliate of Merck KGaA, Darmstadt, Germany, 13Department of Neurology, Medical University of Lublin, Lublin, Poland
PP.060
PP.060 - Digital Tools and Artificial Intelligence Applied to the Myasthenia Gravis Core Examination
1George Washington University, Washington, United States, 2University of La Rochelle, La Rochelle, France, 3Massachusetts General Hospital, Boston, USA, 4Duke University, Durham, USA, 5Yale University, New Haven, USA, 6University of Chicago, Chicago, USA, 7University of Alabama at Birmingham, Birmingham, USA, 8Care Constitution, Houston, USA
The work was supported in part by the MGNet a member of the Rare Disease Clinical Research Network Consortium (RDCRN) NIH U54 NS115054
PP.061
PP.061 - Safety and Effectiveness of Nipocalimab in Adolescents in the Open-Label Phase 2/3 VIBRANCE-MG Study
Dr. Jonathan Strober1, Dr. Shawn Black2, Dr. Sindhu Ramchandren3, Dr. Saunder Bernes4, Dr. Akiyuki Uzawa5, Dr. Yasuhiro Kimoto Kimoto6, Dr. Keiko Ishigaki7, Dr. Tuan Vu8, Mr Dan Huang9, Dr. Yaowei Zhu2, Dr. Hong Sun3,
1UCSF Benioff Children’s Hospital San Francisco, CA, USA, 2Janssen Research & Development, LLC, Spring House, PA, USA, 3Janssen Research & Development, LLC, Titusville, NJ, USA, 4Phoenix Children's Hospital, Phoenix, AZ, USA, 5Chiba University Hospital, Chiba, Japan, 6University of Miyazaki Hospital, Miyazaki, Japan, 7Department of Pediatrics, School of Medicine, Tokyo Women's Medical University Hospital, Tokyo, Japan, 8University of South Florida, Tampa, FL, USA, 9Janssen Research & Development, Janssen-Cilag GmbH, Neuss, Germany, 10Janssen Korea, Seul, Democratic People's Republic of Korea
PP.062
PP.062 - Corticosteroid Dose Tapering with Zilucoplan in Patients with Generalised Myasthenia Gravis: 120-Week Follow-up of RAISE-XT
1Nuffield Department Of Clinical Neurosciences, University Of Oxford, Oxford, UK, 2Department of Neurology, The Ohio State University Wexner Medical Center, Columbus, USA, 3Clinical Research Unit, The Montreal Neurological Institute, Montreal, Canada, 4Academic Neuroscience Unit, Sheffield Teaching Hospitals Foundation Trust, Sheffield, UK, 5Sheffield Institute for Translational Neurosciences (SITRAN), University of Sheffield, Sheffield, UK, 6Department of Neurology, Hanamaki General Hospital, Hanamaki, Japan, 7Department of Neurology, University of South Florida Morsani College of Medicine, Tampa, USA, 8UCB Pharma, Monheim, Germany, 9UCB Pharma, Slough, UK, 10UCB Pharma, Bulle, Switzerland, 11UCB Pharma, Brussels, Belgium, 12Department of Neurology, The University of North Carolina at Chapel Hill, Chapel Hill, United States
PP.063
PP.063 - Glucocorticoid Burden and Toxicities Among Patients With Generalized Myasthenia Gravis: Findings From an Observational Cohort
Nicholas J Silvestri1,
1University of Buffalo, Buffalo, United States, 2Amgen Inc., Thousand Oaks, United States, 3Analysis Group Inc., Canada
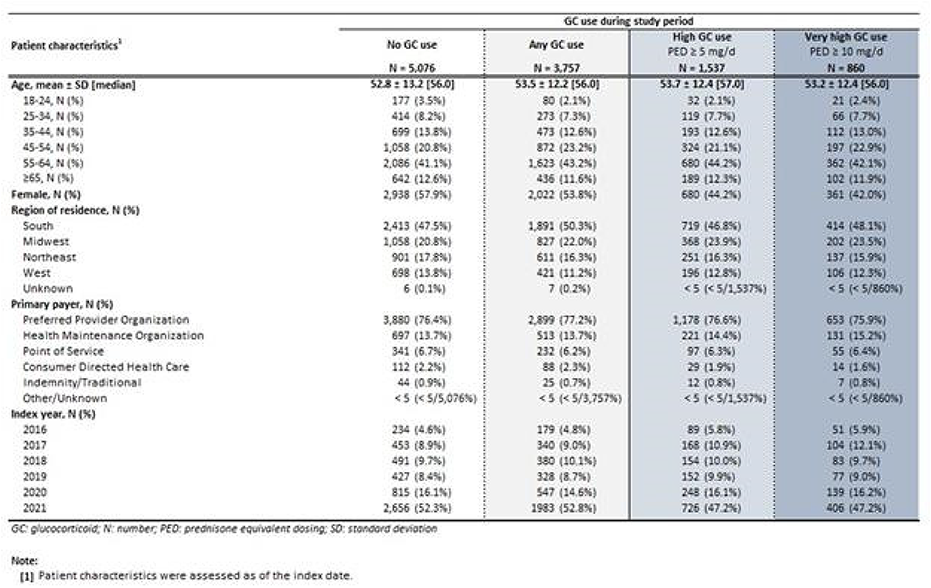
PP.064
PP.064 - Evolving Global Epidemiology of Myasthenia Gravis: Results from a Systematic Literature Review
Moon Seok Kim1,
1Amgen Inc., Thousand Oaks, United States, 2Clarivate Analytics Ltd., London, UK
Despite geographic differences, studies consistently demonstrated an increase in MG incidence and prevalence over time. MG prevalence increased 7-fold from 1934 to 2021, more than doubling in the last 20 years. The substantial increase may partially be attributed to prolonged MG survival: in-hospital MG mortality considerably decreased from 15.4% in 1992 to 12.6% in 2002 and further to 1.8% in 2019. Notably, the in-hospital mortality rate in Australia for MG was 5.3%. Respiratory failure and age were the most important predictors of death. Comorbidities among patients with MG were common and frequently included depression and other autoimmune conditions such as thyroid disease.
PP.065
PP.065 - Long-Term Safety and Efficacy of Efgartigimod in Generalized Myasthenia Gravis During the ADAPT+ Study
1Department of Neurology, School of Medicine, International University of Health and Welfare, Tokyo, Japan, 2Department of Neurology, The University of North Carolina, Chapel Hill, USA, 3University of Kansas Medical Center, Kansas City, USA, 4Ellen & Martin Prosserman Centre for Neuromuscular Diseases, University Health Network, Toronto, Canada, 5University of Toronto, Toronto, Canada, 6Penn Neuroscience Center, University of Pennsylvania, Philadelphia, USA, 7Neurology Clinic, Clinical Center of Serbia, University of Belgrade, Belgrade, Serbia, 8Department of Neurology, Ghent University Hospital, Ghent, Belgium, 9Department of Neurology and NeuroCure Clinical Research Center, Charité – Universitätsmedizin Berlin, Berlin, Germany, 10Keck School of Medicine, University of Southern California, Los Angeles, USA, 11Department of Neurology, University of South Florida, Morsani College of Medicine, Tampa, USA, 12argenx, Ghent, Belgium, 13Department of Neurology, Hanamaki General Hospital, Hanamaki, Japan, 14Department of Neuroimmunology and Neuromuscular Diseases, Fondazione IRCCS Istituto Neurologico Carlo Besta, Milan, Italy
PP.066
PP.066 - Therapeutic Relevance of Regulatory T-Cell Glycosylation in Myasthenia Gravis
Pedro Carneiro1, Manuel M Vicente1,2, Maria Isabel Leite3,
1I3S Institute for Research and Innovation in Health, Porto, Portugal, 2Hannover Medical School, Hannover, Germany, 3John Radcliffe Hospital – Oxford University, Department of Clinical Neurology, United Kingdom, 4Neurology Department, Hospital Santo Antonio, Porto, Portugal, 5Unit for Multidisciplinary Research in Biomedicine, ICBAS - School of Medicine and Biomedical Sciences, University of Porto, Portugal, 6European Reference Network for Neuromuscular Disorders
Regulatory T-cells (Tregs) have proven to be of the utmost importance in the regulation of the immune system, due to their ability to suppress other immune cells. However, Treg dysfunction has been associated with the development of autoimmunity, proving to be a major contributor to the immunopathogenesis of autoimmune diseases. Myasthenia Gravis (MG) is an autoimmune disease associated with the production of autoantibodies that target components of the neuromuscular junction, causing muscle weakness and fatigue, and in more severe cases, may lead to death. Tregs have been implicated in MG pathogenesis, displaying impaired function, which decreases their suppressive ability over B and conventional T-cells. Glycosylation, which consists in the post-translational addition of glycans to proteins, has been proven to have a key role in the regulation of Treg function. The role of glycans in T-cell, and specifically Treg biology, led us to hypothesize that the glycoreprogramming of Tregs may be used as a new clinical approach to control MG.
We started by analyzing the glycome of Tregs in MG and found that glycosylation pattern alterations feature in MG pathogenesis. In fact, blood samples from MG patients were used for a characterization of Treg glycome by flow cytometry. Results show that Tregs from MG patients exhibit a different glycosylation pattern, when compared to healthy controls, characterized by high levels of high-mannose/less complex N-glycans on the cell surface. Moreover, patients with active disease displayed higher levels of high-mannose/less complex N-glycans, when compared with patients in remission. These results are in line with single-cell RNA sequencing data, showing an increased expression of MGAT1 gene (relevant to the synthesis of high-mannose and hybrid N-glycans) in CD4+ cells of MG patients, in comparison to healthy controls.
This led us to propose that Tregs dysregulation may be related to the glycosylation pattern, which can lead to depletion of Tregs suppressive capacity. This project paves the way for new therapeutic applications in autoimmune diseases.
The authors acknowledge FCT (Portuguese Foundation for Science and Technology) for funding (Grant: 2022.01422.PTDC); Portuguese Society of Neurology (SPN) – (Grant: Bolsa Pereira Monteiro) and PARSUK/FCT/British Embassy Lisbon - UK Science and Innovation Network (Grant: PARSUK Bilateral Research Fund).
PP.067
PP.067 - Burden of Illness Amongst Generalized Myasthenia Gravis Patients: An Asia Pacific Real World Study
1Johnson & Johnson International (Singapore) Pte. Ltd., Singapore, 2Janssen Pharmaceutical K.K., Tokyo, Japan, 3Janssen Korea Ltd., Seoul, South Korea, 4Adelphi Real World, Bollington, United Kingdom
PP.068
PP.068 - Clinical Characteristics and Outcomes for Treated Generalised Myasthenia Gravis Patients in Asia-Pacific: A Retrospective Analysis
1Johnson & Johnson International (Singapore) Pte. Ltd., Singapore, 2Janssen Pharmaceutical K.K., Tokyo, Japan, 3Janssen Korea Ltd., Seoul, South Korea, 4Adelphi Real World, Bollington, United Kingdom
PP.069
PP.069 - Efficacy of Efgartigimod Myasthenia Gravis: A Real-World Case Series
1Division of Neurology, Department of Internal Medicine, Faculty of Medicine, Saga University, Saga, Japan, 2Department of Neurology, Karatsu Red Cross Hospital, Karatsu, Japan, 3Department of Neurology, Saga-Ken Medical Centre Koseikan, Saga, Japan, 4 Department of Neurology, Kouhoukai Takagi Hospital, Okawa, Japan
PP.070
PP.070 - The Burden CIDP Patients Experience in Terms of Utilities: Comparison With the General Population
Ms. Febe Brackx1,
1SHE BV, Brussels, Belgium, 2Argenx, Genth, Belgium
Data from POPUP was used to represent the general population. POPUP is a multinational digital observation study among 9000 representative members of the general population to estimate health-related quality-of-life population norms.
The utility values were calculated based on the EQ-5D-5L questionnaire using the UK value set. Utility values range from 1 (Full health) to -1 (Worst health), with 0 corresponding to death. To check for national variations, utility decrements were also calculated based on the US, Belgian, Canadian, German, Spanish and Italian value set. For each value set, the difference between the mean utility values of the CIDP patients and the subgroup of the POPUP participants of the corresponding nationality was calculated.
Furthermore, it was found that a higher level of disability corresponded to lower utility values. This was true for both non-responders and responders, with the former reporting a mean utility value of 0.616 (SD=0.207), 0.448 (SD=0.235), 0.314 (SD=0.254) and -0.063 (SD=0.250) and the latter reporting 0.690 (SD=0.195), 0.554 (SD=0.153), 0.218 (SD=0.318) and -0.028 (n=1) for INCAT score <=3, 4-5, 6-7 and >7 respectively.
National variations in utility decrements between CIDP patients and the general population were observed. Among non-responders, the decrement ranged from -0.321 (Canadian value set) to -0.482 (US value set). For responders, the decrement ranged from -0.117 (German value set) to -0.234 (US value set). Overall, decrements were smaller for responders than non-responders, regardless of the value set.
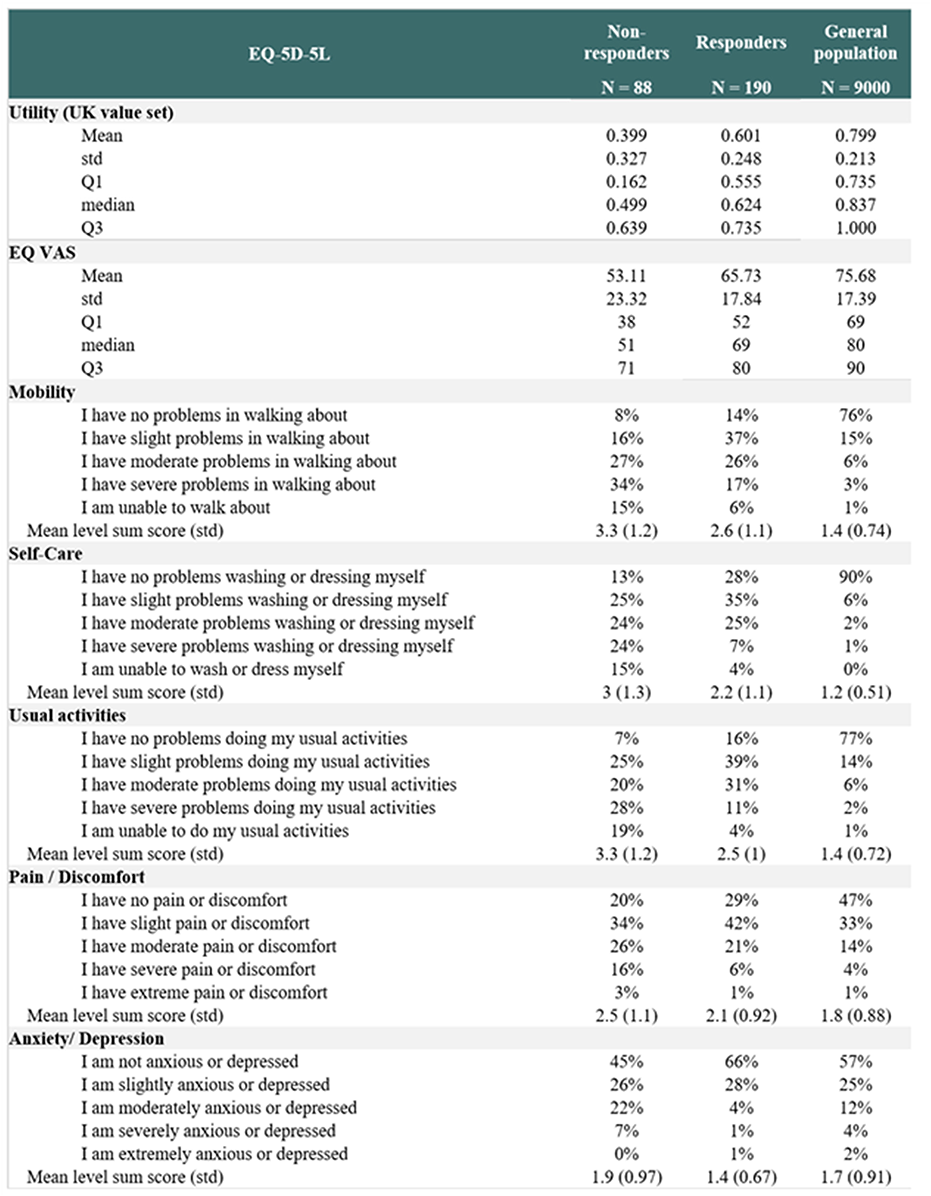
PP.071
PP.071 - Heterogeneity of Eq-5D-5L Utilities Among People Suffering From CIDP
Ms. Febe Brackx1,
1SHE BV, Brussels, Belgium, 2Argenx, Ghent, Belgium
Using ANOVA, we tested for difference in EQ-5D-5L utility values at the end of stage A between subgroups defined by symptom severity, disease duration, age, sex, disease evolution, treatment history, CIDP type and CIDP activity status. The utility values were calculated based on the EQ-5D-5L questionnaire using the UK value set, ranging from 1 (Full health) to -1 (Worst health), with 0 corresponding to death.
No statistically significant difference was found for subgroups defined by the other variables.
For age category, the mean utility values were 0.607 (SD=0.244) for 18-20 years, 0.560 (SD=0.283) for 30-39 years, 0.549 (SD=0.274) for 40-49 years, 0.565 (SD=0.272) for 50-59 years, 0.456 (SD=0.324) for 60-69 years and 0.567 (SD=0.298) for 70+ years (p=0.164).
For sex, the mean utility values were 0.540 (SD=0.273) for men and 0.536 (SD=0.301) for women (p=0.908).
For disease duration, the mean utility values were 0.559 (SD=0.300) for diagnosis <=1 year ago, 0.550 (SD = 0.291) for 1-4 years ago, 0.536 (SD=0.272) for 4-10 years ago, 0.454 (SD=0.310) for >10 years ago (p=0.346).
For disease evolution, the mean utility values were 0.506 (SD=0.281) for progressive disease and 0.572 (SD=0.299) for relapsing disease (p=0.057).
For treatment history, the mean utility values were 0.530 (SD=0.303) for treatment-experienced patients and 0.556 (SD=0.259) for treatment-naïve patients (p=0.500).
For CIDP type, the mean utility values were 0.539 (SD=0.261) for atypical disease and 0.537 (SD=0.298) for typical disease (p=0.959).
For CIDP disease activity status (CDAS) level at screening, the mean utility values were 0.686 (SD=0.271) for level 2, 0.543 (SD=0.288) for level 3, 0.555 (SD=0.363) for level 4 and 0.527 (SD=0.284) for level 5 (p=0.598).
PP.072
PP.072 - The Association Between Utility Scores and Pain, Fatigue and Mental Health in CIDP Patients
Ms. Febe Brackx1,
1SHE BV, Brussels, Belgium, 2Argenx, Ghent, Belgium
Pain was measured using the Brief Pain Inventory – Short Form (BPI-SF), with both the Pain Severity and Pain Interference score ranging from 0 (no pain/interference) to 10 (severe pain/interference). Mental health was measured using the Hospital Anxiety and Depression Scale (HADS), with both the Depression and Anxiety score ranging from 0 (no anxiety/depression) to 21 (severe anxiety/depression). Fatigue was measured using the Rasch-transformed Fatigue Severity Scale (RT-FSS), ranging from 0 (no fatigue) to 21 (severe fatigue). The utility scores were calculated based on the EQ-5D-5L questionnaire using the UK value set, ranging from 1 (full health) to -1, with 0 corresponding to death.
All available data from efgartigimod-treated and placebo-treated patients was pooled to estimate the associations between utility values (dependent variable) and each HRQoL domain separately (independent variable), whilst adjusting for the INCAT score. A linear mixed model was fitted accounting for within-patient correlation.
After controlling for INCAT disability, a significant effect of Pain Severity (slope=-0.035, p<0.0001) and Pain Interference (slope=-0.034, p<0.0001) was observed on the utility value, with a larger pain severity/interference being associated with a lower utility values. No significant interaction effects were found between the pain scores and INCAT.
Both HADS Depression and Anxiety have a negative effect on the utility value, after controlling for INCAT disability. Significant interaction effects were found between the HADS scores and the INCAT score (p=0.020 and p=0.005 for Depression and Anxiety respectively). The larger the INCAT score, the larger the negative effect Depression and Anxiety have on utility values.
Finally, also RT-FSS Fatigue has a negative effect on the utility value, after controlling for INCAT disability. A significant interaction effect was found between Fatigue and INCAT (p=0.033). The larger the INCAT score, the stronger the negative effect of the RT-FSS fatigue score on the utility score.
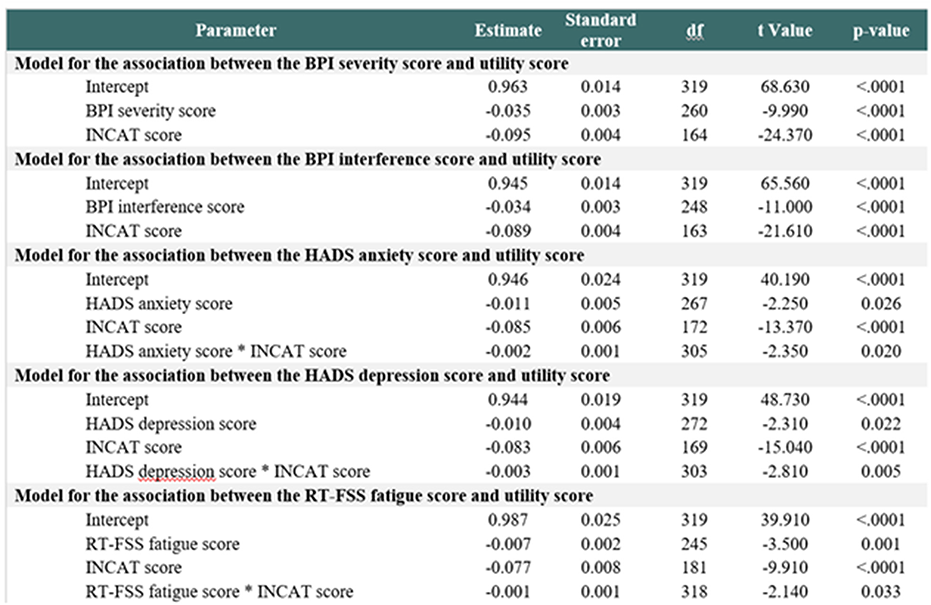
PP.073
PP.073 - Amlexanox: Readthrough Therapeutics Evaluated on Charcot–Marie–Tooth Models of hiPSCs-Derived-Neuronal-Cells Harboring Nonsense Mutations in GDAP1
Dr Nesrine Benslimane1, Camille Loret1, Dr Federica Miressi1, Dr Laurence Richard1, Angélique Nizou1, Dr Ioanna Pyromali1, Dr Pierre-Antoine Faye1,2, Pr Frédéric Favreau1,2, Dr Fabrice Lejeune4,
1Limoges University - UR20218 - NeurIT, Limoges, France, 2Limoges Hospital - Biochemistry and Molecular Gentics Dept, Limoges, France, 3Limoges Hospital - Bioinformatics Dept, Limoges, France, 4CNRS, Inserm, UMR9020-U1277—CANTHER, Lille, France, 5University of Bordeaux, CRISP'edit, TBMCore UAR CNRS 3427, US Inserm 005, Bordeaux, France, 6University of Bordeaux, Modeling transformation and resistance in leukemia, BRIC Inserm U1312, Bordeaux, France
Nonsense mutations are involved in multiple peripheral neuropathies. These mutations induce the presence of a premature termination codon (PTC) at the mRNA level. As a result, a dysfunctional or truncated protein is synthesized, or even absent linked to nonsense-mediated mRNA degradation (NMD) system activation. Readthrough molecules or NMD inhibitors could be innovative therapies in these hereditary neuropathies, particularly molecules harboring the dual activity as amlexanox. Charcot–Marie–Tooth (CMT) is the most common inherited pathology of the peripheral nervous system, affecting 1 in 2500 people worldwide. Nonsense mutations in the GDAP1 gene have been associated with a severe form of CMT, prompting us to investigate the effect of readthrough and NMD inhibitor molecules. Although not clearly defined, GDAP1 could be involved in mitochondrial functions, such as mitophagy. We focused on the homozygous c.581C>G (p.Ser194*) mutation inducing CMT2H using patient human induced pluripotent stem cell (hiPSC)-derived neuronal cells. Treatment during 20 h with 100 µM of amlexanox on this cell model stabilized GDAP1 mRNAs carrying UGA-PTC and induced a restoration of the mitochondrial morphology. These results highlight the potential of readthrough molecules associated to NMD inhibitors for the treatment of genetic alterations in CMT, opening the way for future investigations and a potential therapy.
PP.074
PP.074 - Phase 2 Efficacy and Safety of Riliprubart, a C1s-Complement Inhibitor, in Chronic Inflammatory Demyelinating Polyneuropathy
Luis Querol1,2, Richard A. Lewis3, Hans-Peter Hartung4,5,6,7,
1Neuromuscular Diseases Unit, Department of Neurology, Hospital de la Santa Creu i Sant Pau, Barcelona, Spain, 2Centro para la Investigación Biomédica en Red en Enfermedades Raras (CIBERER), Madrid, Spain, 3Cedars Sinai Medical Center, Los Angeles, United States, 4Department of Neurology, Faculty of Medicine, Heinrich-Heine-University, Düsseldorf, Germany, 5Brain and Mind Center, University of Sydney, Sydney, Australia, 6Department of Neurology, Medical University of Vienna, Vienna, Austria, 7Department of Neurology, Palacky University Olomouc, Olomouc, The Czech Republic, 8Erasmus MC, University Medical Center, Rotterdam, The Netherlands, 9Sanofi R&D, Neurology Development, Cambridge, United States, 10Sanofi, United States, 11Sanofi R&D, Biostatistics and Programming, Bridgewater, United States, 12UCL Queen Square Institute of Neurology, University College London, London, United Kingdom
Figure: Riliprubart targets activated C1s in the classical complement pathway
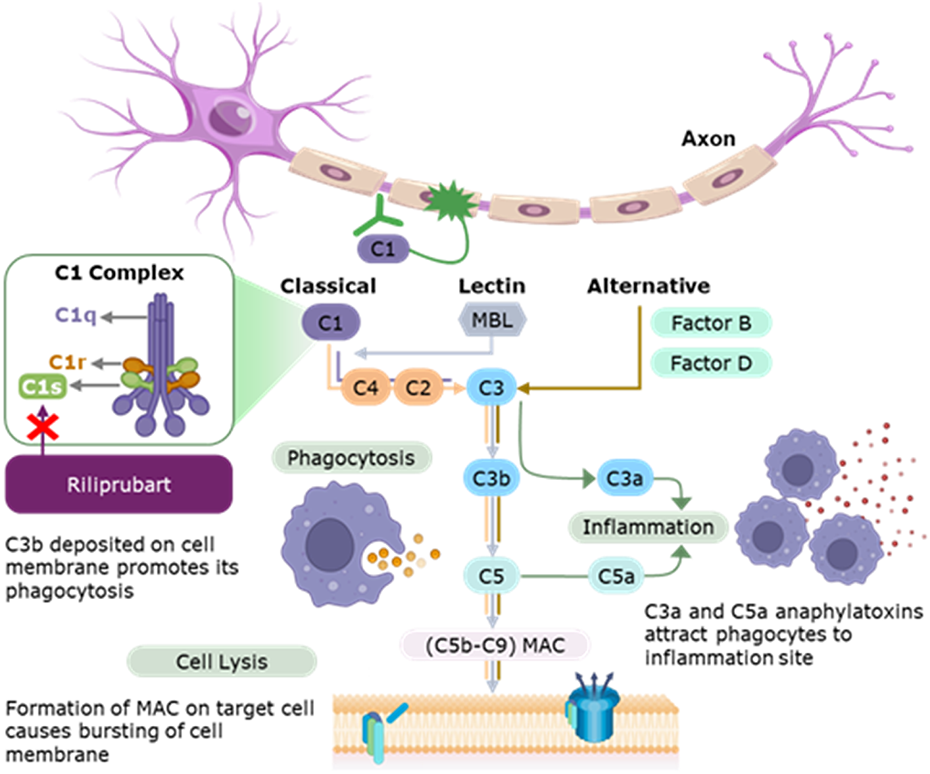
PP.075
PP.075 - Empasiprubart in Multifocal Motor Neuropathy: Initial Safety/Efficacy Data of the Phase 2 ARDA Study
Luis Querol1,2, Chafic Karam3, W. Ludo van der Pol4, Stojan Peric5, Yessar Hussain6,
1Department of Neurology, Neuromuscular Diseases Unit, Hospital de La Santa Creu I Sant Pau, Universitat Autònoma de Barcelona, Barcelona, Spain, 2Centro Para La Investigación Biomédica en Red en Enfermedades Raras (CIBERER), Madrid, Spain, 3Penn Neuroscience Center–Neurology, Hospital of the University of Pennsylvania, Philadelphia, USA, 4Department of Neurology and Neurosurgery, University Medical Center Utrecht, Utrecht, the Netherlands, 5University of Belgrade, Faculty of Medicine, Neurology Clinic, University Clinical Center of Serbia, Belgrade, Serbia, 6Austin Neuromuscular Center, Austin, USA, 7argenx, Ghent, Belgium, 8University of Minnesota, Department of Neurology, Minneapolis, USA
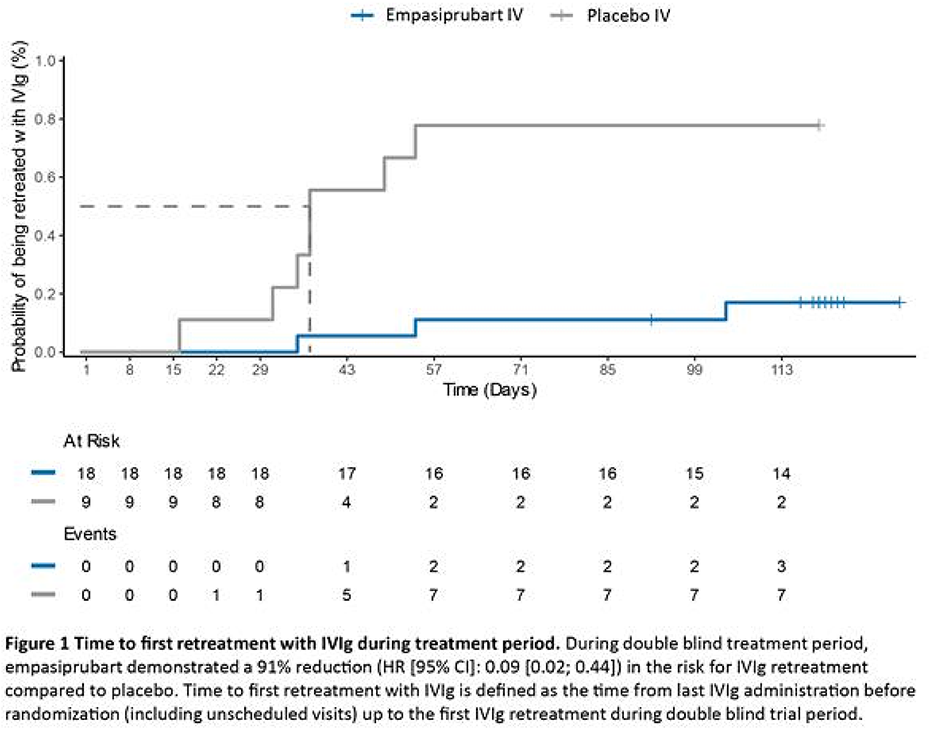
PP.077
PP.077 - Conduction Block and Multifocal Nerve Enlargement in Charcot-Marie-Tooth Disease Caused by a Fibulin-5 Mutation
1Austin Hospital, Melbourne, Australia
We report a 34-year-old Nepalese female with a pathological Fibulin-5 (FBLN5) gene mutation who was misdiagnosed as having an acquired demyelinating neuropathy based on temporal dispersion and conduction block on nerve conduction studies, and discrete nerve enlargements on neuromuscular ultrasound. The patient was initially referred to the neurodiagnostic laboratory for investigation of carpal tunnel syndrome by her general physician based on a three-year history of progressive bilateral hand weakness, she also described a few months of subjective leg weakness. There were no sensory symptoms or cranial nerve involvement. There was no family history of neurological disorders. Physical examination revealed high arched feet and hammertoes. There was distal asymmetrical upper limb weakness, predominantly affecting thumb and finger abduction. Power in the lower limbs was normal. Reflexes were globally reduced. Vibration sensation was mildly reduced in the feet.
Nerve conduction study showed evidence of a generalised demyelinating neuropathy affecting the upper and lower limbs, with median MCV of 20m/s. Temporal dispersion and conduction block was seen in the forearm segment of the right median and bilateral ulnar nerves (Fig 1). Neuromuscular ultrasound showed multifocal nerve enlargement, particularly in the upper limb nerves, with variable size, abnormal echogenicity, fascicular enlargement and areas of abnormal perineural vascularity (Fig 1). CSF analysis showed mildly elevated protein 0.58 g/L, no leukocytes, and matched oligoclonal bands. The patient was diagnosed with an acquired demyelinating neuropathy and treated with IVIg for 6 months without clinical benefit. Subsequent genetic testing revealed a heterozygous pathogenic variant in the FBLN5 gene (c.1117C>T, p(Arg373Cys).
FBLN5 is an extracellular matrix glycoprotein expressed in elastic fibre-rich tissues. It recognized as a rare cause of Charcot-Marie-Tooth disease type 1 (CMT1) with fewer than a hundred cases reported in the literature to date. It has also been reported to be associated with macular degeneration and skin hyper-elasticity. Compared to CMT1A, FBLN5 related neuropathy is commonly associated with sensory symptoms and an upper limb predominance. Electrophysiology studies of reported cases show uniform motor conduction velocity (MCV) slowing in the demyelinating or intermediate range. To our knowledge previous evidence of segmental demyelination has not been described in this condition on nerve conduction studies or neuromuscular ultrasound.
Segmental demyelination has historically been described as a feature of acquired, demyelinating neuropathies, not in inherited neuropathies, which show more uniform demyelination on electrophysiology. The changing landscape of neuromuscular diagnosis in the era of genetic testing has meant that genotype-phenotype correlations are becoming more complex. Temporal dispersion and conduction block have been increasingly described in inherited neuropathies, including SH3TC2, FIG4, MPZ, GDAP1, GJB1, PMP22 and, more recently,
SORD and Connexin-32. This is the first case to demonstrate these features in a FLBN5 neuropathy. The case also supports the growing body of evidence supporting the importance of genetic testing in atypical cases of CIDP despite typical laboratory features of acquired neuropathy.
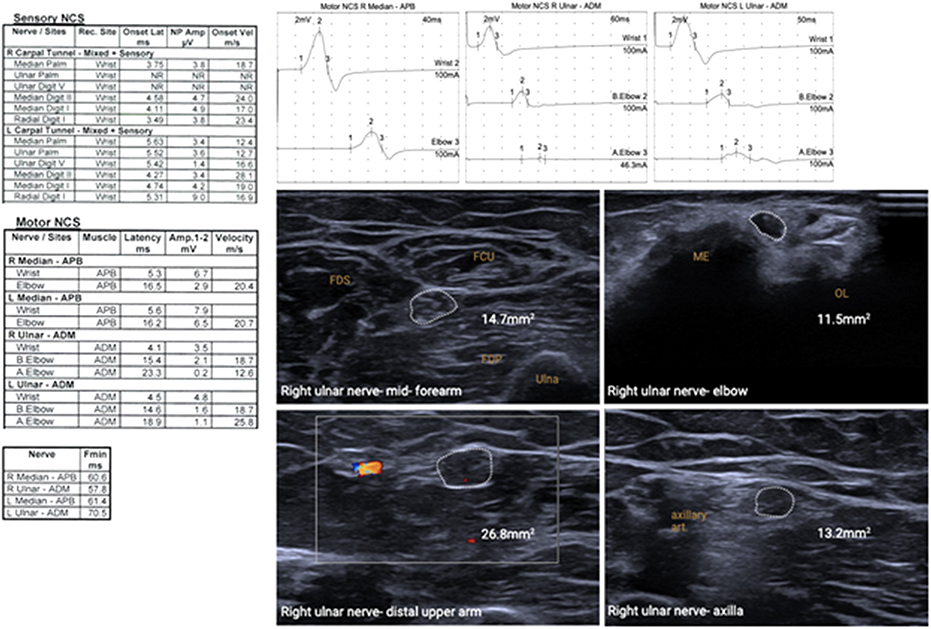
PP.078
PP.078 - Plasmid DNA Encoding HGF Showed Potential in a Phase 1 Clinical Trial for Charcot-Marie-Tooth Disease
1Helixmith Co., Ltd., Republic of Korea, 2Samsung Medical Center, Republic of Korea
Unlike viral gene therapy, a plasmid-based gene medicine can be repeatedly administered because it doesn’t stimulate an immune response. Thanks to this, a plasmid can be readily applied to chronic and inherited diseases. We developed a plasmid that leads to the simultaneous expression of two isoforms of hepatocyte growth factor (HGF-plasmid). This plasmid has demonstrated neurotrophic and angiogenic effects in both nonclinical and clinical studies. Based on these results, HGF-plasmid has been explored in patients with Charcot-Marie-Tooth (CMT) disease type 1A, which accounts for approximately 50% of CMT cases.
In an open-label phase 1 trial, conducted at the Samsung Medical Center in Seoul, Korea by Professor Choi Byung-Ok, Professor of Neurology, Sungkyunkwan University School of Medicine, and Director, Korean Organization for Rare Diseases, twelve subjects were enrolled and received intramuscular injections in both legs on days 0, 14, 90, and 104. Each patient was followed up until day 270 to evaluate safety, tolerability, and efficacy. A total of four adverse events were reported in three subjects. Injection site pruritus and peripheral edema were the only ones assessed as possibly related to the treatment, both of which were mild, and participants recovered. Therefore, dose was maintained throughout the study. No clinically significant changes were observed in serum, urine lab results, or vital signs, and no patients developed antibodies to the HGF protein.
To assess the therapeutic effects of the plasmid, several parameters were investigated: CMT Neuropathy Score version 2 (CMTNS-v2), functional disability scale (FDS), overall neuropathy limitation scale in legs (ONLS-leg), and the 10-meter walk test. The CMTNS-v2 revealed a statistically significant improvement on day 270 compared to baseline (p < 0.01) with a mean decrease of 2.17. The FDS also showed statistically meaningful improvement between the two time points (p < 0.05) with a mean decrease of 0.58. Of the twelve participants, seven showed a one-point improvement while five showed no changes. Although there was no statistically significant change in the ONLS-leg, a one-point improvement was observed in four patients, with no change in eight subjects. The 10-meter walk test showed a slight decrease in walking time (-0.62 seconds) and a slight increase in gait speed (0.06 m/second).
Taken together, repeated intramuscular administrations of HGF-plasmid were well tolerated and showed promising therapeutic potential in patients with CMT type 1A. A larger phase 2 study is being considered for CMT patients having applicable disease scores to verify observations made from this phase 1 study.
PP.079
PP.079 - Two Cases of Familial Amyloidosis with A25T Mutation
1Kagoshima City Hospital, Kagoshima City, Japan, 2Kagoshima University Medical and Dental Hospital, Kagoshima City, Japan
PP.080
PP.080 - A Deep Iintronic Variant in MME Causes Autosomal Recessive Charcot-Marie-Tooth Disease in Two Australian Families
Bianca Grosz1,2,
1Northcott Neuroscience Laboratory, ANZAC Research Institute, Sydney, Australia, 2The University of Sydney, Camperdown, Australia, 3Rare Disease and Functional Genomics Group, Harry Perkins Institute Of Medical Research, Perth, Australia, 4Centre for Medical Research, Faculty of Health and Medical Sciences, The University of Western Australia, Perth, Australia, 5Centre for Population Genomics, Garvan Institute of Medical Research, and UNSW Sydney, Sydney, Australia, 6Centre for Population Genomics, Murdoch Children’s Research Institute, Melbourne, Australia, 7Genomics and Inherited Disease Program, Garvan Institute of Medical Research, Sydney, Australia, 8Faculty of Medicine, University of New South Wales, Sydney, Australia, 9Tasmanian Clinical Genetics Service, Tasmanian Health Service, Hobart, Australia, 10School of Medicine and Menzies Institute for Medical Research, University of Tasmania, Hobart, Australia, 11Molecular Medicine Laboratory and Neurology Department, Concord Repatriation General Hospital, Concord, Australia, 12St Vincent's Healthcare Campus, Faculty of Medicine, UNSW Sydney, Level 5, De Lacy Building, St Vincent's Hospital, Darlinghurst, Australia, 13Brain and Nerve Research Centre, The University of Sydney, Concord, Australia
Loss-of-function variants in the MME gene (membrane metalloendopeptidase) are a known cause of autosomal recessive axonal CMT but are typically exonic or canonical splicing variants. Analysis of whole genome sequence data from two Australian families with autosomal recessive axonal CMT revealed a deep intronic variant in MME (NM_000902.5): c.1188+428A>G. This variant was detected in a homozygous state in four affected members of Family 1, and in a compound heterozygous state with a known pathogenic MME variant (c.467del; p.Pro156Leufs*14) in the proband of Family 2.
PP.081
PP.081 - Opthalmoparesis in Miller Fisher Syndrome: Insights from Imaging and Antibodies against GQ1b Correlation
1Division of Neurology, Department of Medicine, University of Malaya Medical Centre, 59100 Kuala Lumpur, Malaysia
Laboratory investigations, including full blood count, renal function, metabolic panel, and thyroid function tests, were within normal limits. Lumbar puncture showed no evidence of cytoalbuminologic dissociation in the cerebrospinal fluid, with normal cell count and protein levels. Imaging studies of the brain and chest yielded unremarkable results, except for magnetic resonance imaging (MRI) findings indicating T2/FLAIR hyperintensities at bilateral abducens nerves, with notable enhancement observed post-contrast in the left abducens nerve. The neuroimaging findings were notably consistent with the patient's clinical presentation, which included impairment of left eye abduction specifically during leftward gaze, contrasting with the less impaired right eye abduction during rightward gaze. Furthermore, serological testing eventually confirmed the presence of GQ1b-IgG and GT1a-IgG antibodies, supporting the diagnosis of MFS.
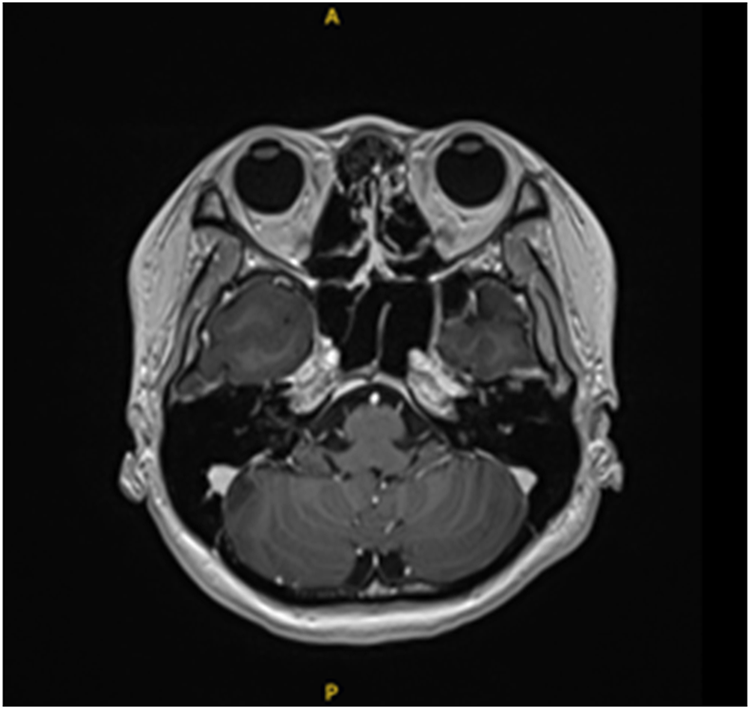
PP.082
PP.082 - Clinical Relevance of Autoantibodies Against the Node of Ranvier in Inflammatory Peripheral Neuropathy Patients
1Kangnam Sacred Heart Hospital, Republic of Korea, 2Seoul National University Hospital, Republic of Korea, 3Yonsei University Hospital, Republic of Korea
Patients with autoimmune nodopathy often exhibit poor responses to conventional treatments such as corticosteroids or IVIG. However, they show favorable responses to immunotherapies like rituximab. Unlike typical CIDP patients, those with autoimmune nodopathy may display ataxia and other distinct clinical features. Moreover, the clinical characteristics vary depending on the presence of specific antibodies such as anti-NF155, NF186, anti-CNTN1, and anti-CASPR1.
In this study, we intend to ascertain the presence of anti-ganglioside antibodies and anti-ranvier node antibodies in patients with inflammatory neuropathy and investigate the clinical characteristics based on the patterns of antibody positivity. We conducted examinations on 179 patients diagnosed with inflammatory peripheral neuropathy at Seoul National University Hospital. For patients exhibiting positivity in the ELISA, we further conducted cell-based assays. Subsequently, we analyzed the clinical presentations and nerve conduction test findings of the patients.
Patients who tested positive for NF-186, CNTN1, and CASPR1 antibodies were diagnosed with CIDP based on clinical and electrophysiological evaluations. One patient positive for NF155 antibodies showed no abnormalities on nerve conduction tests but presented with oculomotor nerve palsy and had concurrent breast cancer. Another patient positive for NF155 antibodies had concomitant MDS and showed polyneuropathy on nerve conduction tests. In addition to paranodal antibody evaluations, ganglioside antibody tests were performed on samples from 154 patients. Among these, 17 patients tested positive for ganglioside antibodies. Of the positive cases, 10 were diagnosed with CIDP, while the remaining 7 exhibited diagnostic features of AIDP, AMAN, or monoclonal gammopathy. In CIDP patients positive for ganglioside antibodies, the therapeutic response did not exhibit substantial differences compared to ganglioside antibody-negative patients. Typically, these patients responded favorably to treatment with IVIG or steroids.In patients positive for paranodal antibodies, although the positivity rate was low, all five patients exhibited a diminished response to the initial CIDP treatment, IVIG. Among them, three patients responded to rituximab and were undergoing regular immunotherapy. In this single-center study, we assessed the antibody test positivity rate in patients with inflammatory peripheral neuropathy and found that those classified as nodopathy demonstrated clinical manifestations and treatment responses consistent with previous research findings. While single-center studies may have limitations in evaluating prevalence and clinical presentations, multi-center studies and additional research are considered necessary.
PP.083
PP.083 - Case of Autoimmune Nodopathy with GBS Like Presentation Post COVID-19 Vaccine
1Mediclinic Middle East, Dubai, United Arab Emirates
43 year old woman, previously healthy apart from hypertension controlled with one medication, presented to the to us with ascending paresthesia, ataxia, back pain and muscle weakness 20 days after receiving the first dose of COVID-19 AstraZeneca vaccine. The patient reported a febrile illness one week after receiving the vaccine and was treated as upper respiratory infection. A week later, she started to have paresthesia and back pain, progressed to weakness and imbalance.
Her physical examination showed moderate proximal weakness, sensory loss of pinprick up to thigh and mid arm and absent reflexes in the lower extremities.
She was admitted with diagnosis of Guillain Barre Syndrome (GBS). CSF showed slightly elevated CSF protein without pleocytosis. Nerve conduction study (NCS) showed prolonged onset latencies and reduced conduction velocities. IVIG 2g/kg was given over 5 days. Patient reported slight improvement and was discharged on day 5.
Three weeks later, the patient reported a fall due to worsening balance and worsening of the neuropathic pain. Repeat NCS showed overall worsening. MRI of the lumbosacral spine showed enhancement of the nerve roots. Diagnosis of GBS with treatment related fluctuation. The patient received a second cycle of IVIG.
Six weeks after the second cycle of IVIG, patient came with significant sensory ataxia and painful paresthesia. Repeat Lumbar puncture showed significant elevation of the CSF protein to 1876 mg/dL. A repeat NCS showed progression with absent sensory responses allover. Significant prolongation of distal latency, slowing of conduction velocities and reduction on CMAP amplitude of motor responses. The diagnosis of chronic inflammatory demyelinating polyradiculoneuropathy (CIDP) with acute GBS like presentation was made. Patient was given pulse steroids and subsequently underwent 5 sessions of plasmapheresis. The patient had remarkable improvement with PLEX in regards to her sensory symptoms and sensory ataxia. One week after completion of PLEX, she could tandem walk and stand on one foot unassisted. Rituximab was started one week after completion of PLEX, 2g given over 2 doses, two weeks apart. She continue to receive another two cycles of Rituximab every 6 months.
Patient is now in remission. NCS was repeated in October 2023, which is two years and 7 months from her initial presentation, it showed normalization of sensory and motor responses as well as F wave latencies.
Serum anti-ganglioside antibody panel, paraneoplastic panel, anti MAG and protein electrophoresis were negative.
Given the acute GBS like presentation and the predominant disabling ataxia, autoimmune nodopathie was considered. Contactin-1 (CNTN1), neurofascin-155 (NF155) antibodies were tested in the serum and were negative. Of note that the sample was sent 7 weeks after the second cycle of IVIG (before PLEX).
Despite the negative serum antibodies, the acute presentation, significant ataxia, intense nerve root enhancement, and the remarkable response to PLEX and Rituximab, we believe this patient had an autoimmune nodopathy (1, 2, 3). As per the second revision of EAN/PNS on diagnosis and treatment CIDP (4), autoimmune nodopathy is considered a separate entity and not a CIDP variant.
Note: NCS graphs and MRI image will be provided.
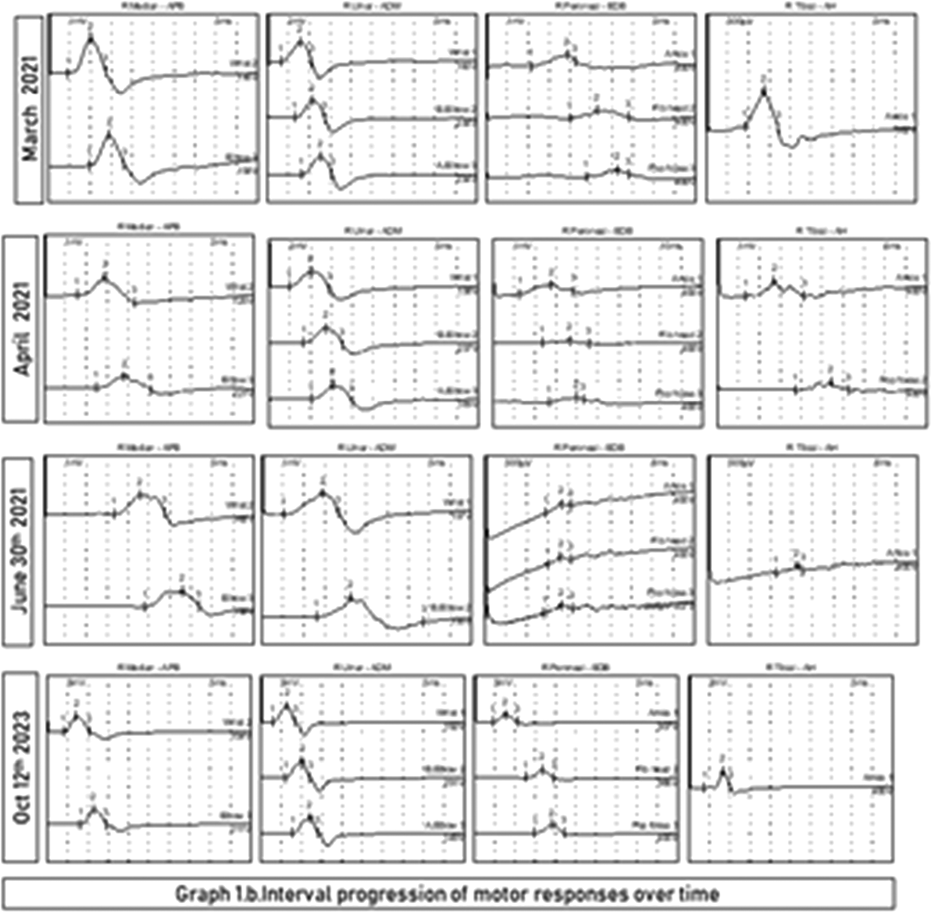
PP.084
PP.084 - Possible Shared Pathogenesis Between Idiopathic Sensory Ganglionopathy and Achalasia: A Case Report
1King Chulalongkorn Memorial hospital, Bangkok, Thailand, 2Faculty of Medicine, Chulalongkorn University, Bangkok, Thailand
We describe a rare case of idiopathic sensory ganglionopathy and achalasia in a 44-year-old Thai male without any known autoimmune comorbidities. He presented with progressive sensory disturbances and dysphagia for over 3 years. Despite multiple treatments, including intravenous immunoglobulin therapy for sensory ganglionopathy, his symptoms persisted. Comprehensive clinical and neurological examinations, alongside specific diagnostic procedures, led to the identification of the two rare conditions. Our case highlights the importance of thorough assessments in managing complex symptomatology and broadens our understanding of idiopathic sensory ganglionopathy and achalasia, particularly when they occur concurrently. We speculate on potential shared pathogenesis, considering autoimmunity involvement in both conditions. However, our patient’s dysphagia did not improve postimmunotherapy, indicating that additional factors should be considered when treating such conditions. Our case underlines the significance of clinical acumen in managing rare and perplexing conditions, paving the way for future investigations into the interconnected nature of autoimmune diseases. Further research into their interconnected nature is encouraged.
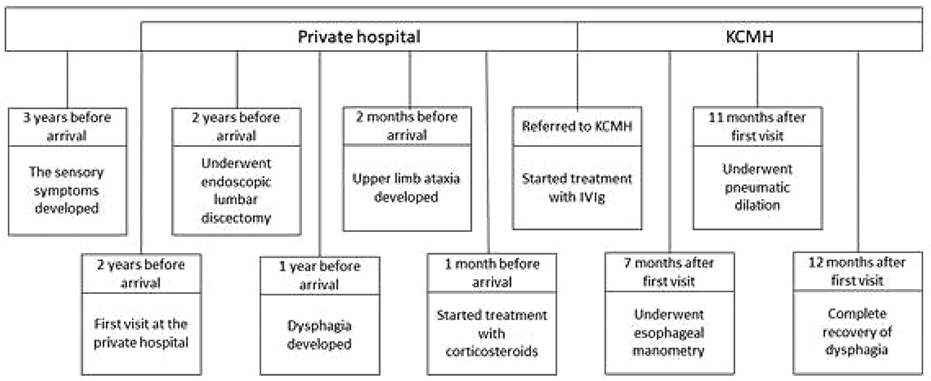
PP.085
PP.085 - Safety Profile of Subcutaneous Efgartigimod PH20 From Clinical Trials in Immunoglobulin G–Mediated Autoimmune Diseases
Pieter A. van Doorn1, Kelly Gwathmey2, Jan L. De Bleecker3, James F. Howard Jr4, Tuan Vu5, Jeffrey A. Allen6, Sofiane Agha7, Ming Jiang7, Peter Ulrichts7,
1Department of Neurology, Erasmus MC, University Medical Center, Rotterdam, the Netherlands, 2Department of Neurology, Virginia Commonwealth University, Richmond, USA, 3Department of Neurology, Ghent University, Ghent, Belgium, 4Department of Neurology, The University of North Carolina at Chapel Hill, Chapel Hill, USA, 5Department of Neurology, University of South Florida Morsani College of Medicine, Tampa, USA, 6Department of Neurology, Section of Neuromuscular Medicine, University of Minnesota, Minneapolis, USA, 7argenx, Ghent, Belgium, 8School of Medicine, Duke University, Durham, USA, 9Department of Neurology, Cedars-Sinai Medical Center, Los Angeles, USA
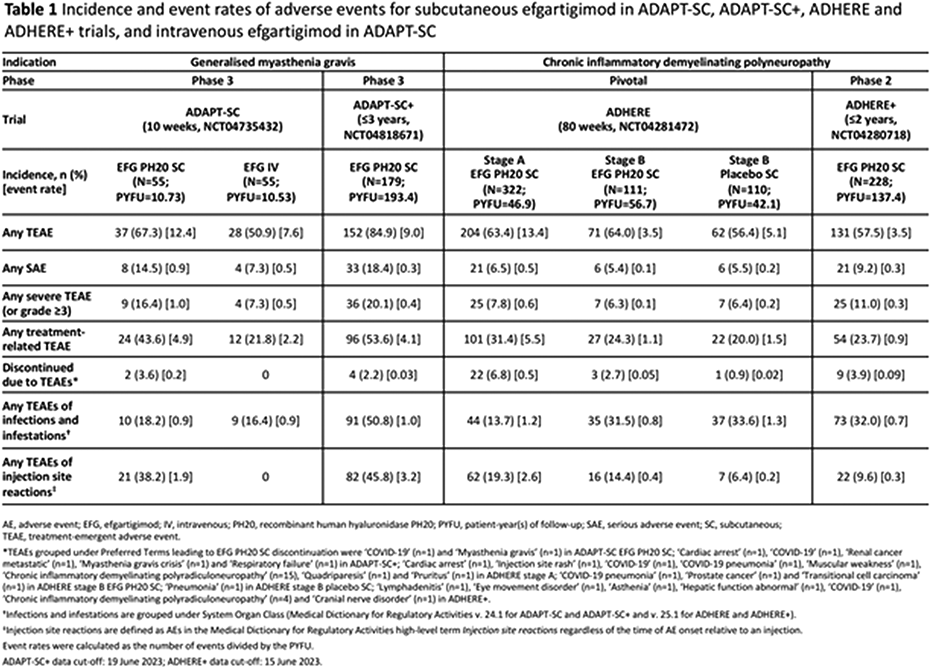
PP.086
PP.086 - Mycophenolate Mofetil for Treatment of Autoimmune Nodopathy
1Seoul National University College Of Medicine, Republic of Korea
PP.087
PP.087 - Validation of the Korean Version of Inflammatory Rasch-Built Overall Disability Scale
Mrs. Woohee Ju1,
1Seoul National University College Of Medicine, Seoul, Republic of Korea
PP.088
PP.088 - Timing of Intravenous Immunoglobulin Treatment and Outcome in Guillain-Barré Syndrome: Is Time Nerve?
1Seoul National University College Of Medicine, Seoul, Republic of Korea, 2Aston Medical School, School of Health and Life Sciences, Birmingham, UK
Introduction/Aims
Despite standard treatment, a considerable proportion of Guillain-Barré syndrome (GBS) patients experience poor recovery, highlighting an unmet therapeutic need. There is a lack of evidence that treatment timing affects recovery. This study aims to investigate the effects of IVIg timing on disability and speed of recovery in GBS.
PP.089
PP.089 - Unilateral Phrenic Nerve Paralysis in Guillain-Barre Syndrome
1Department of Neurology, Dr. Cipto Mangunkusumo National Hospital, Indonesia, 2Department of Neurology, Faculty of Medicine, Universitas Indonesia, Indonesia, 3Department of Neurology, Universitas Indonesia Hospital, Depok, Indonesia
Guillain-Barré syndrome (GBS) is an immune-mediated polyneuropathy characterized by progressive and symmetric muscle weakness with or without sensory deficits. Although GBS is a clinical diagnosis, GBS has several subtypes, the most common being acute inflammatory demyelinating polyradiculoneuropathy (AIDP).
We presented a 29-year-old male with GBS suspected AIDP subtype with a 3-week history of progressive, ascending numbness and weakness of extremities, and bilateral facial palsy. Clinical examination revealed normal vital signs, MRC sum score of 48, “stocking and gloves'' pattern hypoesthesia, hyporeflexia, reduced Single Breath Count Test (SBCT) of 17, mEGOS score of 3, mEGRIS score of 7, GBS disability scale of 4 and normal chest radiograph. The CSF analysis depicted elevated protein levels with cytoprotein dissociation. Nerve conduction study (NCS) could not be performed since at the 2nd day of admission due to clinical deterioration characterized by increased muscle weakness and shortness of breath, therefore the patient was admitted to the ICU and intubated. The therapeutic plasma exchange (TPEx) was initiated shortly after. Despite the first course of TPEx, hypotension with fluctuations in blood pressure were observed over a span of 3 days. Blood pressure stabilized a day after the administration of the second course of TPEx. Elevated right hemidiaphragm indicating right phrenic nerve paralysis was observed on chest radiograph during a one-week follow-up. The patient underwent a total of 5 courses of TPEx over a span of 2 weeks in the ICU, had significant clinical improvement and was discharged 9 days afterwards with only mild muscle weakness with an MRC sum score of 58 out of 60, hypoesthesia and an SBCT of 22.
Phrenic nerve paralysis has been observed in GBS, and marks as a clinical predictor of respiratory muscle weakness and respiratory distress. Phrenic nerve paralysis may be diagnosed with chest radiography, however transcutaneous electrical phrenic nerve stimulation remains the gold standard for establishing the diagnosis. Assessment and decision-making for ICU admission is a critical step in GBS management, and this patient was assessed with a moderate risk (20 – 70%) risk due to having GBS disability score of 4 and elevated liver function. Clinical monitoring shows reduced SBCT of <20 with clinical deterioration, and evidence of infection of chest X-ray are indicative for ICU admission and intubation. In this case, at the onset of respiratory failure, there was no sign of phrenic nerve paralysis on chest radiograph, and it was only seen one week after the onset of respiratory distress, which could imply that phrenic nerve paralysis may not necessarily trigger respiratory distress, however could be observed after the onset of respiratory distress. Clinical scores like mEGOS and mEGRIS are potent tools for determining patient prognosis and the need for assisted mechanical ventilation respectively. It is imperative to remain vigilant to clinical deterioration such as dysautonomia and respiratory insufficiency despite the administration of immunotherapy.
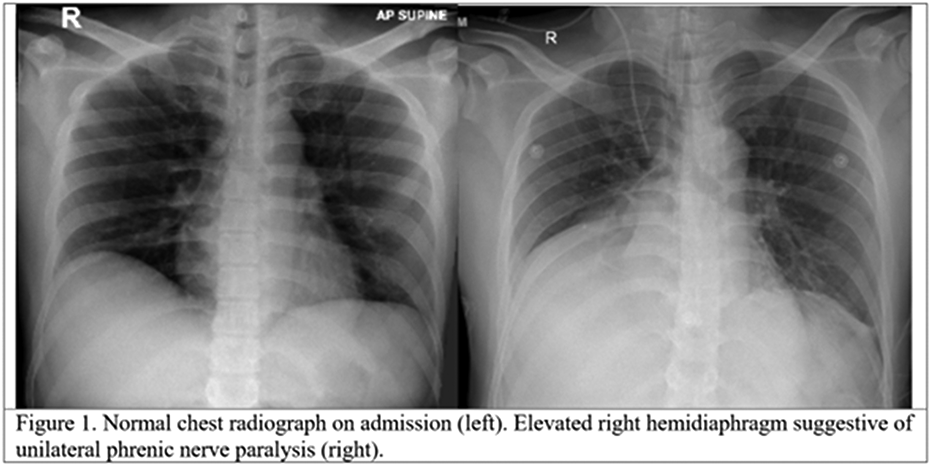
PP.090
PP.090 - Navigating the Enigmatic Realm: Morvan's Syndrome in the Presence of B-cell Non-Hodgkin’s Lymphoma
1Senior Resident, Department of Neurology, Manipal Hospitals, Bengaluru, India, 2Consultant, Department of Neurology, Manipal Hospitals, Bengaluru, India
This abstract outlines a case report detailing presentation and management of Morvan's syndrome in a male patient with concurrent B cell Non-Hodgkin’s Lymphoma. Morvan's syndrome, initially coined as "la choree fibrillaire" by Dr. Augustine Marie Morovan, is characterized by a combination of peripheral nerve hyper-excitability syndrome, autonomic dysfunction, and encephalopathy, commonly associated with antibodies targeting voltage-gated potassium channels such as CASPR 2 and LGI1.
The patient, a male in his late 60s with a history of ankylosing spondylitis, peripheral neuropathy, and recent diagnosis of B cell Non-Hodgkin’s Lymphoma, presented with tremors, muscle twitching, increased sweating, confusion, and visual hallucinations. Neurological examination revealed tremors in the upper limbs, generalized myokymia, and ataxic gait. Electrophysiological studies demonstrated bilateral motor and sensory axonopathy of the lower limbs, alongside electromyographic findings consistent with doublets and triplets. Serological testing for autoimmune encephalitis and paraneoplastic panel done by indirect immunofluorescence revealed strong CASPR 2 antibody positivity. PET CT whole body revealed no active metabolic activity. MRI brain showed no acute abnormality.
Treatment initially comprised intravenous methylprednisolone, providing transient symptoms relief. However, symptoms worsened and later developed segmental orthostatic myoclonus of lower limbs, prompting the initiation of intravenous immunoglobulin and subsequent rituximab therapy, which led to significant improvement in symptoms.
Discussion revolves around the frequent association of Morvan's syndrome with thymoma, although other malignancies including B cell Non-Hodgkin’s Lymphoma have been reported. The case prompts exploration into the potential link between CASPR 2 antibodies and aberrant lymphocyte proliferation. It also underscores the challenges in managing Morvan's syndrome associated with malignancy, emphasizing the importance of recognizing poor initial response to immunotherapy and advocating for more aggressive therapeutic interventions.
In conclusion, this case report contributes valuable insights into the clinical presentation, diagnostic approach, and treatment strategies for Morvan's syndrome in the context of underlying malignancy, emphasizing the necessity for tailored and intensive therapeutic approaches.
PP.091
PP.091 - Outcomes and Treatment Use in Patients With CIDP Switching to fSCIG 10%: Non-Interventional Study Design
Dr. Miriam L Zichlin1, Dr. Joanna Bancerek2, Dr. Colin Anderson-Smits1, Dr. Hakan Ay1, Dr. Jamie Wood1,
1Takeda Development Center Americas, Inc., Cambridge, United States, 2Baxalta Innovations GmbH, a Takeda company, Vienna, Austria
This abstract was first submitted to the American Association of Neuromuscular & Electrodiagnostic Medicine 2024 Annual Meeting. This study was funded by Takeda Development Center Americas, Inc., and medical writing support was funded by Takeda Pharmaceuticals International AG.
PP.092
PP.092 - Clinical Electrodiagnostic Mismatch in Patients with Chronic Inflammatory Demyelinting Polyradiculoneuropathy (CIDP)
1Alfred Health, Melbourne, Australia, 2Royal Melbourne Hospital, Melbourne, Australia, 3Cabrini Health, Melbourne, Australia, 4Box Hill Hospital, Melbourne, Australia, 5Ballarat Hospital, Ballarat, Australia
Other than a shorter disease duration there were no significant differences in patients who meet electrodiagnostic criteria but were not classifiable as definite or possible CIDP (EDX1CIDP0) and patients with CIDP/ Possible CIDP. Interestingly more electrodiagnostic parameters in the EDX1CIDP0 group were significantly different to controls compared to patients with CIDP or possible CIDP.
If a patient meets electrodiagnostic criteria the absence of the appropriate clinical phenotype, should not exclude the diagnosis of CIDP. Clinical phenotypes ascribed to patients with CIDP may help classify patients but may exclude patients from being considered for treatment. Electromyography is an extension of the physical examination and should be used to define the phenotype rather than being secondary to clinical examination findings.
1. Van den Bergh PYK, van Doorn PA, Hadden RDM, et al . European academy of neurology/peripheral nerve society guideline on diagnosis and treatment of chronic inflammatory demyelinating polyradiculoneuropathy: report of a joint task force-second revision. J Peripher Nerv Syst 2021;26:242–68.doi:10.1111/jns.12455 pmid:http://www.ncbi.nlm.nih.gov/pubmed/34085743
2. Doneddu P, De Lorenzo A, Manganelli F. Comparison of the diagnostic accuracy of the 2021 EAN/PNS and 2010 EFNS/PNS diagnostic criteria for chronic inflammatory demyelinating polyradiculoneuropathy. J Neurol Neurosurg Psychiatry 2022;93:1143–50.
3. Rajabally YA, Afzal S, Loo LK, et al. Application of the 2021 EAN/PNS criteria for chronic inflammatory demyelinating polyneuropathy. J Neurol Neurosurg Psychiatry 2022;93:1247–52.
PP.093
PP.093 - Clinical Outcomes, Disease Course, and QoL in Multifocal Motor Neuropathy Patients: iMMersioN, Study in Progress
Stojan Peric1, Luis Querol2,3, Sadiq Altamimi4,
1University of Belgrade, Faculty of Medicine, Neurology Clinic, University Clinical Center of Serbia, Belgrade, Serbia, 2Department of Neurology, Neuromuscular Diseases Unit, Hospital de La Santa Creu I Sant Pau, Universitat Autònoma de Barcelona, Barcelona, Spain, 3Centro Para La Investigación Biomédica en Red en Enfermedades Raras (CIBERER), Madrid, Spain, 4The Neurology Group, Pomona, USA, 5argenx, Ghent, Belgium, 6University of Minnesota, Department of Neurology, Minneapolis, USA
PP.094
PP.094 - Empasiprubart in Multifocal Motor Neuropathy (ARDA Cohort 1): Baseline Characteristics and MMN Confirmation Committee Outcome
Eduardo Nobile-Orazio1, Stojan Peric2, Shahram Attarian3, Thomas Harbo4, Luis Querol5,6, W. Ludo van der Pol7, Chafic Karam8,
1Neuromuscular and Neuroimmunology Unit, IRCCS Humanitas Research Hospital, Department of Medical Biotechnology and Translational Medicine, University of Milan, Milan, Italy, 2Faculty of Medicine, University of Belgrade, Neurology Clinic, University Clinical Center of Serbia, Belgrade, Serbia, 3Referral Centre for Neuromuscular Diseases and ALS, Hôpital La Timone, Marseille, France, 4The Department of Neurology, Aarhus University, Aarhus, Denmark, 5Department of Neurology, Neuromuscular Diseases Unit, Hospital de La Santa Creu I Sant Pau, Universitat Autònoma de Barcelona, Barcelona, Spain, 6Centro Para La Investigación Biomédica en Red en Enfermedades Raras (CIBERER), Madrid, Spain, 7Department of Neurology and Neurosurgery, University Medical Center Utrecht, Utrecht, the Netherlands, 8Penn Neuroscience Center–Neurology, Hospital of the University of Pennsylvania, Philadelphia, USA, 9argenx, Ghent, Belgium, 10Department of Neurology, University of Minnesota, Minneapolis, USA
PP.095
PP.095 - Focal Onset CIDP
1Te Whatu Ora Waikato, Hamilton, New Zealand
Diagnosis of CIDP variants is challenging and often misdiagnosis is frequent, this is due to their heterogeneity in presentation, lack of uniform diagnostic criteria and electrophysiological features for diagnosis and lack of quality evidence to guide their treatment.
PP.096
PP.096 - A subtle Presentation of Anti-Myelin-Associated-Glycoprotein (MAG) Neuropathy Leading to Newly Diagnosed IgM Gammopathy
1Department of Neurology, National Neuroscience Institute (Singapore General Hospital Campus), Singapore
A 59-year-old gentleman was referred to the emergency department for new onset lower limb weakness. He had a history of severe aortic stenosis, ischemic heart disease, hypertension, dyslipidaemia, chronic kidney disease and chronic tophaceous gout. He did not have diabetes mellitus. He had no functional impairment at baseline.
He reported a two-day history of progressively worsening weakness, associated with myalgia, polyarthralgia and pyrexia, affecting all four limbs but most prominent over the bilateral proximal lower limbs. He was unable to ambulate. He had no bulbar, visual or other cranial nerve symptoms, nor symptoms of sphincter involvement. He reported intermittent paraesthesias over the fingers of the right hand for years, but otherwise did not have any other sensory symptoms.
Physical examination revealed weakness of all four limbs predominantly affecting the distal upper limbs and the proximal lower limbs. There was no bulbar nor axial weakness, no muscle wasting and no limb fasciculations. There were no upper motor neuron signs. He did have generalised hyporeflexia as well as reduced pinprick sensation in a stocking pattern affecting the bilateral lower limbs. Proprioception sense was preserved.
Laboratory investigations, imaging, and joint aspiration were diagnostic for a polyarticular gout flare, with simultaneous left wrist cellulitis complicated by abscess formation and tenosynovitis. Myositis-associated and -specific antibodies in the form of anti-Scl-75, anti-OJ and anti-Mi-2b returned positive, but these did not correlate with clinical evidence of myositis nor systemic connective tissue disease. Additionally, the aforementioned findings could not explain the hyporeflexia and sensory findings elicited on objective examination.
A nerve conduction study (NCS) was performed to evaluate for suspected concomitant neuropathy. While technically challenging in view of ongoing soft tissue swelling, the NCS revealed a mixed sensorimotor peripheral neuropathy with mild asymmetry in the lower limbs, with possible conduction block over a non-compressible site affecting the right median nerve. Electromyography was deferred in view of ongoing soft tissue infection and edema. Further investigation revealed positive serum anti-myelin-associated-glycoprotein (MAG) IgM antibodies, as well as an anti-kappa IgM monoclonal band on serum protein electrophoresis with raised serum light chains and mild anaemia, leading to a diagnosis of likely anti-MAG neuropathy in association with a monoclonal gammopathy of undetermined significance (MGUS).
The patient’s weakness significantly improved with management of his gout flare and soft tissue infection, without progression of his sensory deficits, hence treatment of his neuropathy was not required. He was discharged with plans for close outpatient neurological monitoring, including further interval electrophysiological studies, as well as haematological surveillance.
This case illustrates a subtle presentation of anti-MAG neuropathy, with incidental distal sensory-predominant polyneuropathy newly elicited in a patient with multiple clinical “red herrings”. The patient’s presentation is consistent with the insidious onset that is typical for anti-MAG neuropathy. Close neurological monitoring is required given the known natural history of the disease leading to accumulation of disability over time. Additionally, given the known associations with IgM monoclonal gammopathy, close haematological follow-up is also essential. This ensures timely initiation of appropriate neurological and haematological management should clinical progression deem it necessary.
PP.097
PP.097 - Interruptible Demyelination Experimental Studies and Evidence in Human Neuropathies
1Sa Pathology, Adelaide, Australia, 2SA Pathology, Adelaide, Australia
Examination of sural nerves demonstrated demyelinating Schwann cells in 7/12, co-existence of remyelination and active demyelination within one original internode in 3/12, and remyelination with short original internodes in 8/12.
PP.098
PP.098 - Interpretation of Teased Nerve Fibre Preparations
1Sa Pathology, Adelaide, Australia
The teased nerve fibre (TF) technique is the best approach to study peripheral myelinated nerve fibres in their continuity. However, the limitation the interpretation according to surface appearances has long been recognized. Since 1999, sectional studies of TFs for correlating surface appearances with internal structures were performed in human nerve biopsies and animal experiments. Modified criteria for classifying TFs are proposed accordingly. To be noticed, information provided here may help to interpret findings by other pathological examinations when TF preparations are not available.
• Normal TFs: approximately equal internodal length, smooth and regular myelin sheath and similar diameter within and between different internodes without any of the abnormalities in other categories.
• Myelin irregularity: excessive myelin irregularity, wrinkling, and folding of myelin but with the other features of normal TF.
• Demyelination: loss of myelin involves part of or a whole internode. Paranodal demyelination: absence or pale osmic staining of myelin involves paranodal region, not more than 10% internodal length; Partial internodal demyelination: loss of myelin sheath involves paranodal and adjoining internodal region, not more than 50% internodal length; Internodal demyelination: loss of myelin sheath involves 50% to 100% internodal length.
• Remyelination, excessive irregularity of internodal length and/or excessive myelin sheath thickness between internodes.
• Focal swellings.
• Axonal degeneration: linear rows of myelin ovoids and balls along the TF (early stage) or pale amorphous appearance with occasional myelin ovoid (late stage).
• Regeneration: regular short pale internodes; or teased preparation with identifiable thin pale myelin sheath but hardly identified nodes of Ranvier.
• Pale amorphous structure along TFs.
• Mixture of 2 or more changes.
• Others.
PP.099
PP.099 - Painful Legs Moving Toes Syndrome Secondary to Peripheral Nervous System Disorders: A Case Series
1Shree Krishna Hospital Pramkhswami Medical College Bhaikaka University, Anand, India
PP.100
PP.100 - The Australian Neuromuscular Disease Registry - Where we are Now
1Australian Neuromuscular Disease Registry, Murdoch Children's Research Institute, Australia, 2Department of Neurology, Sydney Children's Hospital, Randwick, Australia, 3Discipline of Paediatrics and Child Health, University of NSW, Sydney, Australia, 4Muscular Dystrophy NSW, Parramatta, Australia, 5Department of Health, Perth, Australia, 6Children's Hospital Westmead, Sydney, Australia, 7University of Sydney, Sydney, Australia, 8University Of Queensland, Brisbane, Australia, 9Department of Neurology, Royal Children's Hospital, Parkville, VIC, Australia
586/821 (71.4%) of participants have had data collected longitudinally with 235/821 (28.6%) of participants have contributed data from a single time point. At last update, 499 of participants were children and 322 are adults. Additional demographic, genetic, clinical and management data will be presented on selected disease cohorts. Information on adherence to published standards of care will also be presented.
Challenges for the registry include the need to demonstrate viability and sustainability, stable funding sources, and ‘registry fatigue’.

PP.101
PP.101 - Clinical Characteristics, Treatment Outcome and Predictors of Facial Synkinesis and Hemifacial Spasm After Bell’s Palsy
1Neurological Institute Of Thailand, Bangkok, Thailand
PP.102
PP.102 - DNTH103 Shows Sustainable Inhibition of Complement and Prevents Nerve Conduction Impairment in CIDP Preclinical Model
1University of Toronto, Toronto, Canada, 2Dianthus Therapeutics, Inc., New York, USA
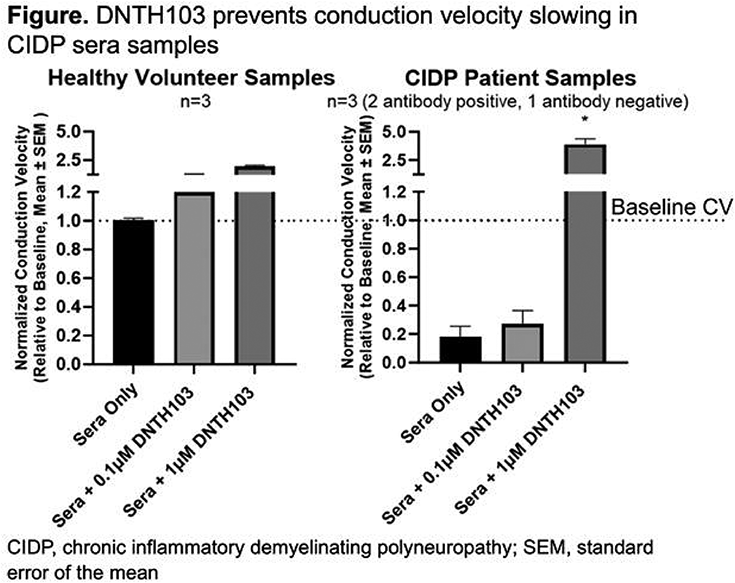
PP.103
PP.103 - Prevalence of Neck Weakness in Motor Neurone Disease - A Retrospective Study
Miss Sally Mathieson1, Mr Tim Sheehy1, Ms Trinh Sia1, Professor Prue Morgan2,
1Calvary Health Care Bethlehem, Caulfield, Australia, 2Monash University, Melbourne, Australia
 Results - 46% of the study’s cohort experience neck weakness. No significant findings were found between the presence of neck weakness and demographics. Significant findings were found with phenotype analysis. The bulbar onset group experienced the highest (56%) and earliest average rates of neck weakness at 23 months after disease onset. Closely followed by those with cervical onset MND who had a prevalence rate of 51.9% experiencing neck weakness. PLS had the lowest (16%) and latest (130 months from disease onset) rate of neck weakness.
Results - 46% of the study’s cohort experience neck weakness. No significant findings were found between the presence of neck weakness and demographics. Significant findings were found with phenotype analysis. The bulbar onset group experienced the highest (56%) and earliest average rates of neck weakness at 23 months after disease onset. Closely followed by those with cervical onset MND who had a prevalence rate of 51.9% experiencing neck weakness. PLS had the lowest (16%) and latest (130 months from disease onset) rate of neck weakness.
PP.104
PP.104 - A Progressive Hemiparetic Variant of Amyotrophic Lateral Sclerosis (ALS) - Mills Syndrome Case Series
1Royal Prince Alfred Hospital, Camperdown, Australia, 2Brain and Mind Centre, Camperdown, Australia, 3Prince of Wales Hospital, Randwick, Australia, 4Neuroscience Research Australia (NeuRA), Randwick, Australia
Today, a distinctively unilateral disease course remains the hallmark of Mills syndrome. Diagnosis is supported by advanced neurophysiology and neuroimaging studies.
It is not currently known why the disease does not progress contralateral to the side of onset, as it would in a typical case of ALS. The underlying pathophysiology of this process is of considerable interest and importance to our understanding of ALS. Further, with few cases of Mills syndrome diagnosed and described in the literature, it is of great significance that we continue to study and report our findings in these patients to further our knowledge of this rare disease.
The patient cohort comprises four males and one female, with a mean age at onset of 61 years. Clinical follow up over eleven years established that all patients have disease, most prominently limb weakness, confined to one side of their body. Spasticity was a key feature in all patients, with asymmetric reflexes, brisk on the symptomatic side, and a normal sensory examination.
Magnetic resonance (MR) tractography established marked asymmetry of the white matter fiber density, corresponding with clinical presentation. Brain imaging with positron emission tomography (PET) scan identified hypometabolism involving the left lateral frontal cortex posteriorly, corresponding to the symptomatic side. Transcranial magnetic stimulation (TMS) showed inexcitability of the motor cortex of the affected side, and motor cortex excitability within normal limits on the contralateral side, consistent with disease confined to one hemisphere. Only one patient had a family history of ALS, in her father, which was a more typical progression of ALS. Genetic studies did not identify a causative gene in this patient.
All 5 patients are still alive, now between 4 and 14 years since symptom onset.
PP.105
PP.105 - CCNF Related Young Onset Amyotrophic Lateral Sclerosis ( ALS )
1Medanta The Medicity, Gurugram, India
On examination he had broken pursuits, mixed dysarthria, tongue fasciculations, spastic tongue with exaggerated jaw jerk. Motor examination revealed atrophy of thenar, hypothenar and first dorsal interosseous bilaterally with spasticity in all four limbs. Power is given in table 1 . All reflexes were exaggerated and plantar bilaterally flexor. Sensory examination was normal. Cerebellar signs were absent. He had a spastic Gait
Whole exome sequencing was done in view of young onset ALS which revealed a heterozygous variant, c.1354C>G p.leu452Val, at exon 12 of CCNF gene.
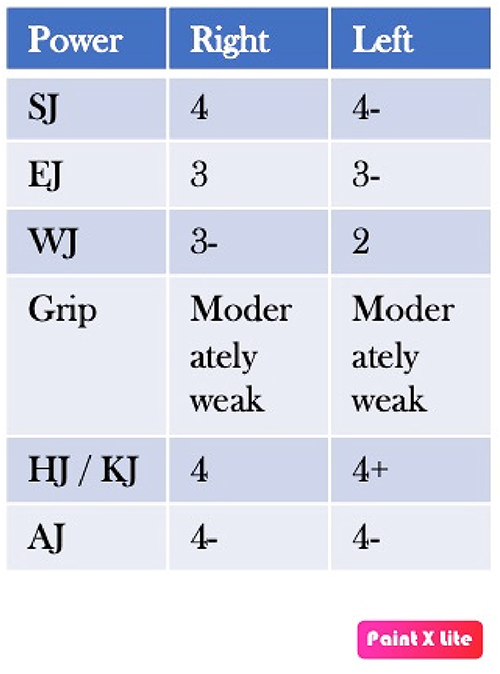
PP.106
PP.106 - PFN1 Mutation’s Role in ALS Pathomechanism: Insights from a Case Study
Dr. Jaehyun Jeon1,
1Department of Neurology, Seoul National University Hospital, Seoul, Republic of Korea, 2Centre for Hospital Medicine, Seoul National University College of Medicine, Seoul National University Hospital, Seoul, Republic of Korea, 3Department of Neurology, Seoul Metropolitan Government Seoul National University Boramae Medical Centre, Seoul, Republic of Korea
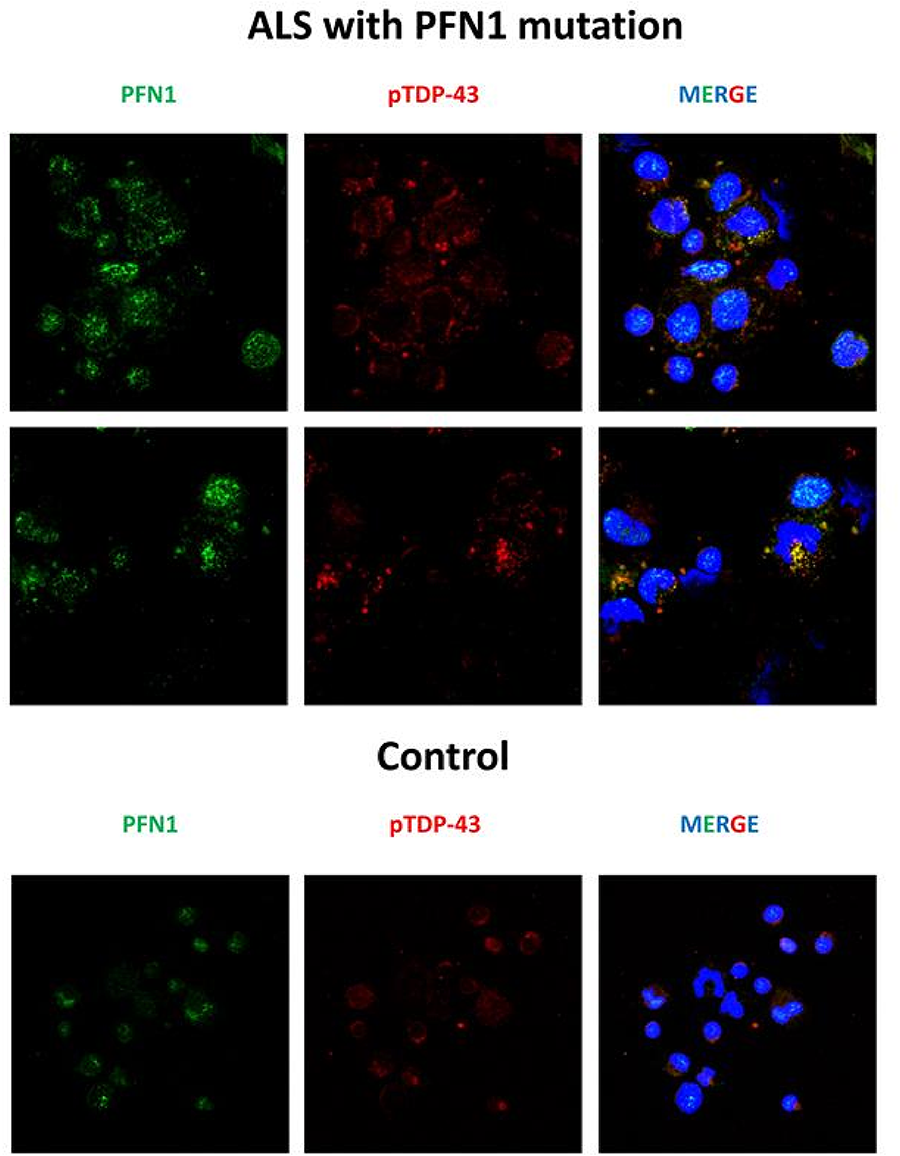
PP.107
PP.107 - Motor Neuron Disease with Vocal Cord Dysfunction
1The Royal Melbourne Hospital, Australia, 2Alfred Hospital, Australia
A 62-year-old right-handed North-Indian man with a thirteen-year history of painless progressive muscle weakness was seen in our clinic. This had started in the lower limbs and gradually progressed to his upper limb. He subsequently developed a hoarse voice and subtle stridor without dyspnoea or bulbar symptoms.
He also had ischemic cardiomyopathy, with a previously implanted automated implantable cardiac defibrillator (AICD). Family history was significant for his mother and brother having similar symptoms with onset in their 40s. Two brothers and two sisters were unaffected.
On examination he had spastic dysarthria and audible stridor at rest without respiratory distress. He had moderate-severe asymmetrical proximal more than distal weakness with right being upper limb more affected than the left and bilateral split hands. In the lower extremities he had moderate proximal weakness. Reflexes were brisk in the upper extremities. He had ankle clonus and left positive Babinski sign. Sensory examination was unremarkable. Serum creatine kinase level was elevated at 539 IU/L.
Electromyography revealed chronic denervation in muscles innervated by the bulbar, cervical, and lumbosacral segments with fasciculation potentials. There was no peripheral neuropathy. Magnetic resonance imaging was precluded by the AICD. Sleep study was normal. Video laryngeal stroboscopy revealed bilateral vocal cord atrophy.
Genetic testing did not detect expansion in androgen receptor CAG repeat. Whole exome sequencing detected a likely pathogenic variant in dynactin subunit 1 DCTN1;M_004082.5 (DCTN1): c.279+1G>C p.(?).
Vocal cord dysfunction is not uncommon in motor neuron disease (MND) (1) but is seldom the prominent feature in phenotypes other than bulbar onset MND. Dynactin 1 (DCTN1) is an axonal transport protein and its deficiency can lead to a number of neurodegenerative conditions including MND (2,3). DCTN1 mutations are typically autosomal dominant with de novo mutations being far less common (3). Disease progression can be variable including slowly progressing illness which is less common (4).
This case highlights a rare form of familial MND with vocal cord involvement as a prominent feature. Assessment of vocal cord function is important in the evaluation of patients with MND.
1. Tomik J, Tomik B, Partyka D, Skladzien J, Szczudlik A. Profile of laryngological abnormalities in patients with amyotrophic lateral sclerosis. The Journal of Laryngology & Otology. 2007;121(11):1064-1069. doi:10.1017/S002221510700610X
2. Puls I, Jonnakuty C, LaMonte BH, Holzbaur EL, Tokito M, Mann E, Floeter MK, Bidus K, Drayna D, Oh SJ, Brown RH Jr, Ludlow CL, Fischbeck KH. Mutant dynactin in motor neuron disease. Nat Genet. 2003 Apr;33(4):455-6. doi: 10.1038/ng1123. Epub 2003 Mar 10. PMID: 12627231.
3. Dulski J, Konno T, Wszolek Z. DCTN1-Related Neurodegeneration. 2010 Sep 30 [Updated 2021 Aug 5]. In: Adam MP, Feldman J, Mirzaa GM, et al., editors. GeneReviews® [Internet]. Seattle (WA): University of Washington, Seattle; 1993-2024. Available from: https://www.ncbi.nlm.nih.gov/books/NBK47027/
4. Tian WT, Liu LH, Zhou HY, Zhang C, Zhan FX, Zhu ZY, Chen SD, Luan XH, Cao L. New phenotype of DCTN1-related spectrum: early-onset dHMN plus congenital foot deformity. Ann Clin Transl Neurol. 2020 Feb;7(2):200-209. doi: 10.1002/acn3.50985. Epub 2020 Feb 5. PMID: 32023010; PMCID: PMC7034498.
PP.109
PP.109 - Rare Forms of Genetically Mediated Amyotrophic Lateral Sclerosis: An Indian Experience
1National Institute of Mental Health and Neurosciences, Bengaluru,Karnataka, India
Out of 15 C9ORF72 genetically proven cases (14-males and 1-female), 5 were familial and 11 were sporadic cases. Eight were limb onset and 7were bulbar onset. FTD-ALS overlap was found in 5 cases. Six patients had follow up data and 5 of them had succumbed to the illness. C9ORF72 is the common cause for genetic ALS. In our cohort, all of these patients had features of sporadic ALS.
Fourteen patients with FIG4 mutations; M:F-10:4. Thirteen had SETX with mean age at onset of 48 years. Twelve had SOD1 with average at onset of 51 years and average duration of 21 months.Ten patients had DCTN1 mutation with average age at onset of 51 years and average duration of illness was 18 months.Nine had OPTN with average age of onset of 50 years and avg duration of 13 months. Eight had FUS with age at onset of 50 years. Eight had NEFH with age at onset of 54 years and average duration of 22 months.Seven patients with ERBB4 mutation with average age at onset of 47 years.There were 5 patients with ANXA11 mutations,mean age at onset of 44 years and mean duration of illness was 11 months. Four had TARDBP with avg age at onset of 50 years and avg duration of 11 months. Three with ATXN2 whose average duration to loss of ambulation after onset was 6 months.Two had SQSTM1. One case with CHMP2B mutation was detected. One patient carried VAPB.
PP.110
PP.110 - Juvenile Amyotrophic Lateral Sclerosis: A Rare and Aggressive Presentation in a 22-year old Filipino Male
1University of the East Ramon Magsaysay Memorial Medical Center, Quezon City, Philippines, 2Cardinal Santos Medical Center, San Juan City, Philippines
Amyotrophic Lateral Sclerosis (ALS) is a rare neurodegenerative disorder primarily affecting adults, but juvenile-onset ALS is exceptionally rare. We report a rare case of a 22 year old Filipino male patient who exhibited early-onset weakness, muscle atrophy, and tongue fasciculations, followed by rapidly progressive dysphagia and respiratory distress. EMG-NCV findings showed evidence for a chronic, active predominantly motor neuronal-axonal loss type of neuropathy involving the tongue and limb muscles bilaterally consistent with a motor neuron disease. The patient was treated with Riluzole with no significant improvement of symptoms. Despite multidisciplinary interventions, the disease rapidly progressed, highlighting the challenges in managing juvenile ALS cases in the Philippine setting where there is limited access to genetic testing. This case report emphasizes the importance of considering ALS in the differential diagnosis of progressive motor dysfunction in younger patients and the complexities involved in their care.
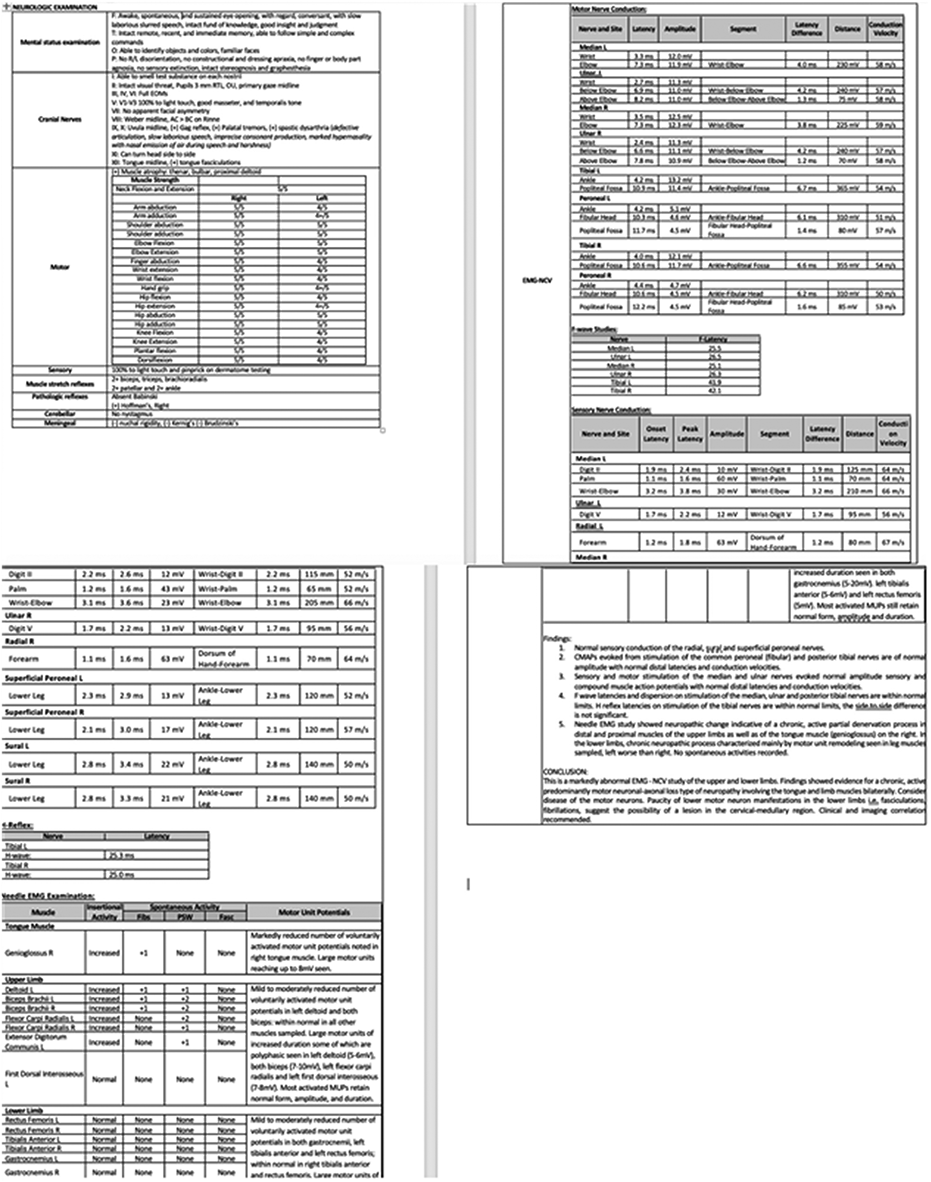
PP.111
PP.111 - Intronic KIF5A TC Variant Modifies Rate of Progression and Survival in Sporadic Amyotrophic Lateral Sclerosis
1Perron Institute for Neurological and Translational Science, Nedlands, Australia, 2Centre for Molecular Medicine and Innovative Therapeutics, Murdoch University, Murdoch, Australia, 3Centre for Neuromuscular and Neurological Disorders, University of Western Australia, Nedlands, Australia, 4School of Health Sciences and Physiotherapy, the University of Notre Dame, Fremantle, Australia, 5Institute for Immunology and Infectious Diseases, Murdoch University, Nedlands, Australia, 6Department of Neurology, Duke University, USA, 7Centre for Medical Research, Harry Perkins Institute of Medical Research, Nedlands, Australia, 8Faculty of Medicine, The University of Notre Dame, Nedlands, Australia, 9Department of Neurology, Fiona Stanley Hospital, Australia
PP.112
PP.112 - Oligodendroglia: ‘Lone Hero’ to Motor Neurons Against Toxic Insults of CSF from Sporadic ALS Patients
1National Institute of Mental Health And Neuro Sciences (NIMHANS), India
Cerebrospinal fluid from Amyotrophic Lateral Sclerosis patients (ALS-CSF) induces marked gliosis and degeneration of motor neurons. Our earlier studies reveal that ALS-CSF induces activation of both, microglia and astroglia skewing them towards detrimental forms with the release of pro-inflammatory cytokines and other neurotoxic substances resulting in obvious neuroinflammatory responses. Amongst the glial cells, the role of oligodendroglia in ALS has been overlooked since demyelination is not a prominent feature of ALS until the end stage of the disease. However, recent studies point towards additional role of oligodendroglia including trophic and metabolic support to the neighbouring neurons. With reduced trophic support from activated astrocytes and microglia as well as altered glucose metabolism in the degenerating motor neurons, it is intriguing to examine the role of oligodendroglia in sporadic ALS. To investigate this, a human oligodendroglial cell line, MO3.13 was exposed to CSF from sporadic ALS patients (ALS-CSF) at 10%v/v for 48hrs and expression of oligodendroglia specific proteins viz. CNPase as well as Olig2 was studied using immunocytochemistry followed by confocal microscopy and Western blotting. ALS-CSF affected the viability of MO3.13 cells as evidenced by live cell imaging and MTT assay. Expression of CNPase as well as Olig2 was found to be significantly reduced in cells exposed to ALS-CSF. Further, to investigate the effect of the observed oligodendroglial changes on motor neurons, NSC-34 motor neuronal cells were co-cultured with MO3.13 cells or supplemented with conditioned medium of the MO3.13 cells which were exposed to ALS-CSF. Live cell imaging experiments reveal better survival of NSC-34 cells upon co-culture with MO3.13 co-cultures as evidenced by the absence of both cytoplasmic vacuolation as well as beading of neurites and better differentiation of the motor neuronal cells. Enhanced lactate levels and increased expression of its transporter, MCT-1 with sustained expression of trophic factors namely, GDNF and BDNF by MO3.13 cells hints towards metabolic and trophic support provided by the surviving oligodendroglia. These findings indicate that oligodendroglial cells are indeed the “lone hero” to the degenerating motor neurons when the astrocytes and microglia turn topsy-turvy.
PP.113
PP.113 - Nuclear Dynamics of Mutant Cu/Zn Superoxide Dismutase in Amyotrophic Lateral Sclerosis
1Tottori University, Yonago, Japan
Cytoplasmic inclusions are observed in motor neurons in amyotrophic lateral sclerosis (ALS) associated with the Cu/Zn superoxide dismutase mutation (mtSOD1). Although these inclusions are a hallmark of the disorder, degeneration is not necessarily initiated in the cytoplasm, nor are these structures the culprit of ALS. We hypothesized that mtSOD1 in the nucleus contributes to motor neuron degeneration. The nucleus stores genetic material and acts as the cell’s control center, and a small fraction of mtSOD1 is reported to be distributed in the nucleus. To confirm this, we explored the roles of mtSOD1 in relation to nuclear proteins, chromosomal DNA, and mRNA expression. An immortalized cell line derived from a transgenic ALS mouse model expressing mtSOD1Leu126delTT with a FLAG sequence was used to guarantee stable immunoprecipitation of mtSOD1-binding molecules in the nucleus using shotgun proteomics and chromatin immunoprecipitation-sequencing (ChIP-seq). We also examined mRNA expression by silencing whole SOD1 (innate mouse Sod1 and mtSOD1) or mtSOD1 alone and comparing these patterns against those in non-silenced counterparts. We identified 392 proteins as mtSOD1-interacting proteins in the nucleus. Gene ontology (GO) revealed these proteins to be enriched for “mRNA processing.” Surprisingly, more than 11% of mtSOD1-interacting proteins were expressed concurrently with previously reported mutant forms of TAR DNA-binding protein 43 (TDP-43)-interacting proteins. ChIP-seq revealed that mtSOD1-interacting DNA portions showed a preference for zinc finger protein-binding motifs. Further, GO analysis of the ChIP-seq data revealed that “mRNA processing” was again enriched among the genes harboring mtSOD1-binding domains. Mouse Sod1 and mtSOD1 mRNA were found to be overexpressed in association with “type 1 IFN response” compared to their silenced counterparts. We revealed that mtSOD1 interacted with nuclear proteins and specific DNA segments and that RNA expression was notably altered when mtSOD1 was silenced. In the GO analyses, molecules related to “mRNA processing” and “type 1 IFN response” were prominent. These data support the hypothesis that compromise of these pathways in neurons and glial cells plays a pivotal role in motor neuron degeneration.
PP.114
PP.114 - A Tricky Tongue Twister: Tongue SCC Mimicking Bulbar MND. A Case and a Discussion
1MND Research Centre, Faculty of Medicine and Human Health Sciences, Macquarie University, Sydney, Australia
We report a case of a 77-year-old man with a 2-year history of dysphagia and dysarthria, presenting a diagnostic challenge.
Initially referred at age 73, he had a medical history of prostate cancer and vitamin D deficiency, with no family history of neurological diseases.
Symptoms began after a radical prostatectomy in 2019, including a sore throat, dry throat, and weight loss.
Despite extensive investigations (CT, barium swallow, thyroid ultrasound, MRI, nerve conduction study, EMG, and blood tests), no definitive diagnosis was made. A provisional diagnosis of bulbar motor neuron disease was given, and the patient was monitored for two years with minimal degeneration.
In 2021, he presented with haemoptysis and a right lower lobe nodule, which responded to antibiotics. However, an incidental tongue lesion was noted during the PEG insertion procedure. Tongue revealed a T3 N1 M1 p16 positive squamous cell carcinoma with lung metastasis.
He underwent chemotherapy and radiotherapy, resulting in stable mass and weight. This case highlights the potential for misdiagnosis, as SCC of the tongue can mimic motor neuron disease. Continuous follow-up is crucial for accurate diagnosis and management.
Misdiagnosis rates of MND can be up to 8%, emphasizing the need for considering a wide differential diagnosis in atypical presentations. The clues in this case were a pure lower motor neuron bulbar disease without any evidence to any other muscle involvement.
PP.115
PP.115 - DCTN1 Mutation Presenting as Vocal Fold Palsy: A Case Report and Genetic Insights
1Department of Neurology, Seoul National University Hospital, Seoul, Republic of Korea, 2Centre for Hospital Medicine, Seoul National University College of Medicine, Seoul National University Hospital, Seoul, Republic of Korea, 3Department of Neurology, Seoul Metropolitan Government Seoul National University Boramae Medical Centre, Seoul, Republic of Korea
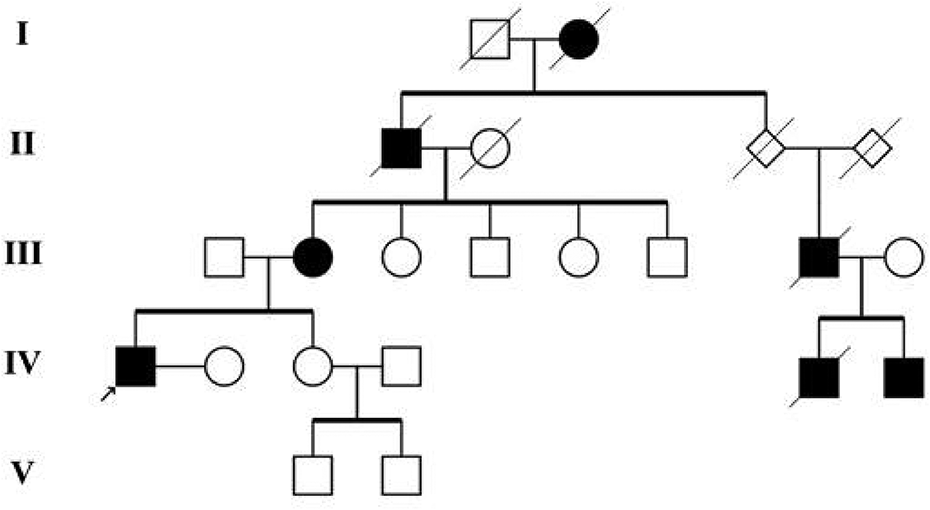
PP.116
PP.116 - Frequency and Evolution of Orthostatic Hypotension in the PRO-ACT Database
Mrs Cecilia Quarracino1, Dr. Francisco Capani, Dr. Matilde Otero-Losada,
1Centro de Altos Estudios en Ciencias Humanas y de la Salud. Universidad Abierta Interamericana., Buenos Aires, Argentina
PP.117
PP.117 - Hirayama Disease with Proximal Muscle Involvement
1AIIMS, Delhi, India, 2AIIMS, Delhi, India
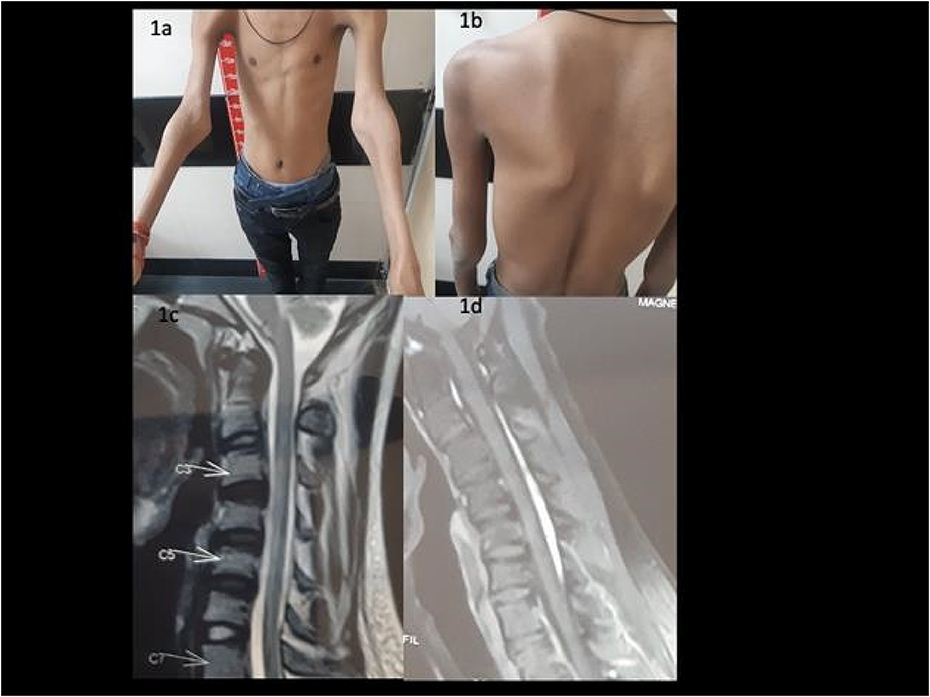
PP.118
PP.118 - Clinical Characteristics of Motor Neuron Disease in Māori and Non-Māori Patients Waikato Hospital, New Zealand
1Te Whatu Ora Waikato, Hamilton, New Zealand
MND is a fatal neurodegenerative condition characterized by progressive weakness, with the most common subtype being amyotrophic lateral sclerosis (ALS). ALS results from the death of both upper and lower motor neurons, while other MND variants affect either upper or lower motor neurons. Clinical features include bulbar weakness with dysarthria and dysphagia, limb weakness, and respiratory muscle weakness. The global incidence of MND is 2 per 100,000 people per year, with a prevalence of around 5 per 100,000 people per year. Most cases are sporadic, with approximately 5% being familial. The median survival is between 20 to 48 months from symptom onset. This observational study aims to characterize the demographics, referral patterns, clinical phenotypes and symptoms of MND in Māori and non- Māori patients in the Waikato region.
We conducted a retrospective analysis of all Māori and non-Māori patients diagnosed with MND and ALS in the Waikato region between 2012 and 2022. Patients were identified through the MND database maintained by the Neurology Department at Waikato Hospital, which serves a population of nearly half a million, including a higher proportion of Māori than the national average. We reviewed all outpatient visits and inpatient reviews within the study period. Diagnoses were confirmed by a neurologist, and subtypes identified included ALS, bulbar palsy and limb onset. Data collected included age at diagnosis, sex, clinical manifestation of MND at presentation, MND subtype, family history, genetic testing, cognitive impairment, and duration of symptoms.
A total of 100 patients diagnosed with MND were included in this study, comprising 9 Māori and 91 non-Māori patients. This study is the first to specifically investigate the clinical characteristics of MND in Māori and non-Māori patients in New Zealand. Notably, the mean age at diagnosis and death did not differ significantly between Māori and non-Māori patients, suggesting similar age-related vulnerability across both groups. However, the shorter mean duration of symptoms in Māori patients (5.67 ± 3.64 months) compared to non-Māori patients (10.29 ± 8.43 months) may indicate differences in disease progression or possibly delays in diagnosis. The higher prevalence of bulbar onset symptoms (55%) over limb onset (45%) in both groups is noteworthy, given the generally poorer prognosis associated with bulbar onset ALS. A summary of the data is shown in Table 1.
Evidently, the high percentage of patients presenting with fasciculations (90%) and muscle wasting (72%) underscores the need for early recognition of these symptoms in the diagnostic process. The majority of the referrals came through the GPs (65%), followed by private practices (11%) and ENT/Gen Med (9% and 7%, respectively). The findings highlight important areas for improvement in clinical practice, including the need for earlier diagnosis, comprehensive family history and genetic testing, and routine cognitive assessments.
When interpreting the results, consider its retrospective nature, potential referral selection bias, and small sample size, which limits the ability to establish casual relationships. Future research with larger patient cohorts and prospective study designs comparing clinical management and survival probability amongst the NZ population is warranted to validate these findings.

PP.119
PP.119 - ARGX-119 in Patients with Amyotrophic Lateral Sclerosis - reALiSe: A Phase 2a Study Design
Ruben P.A. van Eijk1, Boudewijn T.H.M. Sleutjes1, Angela Genge2,
1University Medical Center Utrecht, Utrecht, the Netherlands, 2McGill University, Montreal, Canada, 3argenx, Ghent, Belgium, 4Curare Consulting B.V., Liempde, the Netherlands
PP.120
PP.120 - QRL-201 – a Novel Antisense Oligonucleotide Targeting Stathmin-2 Mis-Splicing in Amyotrophic Lateral Sclerosis
1QurAlis, Cambridge, United States
Phase 1 Study Design
QRL-201-01 is a double-blind, multiple-ascending dose study designed to assess the safety and tolerability of QRL-201 in people living with ALS. In this first-in-human study, up to 64 people living with ALS will receive QRL-201, or matching placebo, in a 6:2 ratio respectively. This study employs sentinel participant dosing and includes multiple safety reviews. The primary endpoint will be incidence of adverse events. The secondary endpoints will be measurements of multiple dose pharmacokinetics (PK). This study will include multiple exploratory endpoints – 1) biomarkers of neuronal loss (neurofilament) and STMN2 biology; 2) clinical outcome measures (ALSFRS-R, ROADS, SVC, HHD, electrophysiology testing); and 3) CSF PK profile. QRL-201-01, also known as the ANQUR study, is ongoing in Canada, the United Kingdom, and multiple European countries. Results from the ANQUR study will inform POC study design.
PP.121
PP.121 - QRL-101 – a KCNQ2/3 Modulator Targeting Hyperexcitability in Amyotrophic Lateral Sclerosis
1QurAlis, Cambridge, United States
Novel Electrophysiology Testing Module
Reducing motor system hyperexcitability is a potential therapeutic strategy in ALS, and as such, electrophysiology endpoints could be utilized as markers of target engagement in clinical development. QurAlis, and its partners, have developed an operational framework to utilize a fit-for-purpose exploratory electrophysiology endpoint for initial clinical trial piloting in the QRL-101 program.
PP.122
PP.122 - Antisense Oligonucleotide Mediated Reduction of C9ORF72 Expansion Containing Transcripts in iPS-Derived Motor Neurons
1Murdoch University, Perth, Australia, 2Perron Institute, Perth, Australia
The pathogenic mechanisms associated with C9ORF72 remain poorly understood, however, three molecular mechanisms are proposed by which C9ORF72 repeat expansion induces neurodegeneration; (1) C9ORF72 loss-of-function (LOF) through haploinsufficiency (1, 2); (2) RNA toxic gain-of-function (GOF) (RNA is produced bidirectionally) (3, 4); and (3) gain-of-function through accumulation of toxic dipeptide repeat peptides (4). Removal of the hexanucleotide repeat expansion (HRE) in C9ORF72 partially ameliorates the GOF mechanisms (5).
Antisense oligonucleotides (ASOs) are single-stranded nucleic acid analogues that can be used to alter gene expression through the modulation of splicing. Morpholinos are chemically modified ASOs that possess a neutral charge and may be used to target expansion-containing C9ORF72 transcripts in ALS and thereby ameliorate GOF mechanisms.
In this project, we employed a steric-blocking ASO strategy to mediate the reduction of expansion containing C9ORF72 transcripts in both C9ORF72 ALS patient dermal fibroblasts and iPSC-derived motor neurons (iPS-MNs).
Morpholinos targeting the C9ORF72 HRE were electroporated into patient fibroblasts and iPS-MNs. Gene expression analysis of C9ORF72 transcript variant 3 (V3)(expansion containing), variant 2 (V2) and total levels of transcript variants (All V) were assayed by digital droplet PCR. Changes in levels of C9ORF72 HRE RNA foci were measured in situ using complementary probes and a Basescope assay.
Following transfection of morpholinos into patient dermal fibroblasts and iPS-MNs, gene expression analysis revealed that morpholinos could selectively reduce levels of mRNA containing the expansion (V3) by >90% compared with the sham control. Reduction of V3 was comparable with levels of reduction seen following treatment with RNase H ASO positive control. Treatment with morpholinos did not results in marked changes to alternative C9ORF72 transcript variants (V2 and All V).
Our data demonstrates that steric blocking morpholinos can effectively reduce levels of expansion containing transcripts V3, without reducing overall expression of C9ORF72 V2 transcripts. Further studies are required to elucidate the functional consequences of morpholino mediated V3 reduction in iPS-derived and mouse models, such as reduced RNA foci and functional and phenotypic improvement. This work has the potential to deliver a treatment for C9ORF72 ALS and a safer alternative to RNase H ASOs.
PP.123
PP.123 - Allele Selective and Splice-Switching FUS Targeted Antisense Oligonucleotide Therapeutic Development for ALS
1Centre for Molecular Medicine and Innovative Therapeutics, Murdoch University, Perth, Australia, 2The Perron Institute for Neurological and Translational Science, Perth, Australia, 3The University of Western Australia, Perth, Australia, 4Duke University, Durham, USA
Funding for part of this study was provided by Motor Neurone Disease Research Australia (MNDRA) in the form of a Daniel McLoone MND Research Grant. TMOs were provided by Professor Marvin Caruthers.
PP.124
PP.124 - Co-Development of Genetic Markers and Antisense Oligonucleotides to Restore Axonal Health in MND
1The University of Notre Dame, Fremantle, Australia, 2Perron Institute for Neurological and Translational Science, Nedlands, Australia, 3Centre for Molecular Medicine and Innovative Therapeutics Murdoch University, Nedlands, Australia, 4The Florey Insitute, Nedlands, Australia
Motor neurone disease, also known as amyotrophic lateral sclerosis (ALS), is a devastating and fatal neurodegenerative disease with little to no effective treatments to slow or halt the disease progression. ALS rapidly affects people’s ability to control their muscles until they eventually lose the ability to walk, speak, swallow, and breathe. At present, there is no way to predict which patients will be more likely to respond to current or newly developed treatments. The major barrier contributing to the lack of available treatments and the high failure rate of ALS clinical trials, is the extremely complex underlying nature of this disease. With approximately 90% of cases being classed as sporadic, meaning they have no known genetic cause, it is difficult to predict how fast each person’s disease will progress and exactly which underlying cellular mechanisms are driving each person’s disease. Therefore, there is an urgent need to develop both tools to identify ALS patients more likely to respond to specific treatments, and novel therapies for ALS patients.
This project focuses on further investigating genetic markers in genes that play a critical role in maintaining nerve function. In combination with this, we will also develop two molecular therapies to help restore nerve function by down regulating genes that prevent nerve regeneration when cells are damaged in the central nervous system. We will assess the genetic markers and molecular therapies in parallel, and this will enable us to determine whether the genetic markers can identify subgroups of patients with a faster functional nerve decline, that may be more amenable to a nerve targeted therapy. This unique strategy will provide a paradigm shift in the approach to patient stratification in future clinical trials, allowing the prioritisation of participants that have a similar underlying disease mechanism, and are therefore more likely to respond to a precision nerve targeted treatment.
PP.125
PP.125 - Troponin T Correlates with Denervation Activity in Patients with Amyotrophic Lateral Sclerosis
1Örebro University Hospital, Örebro, Sweden
Age at diagnosis was a significant predictor of survival in univariate and multivariate analysis (HR= 1.00016, p= <0.001 and HR 1.00014, p=0.012, respectively). Non-spinal onset was a significant predictor of survival in univariate and multivariate analysis (HR 4.293, p=<0.001 and HR 5.142, p=<0.001, respectively).
Numerical hs-cTnT was associated with shorter survival in the univariate Cox regression model (HR_1.019, p=0.009) and in the full multivariable Cox model (HR=1.024, p=0.003). High (≥15 nanogram/L) vs normal hs-cTnT was not a predictor of survival in the univariate Cox regression analysis (HR 1.709, p=0.094).
PP.126
PP.126 - Serum Chitotriosidase as an Early Biomarker of Sporadic ALS: Can it Replace CSF
1Dept of Neurophysiology, National Institute Of Mental Health & Neurosciences, Bangalore, India, 2Dept of Neurology, National Institute Of Mental Health & Neurosciences, Bangalore, India
The cerebrospinal fluid of individuals with amyotrophic lateral sclerosis (ALS-CSF) triggers neurodegeneration of motor neurons and induces gliosis in the in-vitro and in-vivo models of sporadic Amyotrophic Lateral Sclerosis (ALS). Diagnosing ALS, a syndrome characterised by motor neuron degeneration and muscular atrophy, is difficult due to delayed symptom onset and potential overlap with other neurodegenerative conditions. The identification and development of suitable biomarkers may increase treatment options by allowing for quick diagnosis, monitoring illness progression, and thereby providing precise pathophysiology. Our lab pioneered to report 20-fold increased levels of chitotriosidase (CHIT-1) and enzyme activity in ALS-CSF utilising proteomics/ELISA in a large cohort of 158 samples. Further, a significant negative correlation was observed between CHIT-1 levels and disease duration, indicating higher levels of CHIT-1 in the CSF of individuals with a shorter disease duration highlighting its utility as early diagnostic biomarker for ALS. Since lumbar puncture if technically demanding and more invasive, here, we investigated a less intrusive procedure in which CHIT-1 and related proteins such as CHI3L1 and Osteopontin (SPP) levels were assessed in serum of ALS patients' using sandwich ELISA. The data was compared to protein levels in healthy individuals and disease control groups, such as NALS (Other Neurological Non-Neurodegenerative Diseases) and ONDD (Other Neurodegenerative Disorders). Additionally, Serum CHIT-1 levels were objectively measured longitudinally also at three time points (1st: at the time of recruitment, 2nd: 3-6 months after recruitment and 3rd: after 12-18 months; n= 6). Further, the serum CHIT-1 levels were correlated with corresponding CSF CHIT-1 levels and also with the disease severity. CHIT-1, CHI3L1, and Osteopontin (SPP) levels were measured in the serum of 80 ALS patients (ALS; 21-65 yrs), 35 patients with other neurodegenerative diseases (ONDD; 47-75 yrs), 35 patients with other neurological diseases (NALS; 17-75 yrs) and 35 age-matched controls (NC). Our results indicate a significant increase of serum CHIT-1 levels in ALS patients when compared to NC (***p = 0.0001), NALS (##p = 0.0015), and ONDD ($$p = 0.0062). Significant increase was observed with CHI3L1 levels too in serum of ALS patients on comparison with NC (***p = 0.0003) and NALS (####p < 0.0001). However, no significant difference was observed in serum SPP levels across groups. Interestingly, the serum CHIT-1 levels were higher than CSF CHIT-1 levels of ALS patients (**p < 0.01). Longitudinally, the serum and CSF CHIT-1 levels did not show any significant change across the three time-points.
Our study confirms the effective utility of serum CHIT-1 levels in early diagnosis of ALS and highlights the possibility of replacing CSF. Longitudinally, the sustained enhancement of serum CHIT-1 levels with increasing disease duration not only strengthens its biomarker status but also paves a way to develop new therapeutic interventions.
PP.127
PP.127 - Computed Tomography Based Gynecomastia in SBMA as a Diagnostic Biomarker: A Single Center Retrospective Study
1Department of Neurology, Kyungpook National University Chilgok Hospital, Daegu, Republic of Korea, 2Department of Neurology, Asan Medical Center, University of Ulsan College of Medicine, Seoul, Republic of Korea
Motor neuron disease spectrum disease encompass a spectrum of disorders characterized by motor neuron damage and clinically diagnostic challenges exist due to overlapping symptoms especially in the early stages. Spinal bulbar muscular atrophy (SBMA), also known as Kennedy disease, and amyotrophic lateral sclerosis (ALS) are two such conditions sharing clinical features. The aim of this study was to evaluate the utility of chest tomography (CT) imaging in distinguishing between SBMA and ALS, particularly focusing on radiological gynecomastia as a potential early differential diagnostic marker of SBMA.
The study was a single-center retrospective study involving a review of CT images from genetically confirmed SBMA and sporadic ALS patients as well as a control group. Demographic and clinical data were collected, and chest CT images were analyzed for glandular tissue diameter and morphology of gynecomastia. The clinical parameters included age at imaging, disease duration, body mass index, ALSFRS-R and K-SBMAFRS scores. The laboratory data included hormonal status, creatine kinase (CK), creatinine (Cr) and liver function test. The CAG repeat length and 6-minute walk test results of SBMA were retrieved from the registry. Medical conditions and drugs that are known to influence gynecomastia were also analyzed.
The age at imaging of SBMA patients was 57.4±9.11. The ALSFRS-R and K-SBMAFRS was 38.86±5.28 and 39.55±9.44. The disease duration was 11.8 years. The BMI was 23.14±2.64, CK level was 695.93IU/L and creatinine was 0.50mg/dL. The CAG repeat length was 46.93±3.51. The BMI, functional scales, and CT taking age had no statistical difference between SBMA and ALS group. Clinical gynecomastia was 80% while radiological gynecomastia was 93.3% in SBMA. The mean glandular tissue diameter of the CT imaging in SBMA, ALS and controls were 32.22±12.57mm, 15.91±4.81mm and 15.76±7.26mm and it differentiated SBMA from ALS and controls with statistical significance. There was significantly high prevalence of diffuse glandular morphology pattern in SBMA (50%) while there was high prevalence of nodular morphology in ALS and controls (9.1% and 20%).
CT based radiological gynecomastia effectively differentiated SBMA from ALS. Moreover, radiological gynecomastia seems to be an independent factor not influenced by BMI, age, hormones or medication usage. Our results support the usefulness of radiological gynecomastia as a possible differential diagnostic marker of SBMA especially in the early stages and further in depth studies are needed to establish the underlying pathophysiological role of gynecomastia in SBMA.
PP.128
PP.128 - Unraveling Polyminimyoclonus in Hirayama Disease: A Case Series
1Tunku Abdul Rahman Neurology Institute, Kuala Lumpur General Hospital, Jalan Pahang, Malaysia
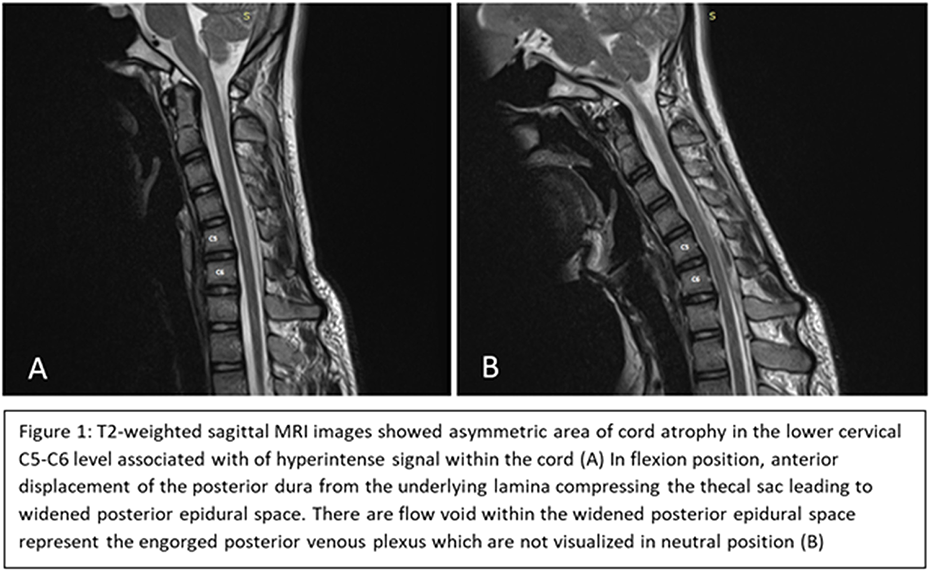
PP.129
PP.129 - Poliomyelitis Sixty Years Prior to Amyotrophic Lateral Sclerosis
1Motor Neuron Disease Research Centre, Macquarie Medical School, Faculty of Medicine, Health and Human Sciences, Sydney, Macquarie University 2109, Australia, 2Brain and Mind Research Centre, Camperdown Sydney, Australia, 3Faculty of Medicine and Health, Camperdown Sydney, Australia, 4Neuroscience Research Australia ( NeuRA), Randwick Sydney, Australia
We present the case of a male with onset in 2007 at 62 years of age with weakness and spasticity in the lower limbs. Investigations resulted in the diagnosis of clinically definite Motor Neuron Disease (MND).
Of significance, the patient had poliomyelitis involving the left lower limb as a child with complete clinical resolution by the age of 11 years. The clinical course was very slow and protracted, with global involvement of both upper and lower motor neurons. Following a fall in 2023 resulting in a flail segment, the patient died from respiratory failure 16 years after symptomatic onset.
Post-mortem examination demonstrated post polio pathology characterised by negative TDP staining and severe loss of Betz cells and anterior horn cells..
This case illustrates post polio syndrome as a possible cause of motor neuron disease. It further highlights the crucial nature of histopathology in understanding the pathogenesis of MND.
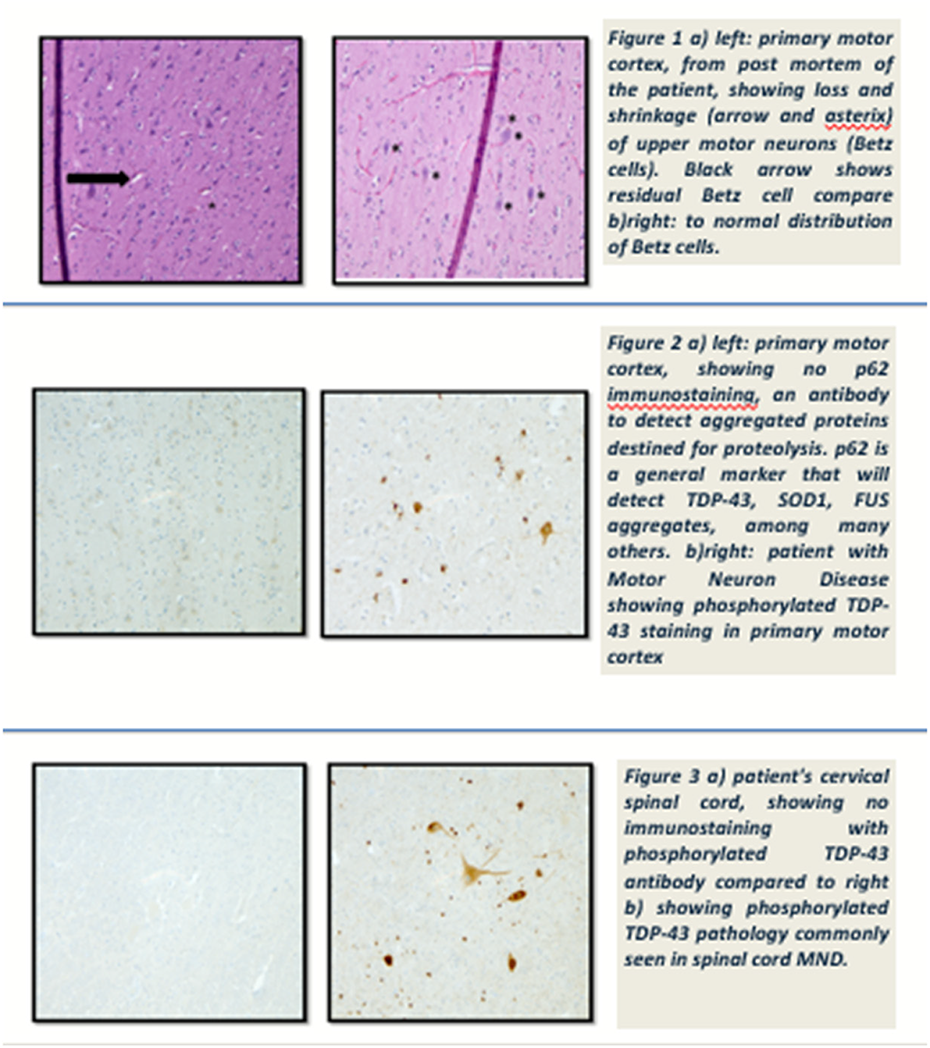
PP.130
PP.130 - Revised Upper Limb Model for Spinal Muscular Atrophy Type 2: Understanding the Natural History
1Hospital Sant Joan De Déu, Barcelona, Spain, 2Hospital Universitario la Fe, Valencia, Spain, 3Hospital Universitario Virgen del Rocío, Sevilla, Spain, 4Hospital Regional Universitario Carlos Haya, Málaga, Spain, 5Hospital Universitario La Paz, Madrid, Spain, 6Hospital Clínico Universitario Virgen de la Arrixaca, Murcia, Spain, 7Hospital Universitario Río Ortega, Valladolid, Spain, 8Institut Recerca Sant Joan de Deu, Barcelona, Spain
Individuals with spinal muscular atrophy (spinal muscular atrophy) type 2 begin to show symptoms between six and 18 months of age and can sit but not walk. Natural history data in SMA type 2 are limited and available studies that longitudinally assess the patters of progression of upper limb motor function in these individuals often have small sample sizes and short follow-up periods. It will be difficult to acquire new systematic longitudinal history data because of the availability of disease modifying therapies. However, while the beneficial effect on motor function is evident in younger and stronger individuals at the start of treatment the effectiveness in severely affected SMA type 2 individuals has not yet been prove.
The aim of our study was described the natural history of the upper limb motor function in paediatric SMA type 2 based in the analysis of a large cohort of non-treated individuals who were assessed longitudinally.
PP.131
PP.131 - Generating Real-World Evidence in Spinal Muscular Atrophy: A Global Disease Registry Strategy
1Biogen, Australia, 2Biogen, Cambridge, USA, 3Biogen, Baar, Switzerland, 4Biogen, Maidenhead, UK
PP.132
PP.132 - Adult Onset SMA Mimicking Myopathy; A Diagnosis a Long Time in the Making
1St Vincent's Health, Melbourne, Melbourne, Australia, 2Austin Health, Melbourne, Australia
SMA is an autosomal, recessive inherited neuromuscular disorder. Type IV (‘adult onset’) SMA is the rarest of the four forms of SMA, with comparatively small numbers in SMA registries world-wide.
In this case, a 67-year-old female, of English/German and Danish ancestry, presented to a new neurologist for review as her previous neurologist had recently retired. She was told she had IBM, based on a muscle biopsy taken over 20 years ago and had been treated with monthly IVIg for many years.
On history, she described being slow at sports as a child and developed significant weakness from the age of 40, initially in the lower limbs, but progressing to the upper limbs. There was no bulbar or respiratory weakness. No other family members had a similar condition. On examination she had mild scapular winging with proximal more than distal limb weakness, worse in the lower limbs. Hip flexors and quadriceps were markedly wasted with relative preservation of hip adduction. Reflexes were present with reinforcement. There were no sensory or cerebellar findings. EMG showed small tibial CMAPs in the lower limbs and EMG widespread neurogenic changes with no spontaneous activity. Her CK was mildly elevated (< 2X ULN), other blood tests were unremarkable.
Genetic testing identified a homozygous SMN1 deletion with 4 copies of SMN2. There were three other variants of uncertain significance which were not felt to be relevant (AMPD1, CHRND and MEGF10).
Her initial muscle biopsy from the vastus lateralis was reviewed and showed fiber hypertrophy, splitting, increased internal nuclei and myocyte degeneration. There were clustered atrophic fibers and slow myosin immunoperoxidase showed slow fiber dominance in the hypertrophic fibers and some rounded atrophy. Fast myosin immunoperoxidase showed a background of focally clustered severely atrophic fibers and some larger fibers reacting with fast myosin. The findings were interpreted as representing a severe, chronic myopathic process on a background of an underlying chronic neuropathic process.
The prolonged pathway to diagnosis in this patient is not surprising and a recent clinical series of adult SMA reported that the mean time to diagnosis is 12.4 years. The muscle biopsy findings are of interest and similar pathological findings have been described in some of the earliest published case reports in adult SMA. Other cases have reported myopathic muscle pathology with protein aggregates or features of limb girdle muscular dystrophy in adult SMA patients. An emerging body of literature confirms that SMN protein has direct effects on skeletal muscle including on myotubule development, mitochondria, contractile protein and neuromuscular junction function. In addition, a murine model with exon 7 deletion in the SMN 1 gene restricted to skeletal muscle, created using the Cre-lox-P system, revealed a muscular dystrophy phenotype.
This case demonstrates some of the diagnostic challenges in adult onset SMA. These patients can phenotypically and pathologically mimic myopathies and historically patients may have been misdiagnosed. The case also highlights the effects of SMN deficiency on skeletal muscle, independent of anterior horn cells which is an area of increasing research interest.
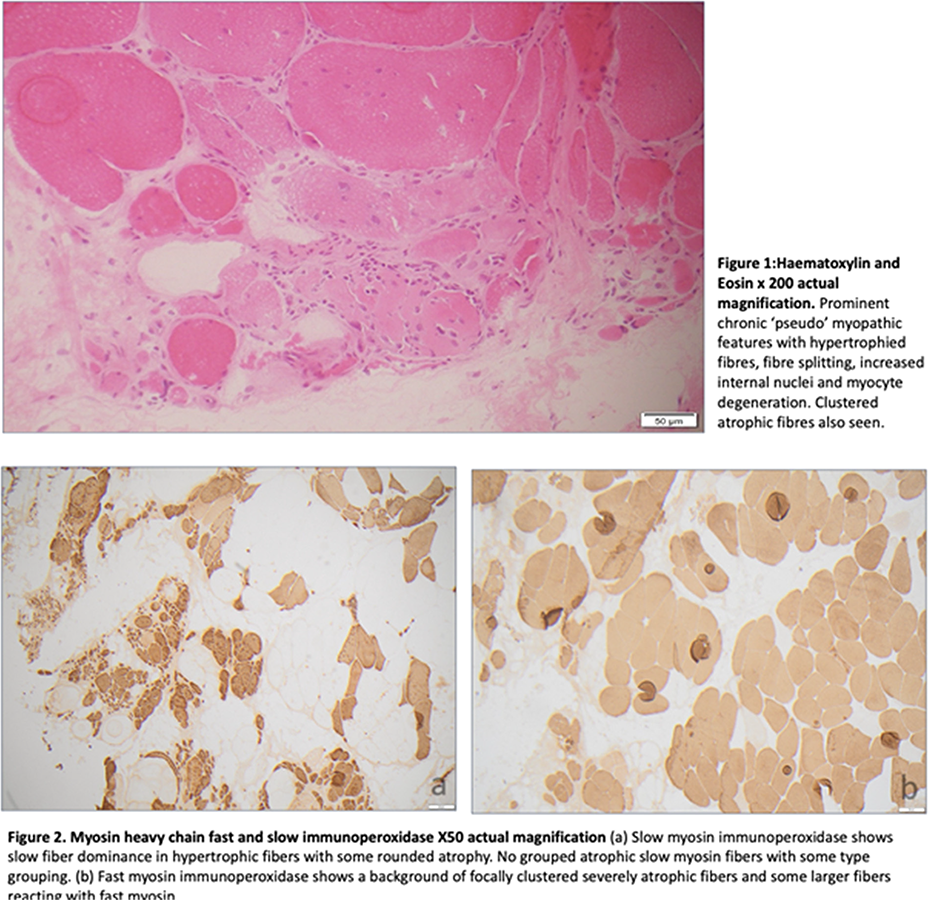
PP.133
PP.133 - Newborn Screening for Spinal Muscular Atrophy: A Pilot Study in Thailand
Prof. Oranee Sanmaneechai Sanmaneechai1, Assoc Prof. Surachai Likasitwattanakul1, Asist Prof Achara Sathienkijkanchai1, Somporn Liammongkolkul1, Theeraphong Pho-iam1
1Faculty Of Medicine Siriraj Hospital Mahidol University, Bangkok, Thailand
PP.134
PP.134 - Nusinersen for Spinal Muscular Atrophy: Experience from Qatar
1Sidra Medicine, Doha, Qatar
PP.135
PP.135 - Real-World Outcomes Following Onasemnogene Abeparvovec in Patients with SMA and Invasive Ventilatory Support
1Center For Experimental Neurotherapeutics, St. Jude Children’s Research Hospital, Memphis, United States, 2Department of Neurology, David Geffen School of Medicine at UCLA, Los Angeles, United States, 3Washington University School of Medicine, St. Louis, United States, 4Nationwide Children’s Hospital, Columbus, United States, 5The Ohio State University College of Medicine, Columbus, United States, 6Kobe University Graduate School of Medicine, Kobe, Japan, 7SMA Europe, Germany, 8Novartis Gene Therapies, Inc., Bannockburn, United States, 9Novartis Gene Therapies Switzerland GmbH, Rotkreuz, Switzerland, 10Department of Paediatrics, MDUK Oxford Neuromuscular Centre & NIHR Oxford Biomedical Research Centre, University of Oxford, Oxford, United Kingdom, 11Department of Pediatrics, Neuromuscular Reference Center, University and University Hospital of Liège, Liège, Belgium
PP.136
PP.136 - Gene Therapy with Onasemnogene-Abeparvovec (Zolgensma®) for Patients with SMA: Real-World Experience from Qatar
1Sidra, Qatar
All cases were diagnosed as having genetically confirmed SMA type 1 or 2 due to homozygous deletions of the SMN1 gene and two to three copies of the SMN2 gene.
37 children (92.5%) were treated within the 2 years eligibility criteria. The other 3 children were treated based on the weight criteria.
14 children were on “bridging therapy” with Nusinersen/Risdiplam until they receive Zolgensma. All, except one, were of type 1.
11 children were on ventilation support (27.5%); 10 were on non-invasive support.
23 children (57.5%) were orally fed.
One child had positive AAV9 antibodies. The test was repeated in 2 months and he seroconverted and did receive the gene therapy, safely.
No serious adverse side effect was observed in all treated children.
Children were enrolled on a consolidated, intensive, 3 months post-therapy rehabilitation program. 4 of the children on ventilatory support were successfully weaned off.
8 different nationalities were treated in Qatar.
Increasing number of children, from different countries, have benefited from this service.
We pioneered the concept of bridging therapy which has helped reducing the lost time waiting for Gene therapy treatment.
Our center was the hub for the SMA patients in the region ensuring children from other countries have access to it. This review has further strengthen the clinical efficacy of gene therapy and emphasize on the importance of early diagnosis of SMA through the national newborn screening program.
PP.137
PP.137 - Development of a National Guideline for the Implementation of Newborn Screening for Spinal Muscular Atrophy
1Sydney Children's Hospital Network and UNSW Medicine, INSW Sydney, Sydney, Australia
PP.138
PP.138 - Use of Gait Analysis and Wearable Metabolic System in Patient with SMA with Disease-Modifying Treatment
1Allied Health Department (Physiotherapy), Hong Kong Children's Hospital, Hong Kong, 2Physiotherapy Department, Tuen Mun Hospital, Hong Kong, 3Department of Paediatrics and Adolescent Medicine, The University of Hong Kong, Hong Kong
PP.140
PP.140 - Switching from Nusinersen to Risdiplam in Spinal Muscular Atrophy-12 Months Follow up in a Romanian Center
1NATIONAL UNIVERSITY HOSPITAL FOR CHILDREN NEUROREHABILITATION "DR NICOLAE ROBANESCU' ", BUCHAREST, Romania, 2University of Medicine and Farmacy “Carol Davila”, BUCHAREST, Romania
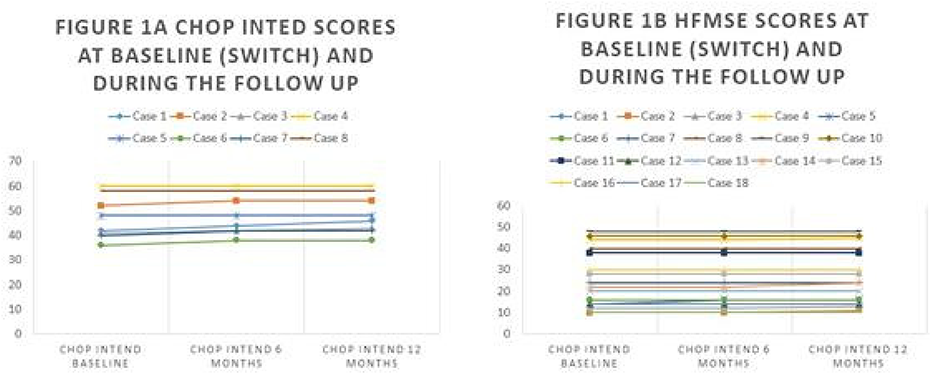
PP.141
PP.141 - Observational Case-Series of Adults with 5q-SMA Attending an Australian Neuromuscular Clinic After Deciding on Treatment
1Dept of Neurology, Royal North Shore Hospital, St Leonards, Australia, 2NeuRA Clinics, Neurodegenerative Service, Prince of Wales Hospital, Randwick, Australia
PP.142
PP.142 - A Systematic Review Describing Nutrition Outcomes of Disease Modifying Therapies in Spinal Muscular Atrophy
1Department of Nutrition, Dietetics and Food, Monash University, Melbourne, Australia, 2Department of Neurology, Royal Children's Hospital, Melbourne, Australia, 3Molecular Therapies Research, Murdoch Children’s Research Institute, Melbourne, Australia
Spinal muscular atrophy (SMA) is a degenerative neuromuscular disorder with profound nutritional implications. Disease modifying therapies (DMT) have improved life expectancy and motor function. This systematic review describes the impact of DMT on nutrition outcomes in SMA.
PP.143
PP.143 - Nutrition Outcomes in Children with Spinal Muscular Atrophy Type 1 Treated with Disease Modifying Therapies
1Department of Nutrition, Dietetics and Food, Monash University, Melbourne, Australia, 2Department of Neurology, Royal Children's Hospital, Melbourne, Australia, 3Molecular Therapies Research, Murdoch Children’s Research Institute, Melbourne, Australia
The nutritional implications of spinal muscular atrophy (SMA) type 1 are profound. Infants with SMA 1 can present with growth failure, gastrointestinal and swallowing dysfunction and require tube feeding. Disease modifying therapies (DMT) have improved life expectancy and motor function; however, few studies have investigated nutrition as a primary outcome of therapy. This retrospective cohort study compares nutrition outcomes of DMT in treated and untreated children with SMA 1.
Median (IQR) weight z-score at six months of age was -0.24 (-1.21, 0.09) for treated participants (n=7) and -2.98 (-3.32, -0.23) for untreated participants (n=7). All treated participants and 89% of untreated participants had dietetic input, enteral nutrition support was the most common intervention. Tube feeding was initiated in 82% of treated and 89% of untreated participants. Median (IQR) age at tube placement was 11.2 (2.2, 20.2) months and 6.2 (2.9, 8.4) months for treated and untreated cohorts respectively. Eighteen percent of treated participants were fed with a jejunal tube and 59% had gastrostomies. All tube-fed untreated participants were fed via nasogastric tubes. Of the fourteen treated tube-fed participants, four later ceased tube feeding, median (IQR) duration of feeding before cessation was 6.5 (1.0, 16.9) months. The median (IQR) number of dietetic reviews was 22 (10, 40) and 5 (4, 7.5) for treated and untreated participants respectively.
PP.144
PP.144 - Real-World Study of Nusinersen Effects in Adults with Spinal Muscular Atrophy Type 2 and 3
1Centre intégré universitaire de santé et de services sociaux de la Capitale-Nationale, Québec, Canada, Québec, Canada, 2Department of Clinical Neurosciences, Hotchkiss Brain Institute, University of Calgary, Calgary, Canada, Calgary, Canada, 3Groupe de recherche interdisciplinaire sur les maladies neuromusculaires (GRIMN), Centre intégré universitaire de santé et de services sociaux du Saguenay–Lac-St-Jean, Québec, Canada, Saguenay, Canada
PP.145
PP.145 - Cost Effectiveness of Newborn Screening for Spinal Muscular Atrophy in Australian Hospitals
1Department of Neurology, The Royal Children's Hospital, Melbourne, Australia, 2Department of Neurology, Sydney Children’s Hospital Network, Sydney, Australia, 3University of New South Wales, Sydney, Australia, 4Department of Neurology and Department of Metabolic Medicine, Perth Children's Hospital, Perth, Australia, 5School of Paediatrics and Child Health, University of Western Australia, Perth, Australia, 6Murdoch Children’s Research Institute, Parkville, Australia, 7Department of Paediatrics, The University of Melbourne, Parkville, Australia, 8Department of Neurology, Women’s & Children’s Hospital, Adelaide, Australia, 9Novartis Gene Therapies Switzerland GmbH, Rotkreuz, Switzerland, 10HTANALYSTS, Sydney, Australia, 11The George Institute for Global Health, Sydney, Australia, 12School of Medicine, University of Adelaide, Adelaide, Australia
PP.147
PP.147 - Genetic Testing in Neuromuscular Disorders at a Quaternary Referrals Centre in Melbourne, Victoria
1Alfred Health, MELBOURNE, Australia, 2Royal Melboune Hospital, Melbourne, Australia
Diagnosis was established in 57% of patients who had a SB or CMA, 35% patients who had GPS, 25% of patients who underwent WES and no patients on WGS. For patients with no prior genetic testing (n=15), diagnosis was made on SB in 4 patients, GPS in 4 patients and WES in 1 patient. For those with prior negative/inconclusive genetic testing (n=14), only 3 PV were identified through GPS. In patients with multiple genetic tests (n=6), where molecular techniques were combined with GPS, only one PV was identified. WGS was performed in three patients, but despite this no PV was identified.
1. Rosenberg A, et al. An evaluation of clinical presentation and genetic testing approaches for patients with neuromuscular disorders. Am. J. Med. Genet. Part A 2023, 191, 2679–2692.
2. Barp A, Mosca L, Sansone VA. Facilitations and Hurdles of Genetic Testing in Neuromuscular Disorders. Diagnostics (Basel). 2021 Apr 14;11(4):701.
3. Krenn M, et al. Next-generation sequencing and comprehensive data reassessment in 263 adult patients with neuromuscular disorders: insights into the gray zone of molecular diagnoses. J Neurol. 2024 Apr;271(4):1937-1946.
PP.148
PP.148 - Detection of STR Expansions on a Targeted Gene Panel using STRipy Improves Ataxia Diagnostics
1Centre for Medical Research, University of Western Australia, Harry Perkins Institute of Medical Research, Perth, Australia, 2Department of Diagnostic Genomics, Department of Health, PathWest Laboratory Medicine, QEII Medical Centre, Nedlands, Australia
Short tandem repeats (STRs) are repetitive DNA sequences with motifs between 2-6 bp that comprise approximately 7% of the human genome. Expansions of STR regions give rise to over 50 diseases with the majority demonstrating primary neurological or neuromuscular presentations. Despite the prevalence of STR expansion disorders, genetically diagnosing these conditions is complicated by a lack of efficient and comprehensive diagnostic screening approaches. For this reason, considerable efforts have been made to develop tools able to genotype STR expansions in short-read sequencing data. Recently, a tool called STRipy was released which provides a graphical user interface able to genotype loci from a catalogue of all known pathogenic STR expansions.
We tested the capacity for STRipy to analyse short-read sequencing data from comprehensive targeted gene panels for neuromuscular disorders within the Department of Diagnostic Genomics, PathWest. There are two primary panels, neurological and muscle, based on whether the gene is linked to primary neurological or muscular disease.
We tested STRipy on two versions of the ataxia neurological subpanel: version 6 and 7. Version 6 already included probes covering STR expansion loci contained within coding regions for four ataxia genes: CACNA1A, PPP2R2B, TBP, and NOP56. Additional probes targeting all relevant STR expansion loci were designed and included in version 7. All STR expansions detected by STRipy were validated and sized using NATA accredited PCR-based diagnostic techniques.
We tested a total of 418 and 67 patients with ataxia on version 6 and 7 of the ataxia subpanels, respectively. For version 6, 59 patients (14.1%) had reportable pathogenic variants, including nine patients with pathogenic repeat expansions detected by STRipy. This included expansions in CACNA1A (4), NOP56 (1), and two loci not included in the panel, ATXN1 (2), and ATXN3 (2), which we hypothesise is from nonspecific binding of probes containing large stretches of CAG repeats. For version 7, 15 patients (22.4%) had reportable pathogenic variants, including three repeat expansions detected using STRipy in ATXN1, ATXN2, and ATXN3. Therefore, STRipy contributed 15.3% and 20% of the solved cases from version 6 and 7 of the ataxia subpanel, respectively. STRipy performed best for loci with short pathogenic repeat thresholds, which is expected because the underlying genotyper (ExpansionHunter) can accurately size repeat expansions when there are reads spanning the repeat locus. STRipy could not provide accurate genotyping results for large complex repeat expansion loci. However, we demonstrate that STRipy can be used on a screening basis for some large pathogenic expansions, such as FGF14.
Here, we demonstrate that screening pathogenic STR expansions using STRipy on a neurological targeted gene panel improves the genetic diagnosis of ataxias in a clinical diagnostic laboratory. STRipy offers a simple computational approach that can comprehensively screen many loci in a single test. This is particularly beneficial for diagnosing atypical phenotypes or cases with onset earlier than expected, which can be troublesome to diagnose using labour-intensive single loci tests. Work is ongoing to explore the diagnostic utility of STRipy in the context of muscle related STR expansion disorders using the muscle panel.
PP.150
PP.150 - A Heterozygous AKR1B1 Missense Variant Segregating with Muscle Cramps in a Large Pedigree
1University Of Gothenburg, Gothenburg, Sweden, 2Capio Neuro Center, Carlanderska Hospital, Gothenburg, Sweden, 3Neuromuscular Centre, Department of Neurology, Sahlgrenska University Hospital, Gothenburg, Sweden, 4Department of Pediatrics, University of Gothenburg, Gothenburg, Sweden
In this study, we present a large Swedish family of four generations with 12 individuals suffering from painful muscle cramps with autosomal dominant inheritance. The muscle cramps usually start in childhood around 3-6 years of age and affect both large and small muscle groups, frequently in the calves, fingers and jaws. They occur spontaneously but may be provoked by exercise, even in non-exercised muscles. The cramps last from several minutes up to a few hours. Some of the affected individuals show slight muscle weakness. Several anti-epileptic drugs have been tried with minor or no effect.
Electromyography was performed in two patients showing hyperexcitability in motor axons, similar to cramp fasciculation syndrome (CFS), but no signs of myotonia or neuromyotonia. Muscle biopsy was performed in two patients demonstrating unspecific myopathic changes such as fiber size variation, few internalized nuclei, structural alterations, and few fibers lacking glycogen, but normal caveolin-3 staining.
Linkage analysis identified a 4.6 Mb chromosomal region with a maximum 2-point LOD score of 3.2 on chromosome 7. Further genetic analysis identified a missense variant in AKR1B1 located in the candidate region segregating with the disease. The variant c.61T>A was predicted by in silico programs to be damaging, CADD score 27, not identified in gnomAD and located in the NADP-dependent oxidoreductase domain. At protein level the conserved amino acid Tryptofan (W) at position 21 is replaced by an Arginine (R) (p.W21R).
AKR1B1, also known as aldose reductase, with highest expression in the adrenal gland. This enzyme plays a crucial role in several biochemical pathways, particularly in the metabolism of glucose and other sugars. The primary function of AKR1B1 is to catalyze the reduction of glucose to sorbitol, which is then further converted to fructose. This enzymatic reaction is important for the metabolism of glucose in various tissues. AKR1B1 has been implicated in various diabetic complications and other diseases where sugar metabolism plays a significant role.
Functional analysis by using recombinant AKR1B1 protein (W21R and wild type) was performed, and the allelic variant W21R presented very little residual enzyme activity using glyceraldehyde as substrate.
In conclusion, we present a large family suffering from autosomal dominant inheritance of muscle cramps and our studies suggest that a missense variant in AKR1B1 is causative of this disorder.
PP.151
PP.151 - Estimating Prevalence for Autosomal Recessive Neuromuscular Disorders in the Korean Population
1Department Of Neurology, Gangnam Severance Hospital, Yonsei University College Of Medicine, Seoul, Republic of Korea
Genetic neuromuscular disorders are clinically and genetically heterogeneous disorders that primarily affect peripheral nerve, muscle, and neuromuscular junctions. This study aimed to identify pathogenic variants, calculate carrier frequency, and predict the genetic prevalence of autosomal recessive neuromuscular diseases (AR-NMDs) in Korean population.
Typically, epidemiological studies for rare diseases utilize the clinical and genetic data of patients. However, recent research has been conducted to estimate the prevalence of AR-NMDs by calculating the carrier frequency from extensive genetic data of the general population. In our study, we analyzed carrier frequency and predicted genetic prevalence of NMDs using genetic information from genomic database of 916 individuals from the Korean general population. We selected 267 genes associated with autosomal recessive NMDs. We identified pathogenic/likely-pathogenic variants according to the guidelines of American College of Medical Genetics (ACMG) and Genomics and the Association for Molecular Pathology. Then, we assessed carrier frequency and predicted genetic prevalence of pathogenic variants in AR-NMDs.
After identifying the pathogenic variants using an algorithm, we calculated the carrier frequency and predicted the genetic prevalence of AR-NMDs in Korea. In total, 165 pathogenic variants were identified, including 76 literature verified and 89 manually verified variants. In a detailed evaluation of variant distribution within our study, we observed a predominance of truncating mutations among the manually classified pathogenic variants associated with AR-NMDs in the Korean population according to ACMG criteria. In Korea population, the carrier frequency of AR-NMDs is 27%. The predicted genetic prevalence of AR-NMDs was estimated to be 38 cases per 100,000 individuals in Korea population. The AR-NMD gene with the highest carrier frequency was DYSF (1.6%), followed by GAA (1.5%) and variant with the highest allele frequency was c.1250C>T in HEXB with 0.0076 in the Korea population.
Our study suggest that 27% of the Korean population are healthy carriers of at least one pathogenic variant that can causes AR-NMDs. Additionally, our study is the first study to analyze the prevalence of neuromuscular disorders in the Korean population according to causative genes.
PP.152
PP.152 - Detection of Structural Variation for Mitochondrial Genomes Through Long-Read Sequencing
1Department of Medical Genetics, National Taiwan Univeristy Hospital, Taipei, Taiwan, 2Department of Pediatrics, National Taiwan Univeristy Hospital, Taipei, Taiwan, 3Department of Neurology, National Taiwan Univeristy Hospital, Taipei, Taiwan, 4Department of Medical ResearchNeurology, National Taiwan Univeristy Hospital, Taipei, Taiwan
PP.153
PP.153 - Major Genetic Mutations Identified in Neuromuscular Disorders from a Single Center in Southern India
1National Institute of Mental Health and Neurosciences, Bengaluru,Karnataka, India
PP.154
PP.154 - RFC1 and FGF14 Diagnostics: Learnings from an Australasian Cohort
1Harry Perkins Institute of Medical Research, Centre for Medical Research, University of Western Australia, Perth, Australia, 2Neurogenetics Laboratory, Department of Diagnostic Genomics,PathWest, Perth, Australia, 3Genomics Pillar, Garvan Institute of Medical Research, Sydney, Australia, 4Centre for Population Genomics, Garvan Institute of Medical Research and Murdoch Children’s Research Institute, Sydney, Australia, 5School of Clinical Medicine, Faculty of Medicine and Health, University of New South Wales, Sydney, Australia, 6School of Computer Science and Engineering, University of New South Wales, Sydney, Australia, 7Department of Neurology, Royal Adelaide Hospital, Adelaide, Australia, 8Adelaide Medical School, The University of Adelaide, Adelaide, Australia, 9UWA Medical School, University of Western Australia, Perth, Australia, 10Neurology and Stroke Unit, Fiona Stanley Hospital, Perth, Australia, 11University of Queensland, Centre for Clinical Research, Herston, Australia, 12Department of Neurology, Fiona Stanley Hospital, Perth, Australia, 13Department of Neurosciences, Griffith University, Sunshine Coast University Hospital, Mount Gravatt, Australia, 14Neurogenetic Unit, Royal Perth Hospital, Perth, Australia, 15Neurology Department, Auckland City Hospital, Auckland, New Zealand, 16Centre for Brain Research Neurogenetics Research Clinic, University of Auckland, Auckland, New Zealand, 17Program in Medical and Population Genetics, Broad Institute of MIT and Harvard, Cambridge, USA
Over 50 neurological and neuromuscular diseases are currently associated with short tandem repeat expansions. As the number and complexity of these disorders continues to grow, accurate detection and sizing of these expansions will be essential for neurogenetic diagnostics. Repeat expansion disorders contribute to several ataxia phenotypes. Here we present our work uncovering the complexities and diagnostic implementation of two of the more common ataxia-associated repeat expansion disorders.
Cerebellar ataxia, neuropathy and vestibular areflexia syndrome (CANVAS) is a progressive, generally late-onset, neurological disorder associated with biallelic pentanucleotide expansions in intron 2 of RFC1. The locus exhibits substantial genetic variability, with multiple pathogenic and benign pentanucleotide repeat alleles previously identified. To assess prevalence and further investigate this heterogeneity we screened a cohort of 242 Australasian patients with neurological disease for biallelic RFC1 expansions. A combination of flanking and repeat-primed PCR (RP-PCR) was used to detect known pathogenic expansions within 15.3% (n = 37) of cases. Patients with inconclusive RP-PCR results also underwent targeted long-read sequencing. This uncovered a high proportion of complex alleles comprised of multiple distinct repeat motifs and identified three novel motifs within our Australasian cohort.
Large GAA expansions in the first intron of the FGF14 gene have recently been associated with late onset cerebellar ataxias. Long-read sequencing of the locus during the initial study identified the presence of large non-pathogenic expansions containing the alternate repeat motifs. We implemented a combination of flanking and long-range PCR to facilitate accurate sizing of normal and expanded alleles. Additionally, bidirectional RP-PCR was used to ensure the expanded allele consisted only of pure GAA repeats. An initial cohort of 11 patients with late onset ataxia phenotypes were screened. Large pathogenic expansions were detected in two patients.
To facilitate high throughput screening and increase diagnostic yield, additional probes for both RFC1 and FGF14 repeat loci were incorporated into the targeted neurological disease gene panel at PathWest. Through manual inspection of the binary alignment map (BAM) files and implementation of the program STRipy, we were able to identify patients with potential expansions. These samples were then cascaded through the PCR-based assays to confirm the presence of pathogenic alleles.
Our results show that pathogenic expansions in both RFC1 and FGF14 make a substantial contribution to neurological disease in the Australasian population. We outline workflows for molecular diagnostic testing that accommodates the genetic heterogeneity observed and incorporates screening of massively parallel sequencing data to improve diagnostic yield for patients.
PP.155
PP.155 - Diagnostic Yield of Whole Exome Sequencing for Neuromuscular Disorders: A Review of 12 Months’ Data
1NSW Health Pathology, Australia, 2School of Medical Sciences, University of Sydney, Australia, 3Sydney Local Health District, NSW Health, Australia
Whole exome sequencing (WES) has become the most efficient diagnostic tool in clinical diagnostic setting for neuromuscular disorders. Testing has become more accessible in Australia since the introduction of Medicare funded testing from late 2022. We conduct a review of 12 months’ data of our gene panel analysis from WES data in our laboratory using the Twist Alliance VCGS exome capture kit on the Illumina NovaSeq 6000 sequencing system. The gene panels range from neuropathies, neuromuscular diseases, ataxia, spastic paraplegia, dystonia and motor neurone disease. Gene list for each panel was established using literature search and public databases.
We have reported 475 cases and identified 167 patients with pathogenic/likely pathogenic variants/clinically relevant variants of uncertain significance (VUS). The diagnostic yield is around 35%, in line with literature. Most variants were identified under comprehensive neuromuscular gene panel and neuropathy gene panel.
Detected variants include single nucleotide variants, indels, copy number variations and short repeat expansion variants . Copy number variations such as PMP22, SPG7 and SPAST exonic deletions and duplications were confirmed by multiplex ligation-dependent probe amplification. Short repeat expansion variants, such as PABPN1 were validated with in-house fragment analysis. Family segregation, combined with clinical assessment were carried out to support the assessment of VUS if available. Selected variants were submitted to ClinVar, so far with 11 variants not previously submitted, and four variants with conflicting classification.
With fast turn-around time under 3 months for positive results, WES has proven to be a powerful first-tier clinical diagnostic tool for the clinical management of patients with suspected inherited neuromuscular disorders, for identification of at-risk family members and to inform reproductive risk. For WES-negative patients, they can be considered for whole genome sequencing where non-coding regions and structure variants can be examined.
PP.156
PP.156 - Muscle Biopsy: When is Still Useful in the Era of Next Generation Sequencing (NGS)
1Campus Pietro d'Abano,Neuromuscular Center,University of Padova, Padova, Italy, 2Institute of Anatomy, Llubljana, Slovenia
Muscle biopsy has been used for diagnostic and research purposes for a long period, while in the meantime muscle genetics has undergone an outstanding molecular revolution helping clinical knowledge. MRI muscle imaging is essential for diagnosing several musculoskeletal afflictions such as degenerative muscle diseases, muscle inflammatory diseases, muscle atrophy, and neurological-related disorders.
From a period where the cause of many neuromuscular disorders was unknown, in 40 years the discovery of many genes and related disorders occurred. The first progress was made through biochemical investigation.
This period started with the first discovery of metabolic disorders such as primary and secondary carnitine deficiency( OCTN2 or ETF- dehydrogenase deficiency), coenzyme Q deficiency, glycogenosis,polyglucosan body diseases, and related enzyme deficiencies that allowed then the treatment of such disorders. A great advancement was made in glycogenosis type 2 (GSD2) an autosomal-recessive disorder caused by the deficiency of the lysosomal enzyme acid α-glucosidase (GAA), which catalyzes the hydrolysis of α-1,4 and α-1,6 linkages of glycogen. GAA deficiency leads to the accumulation of free glycogen in cytoplasm or glycogen trapped in lysosomes and the expression of autophagosomes in various tissues. There is a clinical spectrum of different clinical phenotypes that ranges from the classic rapidly evolving form (Pompe) to a childhood and juvenile form and a more slowly progressive adult form, named "late-onset" Pompe disease (LOPD). Biochemical differences in GSD2 have been documented between infantile, childhood, and adult forms, in studies at Mayo Clinic.
Subsequently, since 2006 Enzyme Replacement Therapy (ERT) has been available and several trials in GSD2 with various types of ERT and chaperones have been done as well as trials with Next Generation ERT. The biopsy for GSD2 is of great importance for a correct diagnosis and is still the golden standard for ERT.
A great development has occurred in the realm of Muscular Dystrophies, with the discovery of dystrophin by reverse genetics in the '90s, since the identification of Becker muscular dystrophy by western blotting and female carriers by immunohistochemistry became possible for whom muscle biopsy is often needed. Various LGMD types can be identified by immunohistochemistry or western blotting. Histochemistry and ultramicroscopy appear useful in detecting cases of floppy infants such as the ones due to congenital fiber disproportion or centronuclear myopathy. Similarly in the case of inflammatory myositis, although MRI muscle imaging gives great information, a muscle biopsy is often necessary to confirm diagnosis before immunosuppressive treatment. Another field includes the Inclusion of Body Myositis and Critical Illness Myopathy for their diagnosis where the ultrastructure demonstrates myosin filament loss (Figure).
In the field of mitochondrial myopathies or neuropathies (MELAS; MINGIE; SANDO etc.), due to the cooperation of two genomes nuclear and mitochondrial, nerve and muscle biopsies for histopathological, ultrastructural, and biochemical measurement of respiratory chain complexes or mitochondrial nuclear-driven disorders are needed.
Examples of biopsies' utility are provided from toxic-metabolic cases such as cocaine inflammatory myopathies, colchicine, or statin myopathies.
The poster illustrates the practical use of muscle biopsy in disorders where it is still needed in the NGS era.

PP.157
PP.157 - NADMED: Novel REDOX Profiling to Enable Awareness of Clinical Relevance of NAD+ and its Derivatives
1Nadmed Ltd, Helsinki, Finland, 2University of Helsinki, Helsinki, Finland
PP.158
PP.158 - Pūnaha Io - the New Zealand NeuroGenetic Registry … AND BIOBANK
1Te Whatu Ora, Te Toka Tumai, Auckland City Hospital, Auckland, New Zealand, 2Centre for Brain Research, Neurogenetics Clinic, Waipapa Taumata Rau, University of Auckland, Auckland, New Zealand, 3Te Whatu Ora - Health New Zealand, Starship Children's Hospital, Auckland, New Zealand, 4Te Whatu Ora - Health New Zealand, Tauranga Hospital, Tauranga, New Zealand
Another barrier to treatment development in rare disease is the lack of biomarkers to track disease progression.
In 2022 The New Zealand Neuromuscular Disease Registry was developed into Pūnaha Io - the New Zealand Neuro-Genetic Registry & Biobank. This represented two changes.
1. A greater awareness of the needs of indigenous people and specifically Māori, the indigenous people of Aotearoa, New Zealand.
2. The development of a Biobank so that researchers could access samples already collected for them in these rare diseases.
Time-stamped clinical data are collected from participants in a number of different ways. Some are seen and assessed annually, some are seen sporadically or in preparation of a clinical trial. Some fill in disease specific questionnaires and some a basic registry-wide minimum dataset. Where possible these data are aligned with internationally agreed datasets.
Participants can also give biological samples, which are then linked to the patient’s condition at that time. Ethics is in place for collection of non-invasice samples (urine and tears), minimally invasive (e.g. blood) and invasive samples (e.g. skin, muscle or CSF).
Samples are deposited in an established biobanking facility, Te Ira Kāwai.. Sample collection, storage, and governance is according to procedures that incorporate Tikanga Māori and acknowledges Te Tiriti o Waitangi.
Muscle conditions:
Muscular dystrophies (346)
Spinal muscular atrophies (78)
Hereditary neuropathies (188)
Myotonic dystrophy (type 1 and 2) (238)
Metabolic myopathies, inflammatory myopathies
CNS diseases:
Huntington’s disease (220)
inherited ataxias (136)
inherited movement disorders, and hereditary spastic paraparesis (40)
Sample collection to date includes
Huntington’s (24)
Friedreich’s ataxia (11)
Spinocerebellar ataxia type 1 (3)
CMT1A (2)
Myotonic dystrophy type 1 (26)
Ultra-rare disorders (12)
Pūnaha Io has recruited patients for four international observational studies for participants with Friedreich’s ataxia, Huntington’s Disease, myotonic dystrophy type 1, CANVAS syndrome, and for clinical trials in myotonic dystrophy, congenital myotonic dystrophy, FSHD, mitochondrial myopathies, Becker muscular dystrophy, and CANVAS syndrome.
Samples have been used for one rare disease and for a study of genetic Parkinson’s disease.
These samples are available to researchers anywhere in the world and we invite anyone interested in these conditions who needs samples to contact us for a collaboration.
PP.159
PP.159 - Screen4Care as a Driver for European-Wide Electronic Health Record-Based Early Disease Detection
1Department of Neurology, University Medical Center Göttingen, Göttingen, Germany, 2Department of Medical Informatics, University medical center Göttingen, Göttingen, Germany, 3Medical Genetics Unit, Department of MedicMedical Genetics Unit, Department of Medical Sciences, University of Ferrara, Ferrara, Italy, 4Children's Hospital of Eastern Ontario Research Institute, Division of Neurology, Department of Medicine, The Ottawa Hospital; and Brain and Mind Research Institute, University of Ottawa, Ottawa, Canada, 5Pfizer Inc., Collegeville, PA 19426, Collegeville, US, 6Translational Molecular Imaging, Max-Planck-Institute for Multidisciplinary Sciences, Göttingen, Göttingen, Germany, 7Department of Pediatrics and Adolescent Medicine; University Hospital Erlangen; Friedrich-Alexander-Universität (FAU) Erlangen-Nürnberg, Erlangen, Germany, 8KPM Center for Public Management, University of Bern, Bern, Switzerland and Swiss Institute for Translational and Entrepreneurial Medicine (sitem-insel), Bern, Switzerland, 9Department of Social Medicine and Public Health, Faculty of Public Health, Medical University of Plovdiv, Plodiv, Bulgaria, 10 University of Southern Denmark, Odense, Denmark, 11Berlin Institute of Health at Charité, Berlin, Germany, 12Experimental and Clinical Research Center, Max Delbrück Center, Berlin, Germany, 13Department of Neuropediatrics and Muscle Disorders, Medical Center University of Freiburg, Faculty of Medicine, Freiburg, Germany, 14University College Dublin, National University of Ireland, Dublin, Ireland, 15Center for Research and Bioethics (CRB), Uppsala University, Uppsala, Sweden, 16Screen4Care consortium, Brussels, European commission
PP.160
PP.160 - Automatic Thresholding Methods for Muscle Ultrasound Echogenicity Analysis in Hereditary Neuromuscular Disorders
Dr. Kanellos C. Spiliopoulos1, Dr. Dimitra Veltsista1,
1Department of Neurology, University of Patras, Patras, Greece
PP.161
PP.161 - Creating an Immune Cell Atlas of the Peripheral Blood for Facioscapulohumeral Muscular Dystrophy (FSHD)
1Murdoch Children's Research Institute, Parkville, Australia, 2Royal Children's Hospital, Melbourne, Australia
The immune system is vital for effective skeletal muscle regeneration, with immune dysfunction known to impair regeneration and impact muscle wasting in chronic muscle disease. An immune infiltrate is present in muscle of FSHD patients, preceding the replacement of fat. However, very little is known about how the immune system influences disease pathology in FSHD. This lack of understanding has limited our ability to provide the best quality of care and hampers the development of the next generation of treatments for patients with FSHD.
To address this, we have recruited 23 patients with childhood onset FSHD through the neuromuscular clinic at Royal Children’s Hospital (Melbourne, Australia), as part of FSHD longitudinal outcome study (iFSHD-LOS). Blood samples have been collected and screened using high-dimensional flow cytometry to identify 55 subtypes of immune cells. Our initial analyses identified alterations in immune cell subtypes in patients with FSHD.
This study aims to create an immune cell atlas of patients with FSHD over the next 5 years using the iFSHD-LOS study. This will involve a yearly blood sample for immune cell phenotyping, transcriptomic and plasma cytokine analyses which will be coupled with key clinical outcome measures to determine severity.
PP.162
PP.162 - Implementation of an Advanced Care Planning Pathway in an Australian Tertiary Hospital Neuromuscular Clinic
1Department of Neurology, Concord Repatriation General Hospital, Sydney, Australia, 2Faculty of Medicine, University of Sydney, Sydney, Australia
There is limited evidence to direct the process of advanced care planning in the adult neuromuscular clinic, for conditions other than amyotrophic lateral sclerosis. In the paediatric literature, there are efforts to find evidence based processes to ensure the physical and emotional wellbeing of patients.1 As the treatment paradigm shifts for our adult cohort, there is a need for evidence to guide the best pathway to advanced care planning in neuromuscular disease. We report the results of a retrospective review of the current experience in an Australian tertiary hospital outpatient clinic. During study period of four months, over 150 patients were seen with a diverse range of conditions including Myotonic Dystrophy, Duchenne Muscular Dystrophy, Facioscapulohumeral Muscular Dystrophy, Spinal Muscular Atrophy, Charcot-Marie Tooth and Spinocerebellar Ataxia. There was a scarcity of documented advanced care plans and the planning experiences for those patients with existing documentation varied. Amongst those with documented advanced care plans there was a lack of consistent indicators for initiating this process. As a result of this review, consensus guidelines were formulated based on expert opinion and implemented in the neuromuscular clinic to address the need for a uniform holistic approach to advanced care planning in the adult neuromuscular cohort.
1. Willis TA, Macfarlane M, Vithlani R, et al. ‘Neuromuscular disease and advanced care plans: traffic light system’, BMJ Supportive & Palliative Care 2021;11:116.
PP.163
PP.163 - Adie's Syndrome: A Timeless Teacher of Neuro-Ophthalmology and Autonomic Disorders
1Shree Krishna Hospital Pramkhswami Medical College Bhaikaka University, Anand, India
1. Explore the historical significance of Adie's syndrome within the realm of classical neurology.
2. Highlight the ongoing relevance of Adie's syndrome in modern neurological practice.
3. Showcase the educational value of the syndrome in teaching the approach to neuro-ophthalmological and autonomic disorders.
1. Unilateral tonic pupil (right pupil 5.5mm, left 3mm)
2. Light-near dissociation
3. Segmental iris sphincter palsy
4. Hypersensitivity to 0.1% pilocarpine
5. Absent ankle reflexes on both sides
These findings closely resemble Adie's original 1932 description, showing the timeless nature of careful clinical observation. The patient's examination became a fascinating journey through neurological history:
- The tonic pupil brought to mind the pioneering work of Saenger (1902) and Strasburger (1902) who first described the condition.
- The light-near dissociation echoed Alexandridis's (1981) clarification of the differential innervation of the iris sphincter.
- The segmental iris palsy recalled Loewenfeld and Thompson's (1967) meticulous iris photography studies.
- Pilocarpine hypersensitivity was reminiscent of Scheie's (1940) research on denervation supersensitivity.
- The absent reflexes harkened back to Holmes (1931) and Adie's integration of pupillary and tendon reflex abnormalities.
This case served as an excellent teaching tool, demonstrating:
1. The importance of a systematic pupillary examination in approaching anisocoria.
2. The value of pharmacological testing in diagnosing pupillary disorders.
3. The significance of integrating seemingly unrelated neurological signs (such as pupillary and reflex abnormalities).
4. The role of careful follow-up in understanding the natural progression of neurological disorders.
As we navigate the complexities of modern neurology, Adie's syndrome stands as a guiding light.
PP.164
PP.164 - Ataxia, Hypo-odontia and Spasticity: a syndrome of Pol3a ( RNA Polymerase III) mutation
1MND Research Centre, Faculty of Medicine and Human Health Sciences, Macquarie University, Sydney, 2109, Australia
She had a slowly progressive course and a few falls over years with no active trauma but a distant left ankle fracture. Her swallow, upper limbs were normal with no fasciculations. Her father passed away with dementia aged 80 with gait issues, but he did not have any clear.
Her past medical history was that of hypercholerestemia and previous quiescent surgically resected breast cancer with no recurrence.
Her examination revealed lower limb spasticity with hyperreflexia with bilateral foot drop with normal upper limb power deep tendon reflexes and a wide based gait.
She had no issues with fertility. She had a history of breast cancer which is under control.
She had a nerve conduction study and EMG which did not reveal any myopathic pattern with normal sural sensory nerve potentials, normal posterior tibialis motor studies, normal common peroneal nerves but left peroneal decrease in amplitude. F waves in tibialis anterior were not prolonged.
Her MRI Revealed Bilateral basal ganglia T2 and T1 Hyperintensities and hippocampal T1 changes.
Of note she had issues with childhood dentition as a child and had poor quality teeth. ( Photo 1).
Genetic testing for hereditary ataxias and motor neuron diseases revealed a mutation in POLR3A in 2 loci: 1309C>T nonsense mutation introducing a stop codon at residue 437. The mutation is felt to be pathogenic.
Most cases are either severe and deadly by age the 3rd decade, and milder forms can happen to 4th or 5th decade. However, no cases in the literature have this mutation in older ages. Milder cases or undiagnosed later-age cases might be possible.
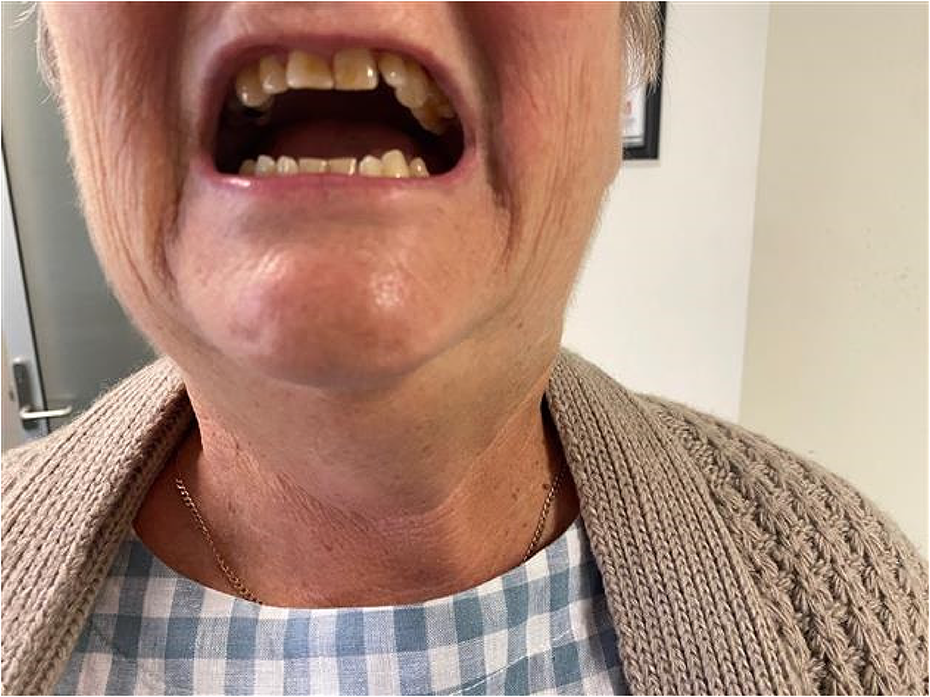
PP.165
PP.165 - Atypical Phenotype of Spinocerebellar Ataxia Type 25?
1Institute of Psychiatry And Neurology, Warsaw, Poland
Neurological examination of the patient showed flaccid paresis of the lower limbs with global atrophy and decreased reflexes, atrophy of hands muscle, shuffling and unstable gait, hushed speech. The slow progression of symptoms during 6 years of assessment was observed, mainly in coordination and imbalance and slight decline of SARA scale scores.
In brain Magnetic resonance imaging (at 26, 29 and 33 years of age) there was no atrophy of the cerebellum and any other changes had not been found. Magnetic resonance imaging of the cervical, thoracic and lumbal column did not revealed cause of the symptoms. Blood α-glucosidase, hexosaminidase A and B levels were normal. In neuropsychological examination no cognitive impairment was noted. ENMG repeated several times revealed axonal motor neuropathy, without myogenic changes. The muscle biopsy showed no changes characteristic for muscular dystrophies, myopathies or connective tissue.
Al-Maadid, Fatima PP.012
Lin, Cheng-Li PP.033
Meinke, Peter PP.042
Mihalatos, Markos OS.08.04
van Doorn, Pieter A. PP.085
Vedartham, Veena PP.046
Vu, Tuan PP.058
Zygmunt, Aldona OS.08.04
Aanismaa, Riikka PP.157
Ab Ghapar, Amirul Asyraf PP.081, PP.128
Aban, Inmaculada PP.060
Abicht, Angela PP.030
Abu-Baker, Aida OS.04.08
Abu-Rub, Mohammad PP.060
Adak, Volkan PP.047
Adams, Justine PP.013
Aggarwal, Rohit PP.028
Agha, Sofiane PP.085
Agolini, Emanuele OS.08.04
Aguzzi, Rasha OS.03.04, PP.058
Ahmed, Rebekah M. PP.104
Ahn, Sohyun PP.082
Akkari, Anthony OS.02.04, PP.111, PP.123, PP.124, SS.16.03
Akkari, P Anthony OS.02.05
Alecu, Iulian OS.06.03
Aleman, Alberto OS.01.08
Alexandri, Nektaria PP.059
Alfano, Lindsay Alfano OS.08.06
Alfano, Lindsay N. OS.04.05
Allan, Georgina OS.04.04
Allen, Jeffrey SS.32.02
Allen, Jeffrey A. OS.05.04, OS.05.08, PP.075, PP.085, PP.093, PP.094
Allen, Jeffrey A. OS.05.05
Allenbach, Yves PP.028
Almendra, Luciano PP.048
Al-Muhaizea, Mohammad OS.06.04
Altaf, Lina PP.034
Altamimi, Sadiq PP.093
Alves, Frauke PP.159
Alzahmi, Fatmah PP.083
Amato, Anthony SS.33.02, TC.04.02, TC.06.03
Amato, M PP.005
Amor, David PL.01.05
Amornvit, Jakkrit PP.084
Amrami, Kimberly K. OS.05.06
Amrami, Kimberly K. OS.05.07
Anbu, Balaji PP.017
Anbu, Balaji OS.04.06
Anderson-Smits, Colin PP.091
Anderton, Ryan PP.111
Andrea, Guja PP.003
Angelini, Corrado PP.050, PP.156, TC.07.05
Annane, Djillali OS.03.04, PP.058
Antozzi, Carlo OS.07.08
Apiwattanakul, Metha PP.023
Aragon Pinto, Catarina OS.05.06
Araújo, Alexandra PQC OS.06.04
Aravindakshan, Rajeev PP.027
Argov, Zohar SS.42.02, TC.01.03
Aronica, Eleonora PP.028
Arthur, Peter PP.011, PP.016
Arvan, Rokhand PP.102
Arvin-Berod, Clémence OS.09.06, PP.071, PP.072
Arvin-Berod, Clémence PP.070
Arvin-Bérod, Clémence PP.093
Asher, Damon R. OS.01.04
Ashhurst, Jasmine PP.104
Atassi, Nazem OS.05.08
Atassi, Nazem PP.074
Atchayaram, Nalini OS.09.08, PP.027
ATCHAYARAM, NALINI PP.026
Attarian, Shahram PP.094
Attarian, Shahram PP.053
Auwarter, Kristen PP.074
Avendaño, Javier PP.049
Awaya, Tomonari OS.02.07
Ay, Hakan PP.091
and the MGBase Study Group, . SS.43.03
Baars, Paul PP.028
Bakker, Anthony PP.016
Balsells, Sol PP.130
Bancerek, Joanna PP.091
Barnett-Tapia, Carolina OS.07.07, PL.04.02, PP.051, SS.43.03
Barthel, B PP.005
Basiri, Keivan PP.052, 076
Basiri, Matin PP.052
Baskar, Dipti PP.041, PP.109, PP.153
Bassez, Guillaume OS.01.05
Basta, Ivana OS.05.05
Bastenier-Boutin, Louis PP.144
Baten, Abdul PP.155
Bautista, Angelo PP.011, PP.016
Bayoumi, Ahmed PP.051
Bedlack, Richard PP.111
Beecher, Grayson OS.05.06
Behera, Alok PP.042
Beltran, Sergi OS.08.04
Ben Omran, Tawfeg PP.136
Ben Omran, Tawfeg PP.012
Benatar, Michael OS.08.07, SS.14.03
Benguerba, Kamal OS.06.05, PP.135
Benguerba, Kamal OS.06.07
Ben-omran, Tawfeg PP.134
Benslimane, Nesrine PP.073
Benusa, Savannah OS.08.03
Berini, Sarah OS.09.03
Berlowitz, David OS.04.03
Bernadete Dutra de Resende, Maria OS.04.06, PP.017
Bernardo, Roberto OS.06.03
Bernardo, Sofia PP.048
Bernes, Saunder OS.06.07
Bernes, Saunder PP.061
Bertini, Enrico OS.06.04, OS.08.04
Bevilacqua Rivas, Jorge OS.01.07
Beydoun, Said PP.065
Bhagat, Savita PP.112
Bielak, Michał PP.047
Billich, Natassja PP.142, PP.143
Birija, Lauretta PP.067, PP.068
Bischof, M. PP.145
Bisson, Camille OS.06.08
Bjaj, Shruti PP.035
Black, Michael PP.148
Black, Shawn PP.061
Blankart, Rudolf PP.159
Blankenbiller, Tricia PP.004
Bleecker, Jan De OS.07.08
Blemker, Silvia OS.08.03
Blum, Stefan SS.44.03
Bo, Ryosuke PP.135
Boehnlein, Marion OS.03.05
Boggs, Bryan PP.120, PP.121
Boon, Andrea HO.03.02, SS.09.04, SS.25.04
Boonmee, Nawarat PP.001
Borojevic, Branko PP.147
Borojevic, Branko PP.024
Boroojerdi, Babak OS.03.06, PP.057, PP.062
Bosch, E. Peter OS.09.03
Bossuyt, Patrick PP.028
Bowden, Emma PP.120, PP.121
Bowman, Tom PP.121
Brackx, Febe OS.09.06, PP.070, PP.071, PP.072
Brais, Bernard SS.05.02
Brajkovich, Elina OS.01.06
Bratkovic, Drago OS.01.03
Brauer, Edward PP.053
Bray, Paula PP.010
Braz, Luis PP.048
Breiner, Ariel OS.01.08
Bricalli, Barbara
Briggs, Caitlin PP.102
Bril, Vera OS.03.05, OS.07.04, OS.07.07, OS.07.08, PP.051, PP.053
Bril, Vera OS.03.04, PP.058, PP.065, SS.46.03
Brum, Marisa PP.048
Brusch, Anna PP.025
Bryen, Samantha PP.080
Buccella, Filippo PP.017
Buccella, Filippo OS.04.06
Buchanan, Christina PP.158
Budikayanti, Astri PP.089
Burden, Steven J PP.049
Bushnag, Areej PP.034
Bustos, Asunción PP.032
Butzkueven, Helmut SS.43.03
Buzkova, Jana PP.157
Buzzard, Katherine SS.43.03
Byrne, Barry J. OS.01.03
Cadour, Stephanie PP.075, PP.094
Cai, Zhao PP.097, PP.098
Cairns, Anita PP.006, PP.013
Callaghan, Brian PL.03.04, SS.28.04, TC.02.03, TC.08.05
Calvo, Rocío PP.130
Campbell, Craig OS.03.03
Campbell-McLean, Carolyn PP.100
Capani, Francisco PP.116
Carlins, John PP.078
Carneiro, Pedro PP.066
Carrera-García, Laura PP.130
Carroll, Antonia SS.26.04
Carroll, Kate PP.010
Caruthers, Marvin H. PP.001
Castelli, Jeff OS.01.03
Cavalli, Michele OS.01.08
Cerletti, Massimiliano PP.039
Chakraborty, Sandipana PP.108
Chan, Fiona SS.35.03
Chan, Helen KF PP.138
Chan, Nerita NC PP.138
Chan, Sophelia HS PP.138
Chan, Stephen WW PP.138
CHANDRIKA1, H PP.126
Chang, Thomas OS.09.05
Chang, Ting OS.05.04
Chao, Chi-Chao OS.02.06, OS.05.03
Chao, Hua-Chuan PP.043
Charfi Triki, Chahnez PP.039
Chary, Sowmya PP.131
Chawla, Tanushree PP.040, PP.105
Chen, Jessie PP.077
Cheong, Pak Leng PP.155
Cheung, Michael OS.04.03
Chien, Yin-Hsiu OS.06.03
Chien, Yin-Hsiu PP.152
Chikkanna, Dr Umesh PP.014
Chin, Michael PP.155
Chintalaphani, Sanjog PP.154
Choi, Byung-ok SS.26.03
Choi, Byung-Ok PP.078
Choi, Seok-Jin PP.106, PP.115
Choi, Young-Chul PP.036, PP.151
Choi, Yun Jung PP.036, PP.151
Chompoopong, Pitcha SS.24.03
Chopra, Abha OS.02.04
Chou, Michael PP.051
Chroni, Elisabeth PP.160
Chrościńska-Krawczyk, Magdalena PP.047
Chu, Shannon PP.155
Chung, Hye Yoon PP.054
Ciafaloni, Emma OS.08.07
Ciafaloni, Emma OS.01.04
Claeys, Kristl G. OS.01.03, OS.04.05
Claeys, Kristl G. PP.053
Clark, D. PP.145
Clark, Damian R PP.013
Clay, Rebecca PP.020
Clayton, Joshua OS.04.04
Clayton, Joshua OS.01.06
Cleland, James PP.158
Clemens, Andreas OS.08.04
Clemens, Paula SS.02.04
Clemens, Paula R. OS.01.03
Cocco, Giovanni PP.083
Cochrane, Thos PP.120
Cole, Rylee PP.022
Coles, Chantal PP.007, PP.161
Coles, Chantal PP.021
Colgan, Samantha PP.022
Collaborators Consortium Latin SEQ, + OS.08.05
Collins, Jessica SS.14.03
Collins, Michael SS.27.02, SS.30.02
Comi, Giacomo OS.04.05
Connolly, Anne OS.06.03
Conte, Raffaele PP.019
Conte, Raffele PP.003
Conyers, Joe PP.067, PP.068
Corbett, Andrew PP.131
Cortés-Vicente, Elena PP.053
COS Consortium, OS.01.07
Costa, Sara PP.048
Côté, Isabelle PP.144
Cotter, Megan PP.147
Cousins, Matthew OS.08.03
Cowley, Lorraine OS.08.05
Crane, Jordan OS.01.06
Crossman, Vanessa PP.007
Cruse, B PP.107
Cruz, Simao PP.048
Cunnigham, Chloe PP.147
Cutter, Gary PP.059, SS.43.03
Dabbous, Omar OS.06.05, OS.06.07
Dangouloff, Tamara OS.06.08, PL.01.04
Darin, Niklas PP.150
Daron, Aurore OS.06.08
Darras, Basil OS.06.03
Darton, Eddie OS.01.04
Das, Animesh PP.117
Dastur, Rashna PP.035
Davidson, Zoe OS.04.03, PP.009, PP.010
Davidson, Zoe PP.143
Davidson, Zoe E PP.142
Davis, Abby PP.022
Davis, Mark PP.006, PP.148, PP.154
Day, Bruce PP.092, PP.147
Day, John OS.06.03
Day, John W. OS.01.07
De Bleecker, Jan L. PP.085
De Bleecker, Jan L. OS.07.06, PP.065
De Roeck, Arne OS.05.05
De Valle, Katy PP.161
De Waele, Liesbeth OS.03.03
Deconinck, Nicolas OS.03.03, OS.07.03
Delicha, Eumorphia Maria PP.057
Della Marina, Adela PP.047
Delmar, Paul OS.06.08
Desai, Devangi PP.099, PP.163
Desai, Soaham PP.099, PP.163
Desai, Urvi PP.057
Desai, Urvi OS.01.07
Deveson, Ira PP.080, PP.154
Dewilde, Sarah OS.09.06, PP.070, PP.071, PP.072
Diasinos, Francine OS.03.04
Diaz Manera, Jordi OS.08.05
Diaz-Manera, Jordi OS.01.05
Díaz-Manera, Jordi OS.01.03
Dill, Larissa OS.02.05
Dimachkie, Mazen PP.022
Dimachkie, Mazen M. OS.01.03, PP.059
Dincq, Stéphanie PP.049, PP.119
Dion, Patrick OS.04.08
Dodd, Nigel OS.06.03
Doerflinger, Melanie OS.09.05
Dofash, Lein OS.04.04, PP.006
Donovan, J PP.005
Donovan, Joanne OS.08.06
Drummond, Kerri OS.07.03
Du, Bing PP.029
DuCharme, Olivia OS.08.03
Duda, Petra W. OS.03.06
Dugar, Ashish OS.03.03
Dyck, P. James B. OS.09.03
Dyck, Peter OS.09.03
Dyck, Peter J. OS.05.06
Dyck, P. James B. OS.05.06
Dyck, P. James B. OS.05.07
Dyck, Peter J. OS.05.07
Dyke, Jason PP.025
Dysgaard, Tina OS.05.04
de Valle, Katy PP.020, PP.021
de Visser, Marianne PP.028
Eagle, Michelle OS.08.06
Eaton, Susan PP.131
Edelweiss, Evelina OS.09.04
Eggenspieler, Damien OS.06.08
Eggers, Christian OS.05.04, OS.05.05
Eichinger, Kate PP.020
Einhorn, Moshe OS.08.04
Elbaum, Daniel PP.120, PP.121
Elkins, Jacob S. OS.01.04
Elkins, Jacob S. PP.015
Ellis, Melina PP.080
Engelstad, JaNean OS.05.06
Engelstad, JaNean OS.09.03
Ennamuri, Sravya PP.015
Eon, Victoria PP.120
Eppie, Yiu PP.100
Erbas, Yasemin PP.135
Eriguchi, Makoto PP.069
Euro, Liliya PP.157
Evans, Rachel OS.06.06
Evers, Sanne PP.028
Expósito-Escudero, Jésica PP.130
Fadli, Nurul PP.089
Fan, Pi-Chuan PP.152
Faragher, Mark PP.092
Farmakidis, Constantine OS.07.08, PP.022
Farnè, Marianna OS.08.04
Farrar, Michelle OS.06.03, OS.06.04, PP.013, PP.100, SS.04.03, SS.13.04
Farwell, Wildon OS.01.05, OS.03.03
Fattal-Valevski, Aviva PP.139
Favreau, Frédéric OS.09.07, PP.073
Fawzi Osman, Mahmoud PP.136
Fawzi Osman, Mahmoud PP.134
Faye, Pierre-Antoine OS.09.07, PP.073
Felix, Ana Carolina PP.048
Feng, Xue OS.08.03
Ferlini, Alessandra OS.08.04, PP.159
Fernandes, Angela PP.066
Fernández-García, Miguel Ángel PP.130
Fernandez-Torron, Roberto OS.01.07
Filosto, Massimiliano PP.019
Finkel, Richard OS.06.05, OS.06.07, PP.135
Finkel, Richard S OS.06.04
Flanigan, Kevin OS.03.03
Fletcher, Sue OS.01.06
Fletcher, Sue PP.122
Flynn, Loren OS.02.04, OS.02.05, PP.123
Flynn, Loren SS.16.03
Folland, Chiara PP.148
Fontoura, Paulo OS.06.04
Fontoura, Paulo OS.01.04
Forbes, Robin PP.013, PP.100
Forbes, Sean PP.015
Fortunato, Fernanda OS.08.04
Foster, Meredith OS.04.07
Freimer, Miriam OS.03.06, PP.057, PP.062
Frick, Glen OS.03.04, PP.058
Friedman, Seth PP.020
Gadaleta, Giulio PP.019
Gaki, Eleni OS.01.08, OS.06.04
Gamaarachchi, Hasindu PP.154
Gangatharan, Shane SS.08.02
Gangfuss, Andrea OS.04.05
Ganti, Rakhee PP.120, PP.121
Gao, Ping PP.002
Garbey, Marc PP.060
Garcia, David OS.07.05
Garg, Ajay PP.117
Garvey, Anthony Garvey OS.09.05
Gasser, Cyrill OS.06.06
Gattas, Susannah PP.149
Gayfieva, Maryam OS.03.05
Gazzerro, Elizabetta PP.159
Geara, Serge PP.114, PP.129, PP.164
Gelinas, Deborah PP.049, PP.119
Gelinas, Deborah PP.053
Genge, Angela OS.03.06, OS.07.05, PP.062, PP.119, PP.120, PP.121
Ghaoui, Roula PP.154
Ghia, Darshan PP.154
Gibson, Gregor PP.067, PP.068
GIGORAS, FLORIN PETRU PP.140
Girma, Helen PP.060
Gistelinck, Fien PP.056
Goedeker, Natalie PP.135
Goh, Khean Jin PP.081
Goh, Kim Ling PP.045
Gokhale, Sankalp PP.055, PP.102
Golagha, Mahshid PP.052
Gonzalez Chamorro, Alejandro OS.08.05
Gooch, Jonathan PP.131
Gooding, Rebecca PP.148
Goodman, Lizbeth PP.159
Gopalakrishnan, Sathej PP.059
Gordish Dressman, Heather OS.01.07
Goret, Marie OS.09.04
Gorni, Ksenija OS.06.04
Goto, Yuichi OS.02.07
Govindarajan, Raghav PP.057
Goyal, Vinay PP.105
Goyal, Vinay PP.040
Graefe, Adam PP.159
Gray, Emma SS.15.02
Greve, Bernhard OS.03.05
Griffiths, Mandie OS.04.03
Grimson, Fiona OS.03.06, PP.062
Gros-Louis, F. PP.038
Grosskreutz, Julian OS.03.05
Grosz, Bianca PP.080
Grounds, Miranda PP.011
Grounds, Miranda PP.018
Grudniak, Mariusz OS.07.08
Guerin, Annie PP.063
Guglieri, Michela PP.002
Guglieri, Michela, on behalf of DELIVER study investigators OS.03.03
Guidon, Amanda PP.060
Guptill, Jeffrey OS.07.05, OS.07.06, PP.053, PP.065
Guptill, Jeffrey PP.056
Guptill, Jeffrey T. PP.085
Guptill, Jeffrey T. OS.05.04, OS.05.05
Gurfinkel, Yuval OS.02.04
Guridi, Maitea OS.01.04
Guzman, Jennifer PP.078
Gwathmey, Kelly OS.05.05, PP.085
Gwathmey, Kelly PP.053
Habermann, Thomas M. OS.05.07
Habib, Ali A OS.07.04
Habib, Ali A. OS.03.05, PP.053
Hadden, Robert SS.30.02
Hagiwara, Atsuko OS.02.07
Hagiwara, Masatoshi OS.02.07
Hakim, Manfaluthy PP.089
Halani, Hiral PP.035
Hall, Jonathan PP.042
Halliday, Glenda PP.129
Hamwright, Marqus PP.093
Han, Baoguang OS.01.05
Hanajima, Ritsuko PP.113
Hanchaiphiboolkul, Suchat PP.101
Handschin, Christoph PP.047
Hannaford, Andrew PP.162, SS.19.04
Hansson, Mats PP.159
Hara, Nozomi OS.02.04, OS.02.05
Harbo, Thomas PP.094
Hareendran, Asha OS.07.04
Harms, Matt OS.01.07
Harsono, Adrian Ridski PP.089
Hartung, Hans-Peter PP.074
Harvey, Brittany PP.057
Hatch, Maya PP.020
Hatchell, Niall PP.067, PP.068
Hawkins, Neil OS.06.06
Heckmann, Janine SS.43.03
Hedberg-Oldfors, Carola PP.150
Heim, Andrew PP.022
Hemke, Robert PP.028
Henderson, Robert PP.149, PP.154
Henggeler, Caroline PP.031
Herr, Keira PP.067, PP.068
Hewamadduma, Channa OS.03.06
Hewamadduma, Channa OS.05.04, PP.062
Hill, Lorna PP.002
Hilsden, Heather OS.01.07
Hinckley, Sandy PP.120, PP.121
Ho, Stephanie PS PP.138
Hodari, Moana OS.07.06
Hodgkinson, Victoria PP.144
Hoffmann, Sarah OS.07.08
Holdbrook, Fred OS.01.03
Holla, Bharath PP.014
Honda, Hiroki PP.113
Honda, Naoto PP.113
Hong, Yoon-Ho PP.088, PP.106, PP.115
Hoshino, Yuki PP.069
Hosokawa, Motoyasu OS.02.07
Houweling, Peter OS.04.04, PP.007, PP.021, PP.161
Howard, James F. OS.03.06, PP.057
Howard, Mark OS.04.03
Howard Jr, James OS.03.04
Howard Jr, James F. PP.059, PP.085
Howard Jr, James F. OS.07.06, PP.056, PP.058, PP.065
Howard Jr., James PP.062
Howard Jr., James F. OS.08.07
Hristova, Daniela PP.053
Hsieh, Sung-Tsang OS.05.03, TC.04.03
Hsieh, Sung-Tsang OS.02.06
Hsu, Chia-Lang PP.152
Hsueh, Hsueh-wen OS.05.03
Hsueh, Hsueh-Wen PP.152
Hsueh, Sung-Ju OS.02.06
Hu, Menghan PP.002
Huang, Dan PP.061
Hübner, Miriam PP.159
Hughes, Richard A. C. PP.074
Hussain, Yessar PP.075
Hussain, Yessar PP.053
Hussain, Yessar M. OS.05.04
Hussain, Yessar M. OS.05.05
Huszti, Ella OS.07.07
Hutton, Elspeth OS.03.08, PP.092
Huynh, William PP.104
Iacoangeli, Alfredo SS.21.04
Ibrahim, Khalid PP.136
Ide, Toshihiro PP.069
Idiaquez, Juan OS.07.07
Iff, Joel PP.002
Imrell, Sofia PP.125
Indrawati, Luh Ari PP.089
Inoue, Yukako PP.069
Irving, Kathryn PP.013
Ishigaki, Keiko PP.061
Ishikawa, Fumi PP.079
Istas, Geoffrey OS.09.06, PP.070, PP.071, PP.072
Istas, Geoffrey OS.05.05
Iwasaki, Megumi PP.069
Iyadurai, Stanley PP.047
Jaber, Birgit OS.06.04
Jack, Dominic PP.059
Jain, Gayatri PP.077, PP.132
Jalali, Sedi PP.161
Jalilitabaei, Parastoo PP.162
James, Ian PP.111
James, Meredith OS.04.05
James, Meredith K. OS.01.07, OS.08.06
Jansson, Sonja PP.157
Javor, Andrija PP.059
Jehl, Jérémy OS.09.04
Jeon, Jaehyun PP.106
Jeong, Jae-gyun PP.078
Jiang, Ming PP.085
Jimenez, Rosa H. PP.053, PP.056
Johari, Mridul OS.04.04, SS.01.02
Johnson, Kristina PP.120, PP.121
Johnson, Nicole PP.131
Johnson, Shelley PP.017
Johnson, Shelley OS.04.06
Johnston, Patrick B. OS.05.07
Jones, Elizabeth OS.04.03
Jones, Kristi PP.013, PP.100
Jordan, Nerissa PP.154
Ju, Woohee PP.086, PP.087
Juel, Vern PP.060
Jun, Jaehyun PP.115
Jurkovic, Danijel PP.058
K, Karthik OS.09.08, PP.027
Kaminski, Henry PP.059, PP.060, SS.41.03, SS.43.02, SS.43.03
Kamperman, Renske PP.028
Kaneb, Hannah PP.121
Kang, Min K. PP.057
Kang, Minsung OS.02.02, PP.127
Kang, Peter B. PP.006
Kangas, Nina PP.131
Kao, Justin OS.09.05
Kapoor, Mahima PP.092, PP.147
Kaprielian, Roger OS.07.05
Karam, Chafic PP.075, PP.094
Karam, Chafic PP.065
Karcher, Keith OS.07.08
Kariyawasam, D.S. PP.145
Kasahara-Kiritani, Mami PP.067, PP.068
Katsuno, Masahisa OS.03.04, PP.058
Katz, Matthew PP.149
Katz, Natalie PP.020
Katzberg, Hans OS.07.07, PP.102
Katzenellenbogen, Sharona PP.139
Kava, M.P. PP.145
Kawai, Rira PP.079
KEERTHIPRIYA, M. S. PP.126
Keerti Priya, Madassu PP.109
Kennedy, Rachel PP.010
Kennerson, Marina PP.080, SS.26.03
Kennerson, Marina PP.155
Kenney, Allison OS.08.03
Khadilkar, Satish PP.035
Khan, Shaida PP.057
Kharma, Nawwaf OS.04.08
Kharrazi, Sohail OS.01.06
Khatri, Bhupendra PP.057
Khurana, Arushi OS.05.07
Kiernan, Matthew SS.19.02, TC.08.02
Kiernan, Matthew C. PP.104
Kilburn, N PP.005
Kim, Hyunjin OS.02.02, PP.127
Kim, Jong-Su PP.106, PP.115
Kim, Moon Seok PP.064
Kim, Seung Woo PP.054
Kim, Sohyeon OS.02.02
Kim, Soo-hyun PP.036, PP.151
Kimoto, Yasuhiro Kimoto PP.061
King, Anna SS.14.03
King, Rebecca L. OS.05.07
Kira, Jun-ichi SS.34.03
Kiriaev, Leonit PP.007
Kirschner, Janbernd PP.159
Kirschner, Janbernd OS.08.04
Kishnani, Priya S OS.04.07
Kishnani, Priya S. OS.01.03
Klaic, Marlena PP.010
Klein, Christopher OS.09.03
Klein, Christopher OS.05.06
Klein, Christopher J. OS.05.07
Kletzl, Heidemarie OS.06.04
Knieling, Ferdinand PP.159
Kodambail, Ananthapadhmanabha PP.109
Koelman, Johannes PP.028
Koike, Haruki PP.069
Kokaliaris, Christos OS.06.06
Koks, Sulev SS.21.03
Koks, Sulev OS.02.03
Komaki, Hirofumi OS.01.04
Koopmans, Petra OS.09.06, PP.070, PP.071, PP.072
Korajkic, Nadja PP.107
Kornberg, Andrew PP.013
Kornblum, Cornelia PP.030
Korobko, Denis OS.07.06
Koster, Matthew OS.09.03
Krefting, Dagmar PP.159
Krishna, Lekshmi PP.108
Krishnan, Dhayalen PP.128
Kubo, Jyunnpei PP.079
Kumar, Kishore PP.080
Kumar, Vijay OS.09.08
Kuntz, Nancy SS.39.02
Kunz, Wolfram S. PP.030
Kurosawa, Ryo OS.02.07
Kushlaf, Hani OS.01.03
Kusner, Linda SS.37.02, SS.37.03
Kuwabara, Satoshi OS.05.05
Kuwabara, Satoshi OS.05.04
kariyawasam, Didu Sanduni PP.137
Lai, Joyce HY PP.138
Laing, Nigel OS.01.06, OS.04.04, OS.09.05, PL.01.03, PP.006, PP.080, PP.111, PP.148, PP.154, TC.03.02
Lakhotia, Arpita OS.06.07
Lamb, Christopher OS.05.06
Lamont, Phillipa PP.154
Lao, Miko LM PP.138
Laporte, Jocelyn OS.09.04
Larcher, Leon PP.122
Larcher, Leon OS.02.05
Laurence, Craig PP.037
Lauria, Giuseppe OS.05.04
Laverty, Chamindra G. OS.04.05
Lavi, Revital PP.139
Lavrov, Andreea PP.057
Law, Meng OS.03.08
Le Bolay, Claire PP.059
Le Masson, Gwendal OS.05.04
LEANCA, MADALINA PP.008, PP.140
Leaver, Sharon PP.141
Lee, Aven SS.16.04
Lee, Bolam PP.061
Lee, Ni-chung PP.152
Leite, M. Isabel OS.03.06, OS.08.07
Leite, Maria Isabel PP.062, PP.066
Lejeune, Fabrice PP.073
LEKSHMI, KRISHNA PP.126
Lemaire, Jeannine PP.032
Leon-Astudillo, Carmen OS.01.04
Leong, Wai PP.025
Lesport, Quentin PP.060
Leung, Doris PP.020, SS.03.03, SS.09.03
Levine, Todd PP.057
Lewis, Lisa PP.055
Lewis, Richard A. OS.05.08, PP.074, PP.085
Lewis, Richard A. OS.05.05
Leypoldt, Frank OS.05.04
Li, Yuebing OS.07.06
Lia, Anne Sophie OS.09.07, PP.073
Liammongkolkul, Somporn PP.133
Lian, Pengchao PP.029
Liang, Christina PP.141
Likasitwattanakul, Surachai PP.133
Lilleker, James OS.01.05
Lin, Jie OS.05.04
Lin, Kon-Ping PP.043
Lin, Yen-Hung OS.05.03
Lindberg, Christopher PP.150
Ling, Leona E. OS.07.08
Lipowska, Marta OS.05.04
Liu, Emelline PP.017
Liu, Emelline OS.04.06
Liu, Li PP.085
Liu, Mingyao PP.029
Lo, Daryl PP.096
Lochmuller, Hanns OS.01.08, PP.041, PP.049
Lochmüller, Angela PP.037
Lochmüller, Hanns PP.159
Locklin, Jason OS.07.05
López Lobato, Mercedes PP.130
Lorentzos, Michelle OS.03.03
Loret, Camille OS.09.07, PP.073
Lovlom, Leif Erik OS.07.07
Lowes, Linda OS.01.07, OS.08.06
Lowes, Linda P. OS.04.05
Lucia, Alejandro PP.032
Luikinga, Sophia SS.18.03
Luo, Xiaodong OS.05.08
Luo, Xiaodong PP.074
Lyengar, Krishnan PP.006
Lynch, Matthew PP.006
MacDougall, J PP.005
MacDougall, James MacDougall OS.08.06
Macon, William R. OS.05.07
Macwan, Samir PP.057
Magargee, Scott OS.08.03
Mahajan, Anadi OS.06.06
Maki, Yoshimitsu PP.079
Malfatti, Edoardo OS.01.06
Mandrekar, Jay OS.05.07
Manera, Jordi Diaz OS.04.05
Manfrini, Marianna PP.015
Maniaol, Angelina OS.03.06
Mansukhani, Khushnuma PP.035
Mantegazza, Renato OS.03.05, OS.03.06, PP.059, PP.065
Mantegazza, Renato OS.03.04, OS.07.06, PP.053, PP.058
Manzur, Adnan PP.037
Margutti, Alice OS.08.04
Martin, Sylvia PP.159
Martinez, Clarida PP.120
Martínez-Salcedo, Eduardo PP.130
Martinez-Thompson, Jennifer M. OS.05.07
Mashchenko, Oleksandr PP.075, PP.094
Mason, Stefanie OS.01.04
Masschaele, Delphine PP.053
Masson, Riccardo OS.06.03
Mastaglia, Frank PP.111
Matalonga, Leslie OS.08.04
Mathews, Katherine OS.06.07
Mathieson, Sally PP.103
Matos, Anabela PP.048
Matthews, Emma SS.11.03
Matyas, Gabor PP.031
Mauermann, Michelle OS.09.03
Mauermann, Michelle L. OS.05.07
Mayhew, Anna OS.01.07
Mayhew, Anna OS.08.06
Mazen, Dimachkie SS.41.02
McCann, Heather PP.129
McCloskey, Conor PP.064
McCombe, Pamela PP.149, SS.16.04, SS.43.04
McDonald, Craig OS.08.06
McDonald, Craig M. OS.01.04
McEniery, David SS.43.04
McIver, Tammy OS.01.08
McLean, Catriona PP.024, PP.132
Medeiros, Luisa PP.048
Mehl, Lesa OS.06.03
Meienberg, Janine PP.031
Meisel, Andreas PP.053, PP.059
Meisel, Andreas OS.03.04, PP.058, PP.065
Mejzini, Rita OS.02.04, OS.02.05, PP.123, SS.16.03
Mełges, Anna PP.047
Melville, Zay OS.09.05
Mendell, Jerry OS.01.07
Mendell, Jerry R. OS.01.04
Mendoza, Meg OS.07.07, PP.051
Menon, Deepak PP.027, SS.46.03
Menon, Parvathi SS.19.03
Menon, Deepak OS.09.08
Mensova, Livie PP.031
Mercuri, Eugenio OS.06.03, OS.07.03, PP.002, PP.017
Mercuri, Eugenio OS.04.06
Mercuri, Eugenio M. OS.01.04
Merkies, Ingemar S.J. OS.05.08
Metz, Craig OS.02.05
Meyer, Stefanie PP.159
Meyers, Andrea PP.063
Meyers, Andrea PP.064
Meznaric, Maria PP.156
Micallef, Ivana N. OS.05.07
Middlehurst, Ben SS.21.02
Min, Younggi PP.086, PP.087, PP.088
Minicuci, Giacomo Maria PP.050
Minks, Eduard OS.07.08
MIREA, ANDRADA PP.008
MIREA, ANDRADA PP.140
Miressi, Federica OS.09.07, PP.073
Mishra, Ram Kinker PP.022
Mitrpant, Chalermchai PP.001
Mix, Chris OS.01.05, OS.03.03
Mole, Trevor OS.09.06, PP.070, PP.071, PP.072
Monahan, Gavin OS.04.04
Mongini, Tiziana PP.019
Morgan, Prue PP.103
Morgan, Sarah PP.025
Morris, Jacob OS.08.03
Morris, Katrina PP.162
Mozaffar, Tahseen OS.01.03, OS.01.07
Mroczek, Magdalena PP.031, PP.047
Munger, Kassandra PP.131
Muntoni, Francesco OS.01.04, OS.04.06, OS.07.03, PP.002, PP.017, PP.037, SS.02.02, SS.12.03
Murai, Hiroyuki PP.065
Murgod, Uday PP.046, PP.090
Murphy, Alexander P. OS.01.04, PP.015
Mythri, R. PP.108
Nagaishi, Yukiko PP.069
Nagpal, Chand PP.089
Nalini, Atchayaram PP.041, PP.108, PP.109, PP.153
Nalini, Atchayaram PP.112
NALINI, ATCHAYARAM PP.126
Narayanan, Ramesh SS.26.03
Nascimento, Andrés PP.130
Nascimento, Andrés OS.01.04
Nascimento Osorio, Andrés PP.017
Nascimento Osorio, Andrés OS.04.06
Nashi, Saraswathi OS.09.08, PP.027
Nashi, Saraswati PP.026, PP.108, PP.153
Natera-de Benito, Daniel PP.130
Nathani, Dev PP.045
Naylor, Maria OS.03.03
NEAGU, ELENA PP.008, PP.140
Needham, Merrilee OS.04.07, PP.025, PP.111
Nelson, Leslie OS.06.04
Ngo, Shyuan SS.18.04
Nguo, Kay PP.009
Nguo, Kay PP.142, PP.143
Ni, Xiao OS.07.03, PP.002
Nicholson, Garth PP.080
Nicolle, Michael OS.03.04, PP.058
Nicotra, Alessia PP.119
Nilsson, Staffan PP.150
Nishide, Masahiro SS.26.03
Nishino, Ichizo OS.02.07
Nizou, Angélique OS.09.07, PP.073
Noakes, Peter PP.018
Nobile-Orazio, Eduardo PP.094
Noguchi, Satoru OS.02.07
Nolting, Axel PP.059
Nordin, Magnus PP.150
North, Kathryn PP.007
Novelli, Antonio OS.08.04
Nowak, Kirsten PP.013
Nowak, Kristen PP.100
Nowak, Richard PP.060
Nowak, Richard J. OS.08.07
Nowak, Richard J. OS.07.08
Nunes, Adonay PP.022
Nury, Marianne PP.144
Nyoungui, Elisabeth PP.159
Ñungo, Carolina PP.130
O’Connor, Kevin C. PP.059
Obrien, Katie PP.142, PP.143
O'Brien, Katie PP.009
Octaviana, Fitri PP.089
Ogasawara, Masashi OS.02.07
Ogawa, Megumu OS.02.07
O'Gorman, Cullen PP.013, PP.100
O'Grady, Gina PP.158
Ohara, Hiroaki OS.02.07
Okumu, Sarah OS.01.08
Oldfors, Anders PP.150, TC.06.04
Olive, Montse OS.01.07
O'Malley, Anna PP.013, PP.100
Omer Ibrahim, Khalid PP.134
Omer Ibrahim, Khalid PP.012
ONOSE, GELU PP.008, PP.140
Opincariu, Irina PP.139
Orogun, Larry OS.07.03, PP.002
Ortez, Carlos PP.130
Ortez Gonzalez, Carlos OS.04.05
Osman, Mahmoud PP.012
Osredkar, Damjan OS.06.08
Otero-Losada, Matilde PP.116
Ottombrino, Silvia OS.08.04
on behalf of the Pompe Registry sites, OS.04.07
on behalf of the RAINBOWFISH Study Group, OS.06.04
Özkaynar, Pinar PP.028
Paik, Julie SS.33.05
Palace, Jacqueline PP.059
Palace, Jaqueline PP.049
Palfreeman, Laura OS.06.04
Pane, Marika OS.01.05
Pang, Yu Zhi PP.096
Pannullo, Francesca OS.07.04
Paradas, Carmen OS.01.07
Paradis, Angela PP.131
Paramalingam, Shereen SS.09.02
Parihar, Jasmine PP.117
Parinello, Guillaume OS.06.08
Parish, Ilia PP.022
Park, Hyung Jun PP.036, PP.151
Park, Jenny Y PP.063
Park, Jenny Y. PP.064
Park, Jin-Mo OS.02.02
Park, Jin-Sung OS.02.02, PP.127
Park, Jin-Sung OS.07.08
Park, Susanna SS.29.02
Parmar, Jevin PP.006, PP.080
Parmova, Olesja PP.031, PP.044
Pascuzzi, Robert M OS.07.04
Pasnoor, Mamatha PP.022
Pasnoor, Mamatha PP.065
Pasutharnchat, Nath PP.084
Patcharatrakul, Tanisa PP.084
Patel, Shilpan OS.09.05
Patterson, Kristina R PP.063
Patterson, Kristina R. PP.064
Pauline Joseph, Joyce PP.128
Pauset, Amandine OS.09.07
Peacock, A. PP.145
Pegoraro, Elena OS.01.07
Pellicci, Daniel PP.161
Pelosi, Luciana OS.09.05
Pereira, Nolette PP.006
Perez-Lloret, Santiago PP.116
Perez-Siles, Gonzalo SS.26.03
Peric, Stojan PP.055, PP.075, PP.093, PP.094
Peric, Stojan PP.065
Perlini, Luca PP.038, PP.039
Persson, Emma PP.075, PP.093, PP.094
Pestronk, Alan OS.01.07
Pfaff, Abigail OS.02.03, SS.21.03, TC.03.04
Pham, Xiuxian PP.024
Phan, H PP.005
Phan, Han OS.03.03, OS.08.06
Philips, Glenn PP.119
Pho-iam, Theeraphong PP.133
Pinho, Salome S PP.066
Pini, Jonathan OS.01.08
Pinós, Tomàs PP.032
Pinto, Catarina Aragon OS.09.03
Pinto, Marcus OS.05.06, OS.09.03
Pinto, Maria Joao PP.048
Pitout, Ianthe PP.122
Po, Kimberly PP.110
Podhorna, Jana PP.085
Podhorna, Jana OS.07.06
Poh, Chien Yen PP.013
Polavarapu, Kiran PP.041, PP.109, PP.153
Poleur, Margaux OS.06.08
Polzer, John PP.121
Polzer, John PP.120
Pompilio, Giulio PP.038
Poonsub, Narisa PP.001
Potter, Rachael A. OS.01.04
Potters, Wouter PP.028
Poulose, Prashanth OS.09.08, PP.027
Pradhan, Raj Kumar PP.112
Prior, David OS.04.03
Priya, Keerthi PP.108
Proud, Crystal OS.01.04
Proud, Crystal M. OS.04.05
Prouzet-Mauléon, Valérie OS.09.07
Puig-Ram, Cristina PP.130
Puma, Angela OS.01.08
Punga, Anna PL.04.03
Pureesatien, Punchai PP.084
Pyromali, Ioanna OS.09.07, PP.073
Qashqari, Hebah PP.034
Qi, Cynthia Z. OS.07.05
Quarracino, Cecilia PP.116
Querol, Luis OS.05.08, PP.074, PP.075, PP.093, PP.094
Querol, Luis OS.05.05
R Yerra, S PP.107
Raaphorst, Joost PP.028
Rabbia, Michael OS.06.04
Rainey, Amy PP.004
Rajabally, Yusuf PP.088
Raju, Dheeraj OS.06.07, PP.135
Raju, T.R. PP.112
Raju, Trichur R PP.108
RAJU, TRICHUR R PP.126
Ram, Ramesh OS.02.04
Ramakrishna, Kishore Kumar PP.014
Ramchandren, Sindhu OS.07.08, PP.061
Rampal, Nishi OS.08.07
Ramya, V. PP.108, PP.112
Rathnakara, Ranjitha PP.090
Ravenscroft, Gianina OS.01.06, OS.04.04, OS.09.05, PP.006, PP.080, PP.148, PP.154, SS.01.03, SS.12.02
Ray, Soma OS.01.05, OS.03.03
Rea, Sarah OS.02.04
Reddel, Stephen SS.43.03
Reddy, Divya PP.004
Reid, Carol OS.01.04, PP.015
Reilly, Mary PL.03.03, SS.26.02
Reimann, Jens PP.030
Reis, PP.080
Reiss, David OS.09.04
Rejdak, Konrad PP.059
Rejmer, Protazy PP.031
Reklis, Lindsay PP.004
Remiche, Gauthier TC.03.05
Reyna, Sandra OS.06.05, PP.135
Reyna, Sandra OS.06.07
Reynolds, Molly PP.162
Ricci, Giulia PP.003, PP.019
Richard, Laurence PP.073
Richards, A PP.107
Riem, Lara OS.08.03
Ringbauer, Manuela PP.091
Ringland, Ciara PP.068
Ringland, Ciara PP.067
Roberts, Mark OS.01.03
Rochefort, Daniel OS.04.08
Rodesch, Matt OS.08.04
Rodino-Klapac, Louise R. OS.01.04, PP.015
Rodrigue, Xavier PP.144
Rodrigues, E PP.107
Rodrigues, Edrich OS.03.08, PP.024, PP.092, PP.147
Rodrigues, Miriam PP.154
Rodrigues, Miriam PP.158
Rodríguez, Gabriel Eduardo PP.116
Rodriguez Gonzalez, Carlos PP.150
Rodrigures, Miriam OS.09.05
Rogers, Mary Louise SS.14.03
Rojana-udomsart, Arada PP.023
Rojavin, Mikhail OS.08.07
Romero, Norma OS.01.06
Roos, Andreas PP.047
Rostedt Punga, Anna OS.08.08
Röttger, Richard PP.159
Rouleau, Guy OS.04.08
Rovini, Amandine OS.09.07
Rowe, Dominic PP.114
Rowe, Dominic B PP.129
Rowe, Dominic Brock PP.164
Rowe, Peter W PP.013
Roxburgh, Richard OS.01.05, OS.09.05, PP.154, PP.158, TC.03.03
Rubin, Devon OS.09.03
Rubino, Samantha PP.121
Rufibach, Laura OS.01.07
Russell, A PP.005
Russo, Ali PP.022
Sacca, Francesco OS.07.06
Sacconi, Sabrina OS.01.08, OS.03.05
Sagi, Liora PP.139
Sahagian, Gregory PP.053
Saier, Christina OS.08.04
Saito, Kayoko OS.06.05
Sako, Yukiya OS.02.07
Salazar, Rachel OS.07.03, PP.002
Salmon, Kristiana PP.120, PP.121
Salmon, Lucinda PP.147
Samba, Djimde PP.146
Samuelsson, Lena PP.150
Sanchez Riera, Carles PP.039
Sanga, Panna OS.07.08
Sanmaneechai, Oranee Sanmaneechai PP.133
Sano, Misako OS.06.05
Sansen, Stefaan OS.08.04
Sansone, Francesco PP.003, PP.019
Sansone, Valeria OS.01.05
Santalla, Alfredo PP.032
Santorelli, Filippo Maria PP.003
Santos, Ernestina PP.048
Santos, Ernestina PP.066
Santos, Telma PP.048
SARASWATI, NASHI PP.126
Sarkar, Nisha PP.108, PP.112
Sarkozy, Anna PP.037
Sasidharan, Sandhya PP.022
Sathienkijkanchai, Achara PP.133
Sathyaprabha, Talakad N. PP.108
Sathyaprabha, Talakad. N. PP.112
Savic, Natasa PP.062
Saw, Jacqui PP.025
Sawahreh, Mohammad PP.012
Schafer, Balazs PP.001
Schara-Schmidt, Ulrike OS.01.04
Schee, Jie Ping PP.081
Schepers, Josef PP.159
Schoser, Benedikt OS.01.03, OS.01.05, OS.04.07
Schoser, Benedikt PP.042
Schultz, David SS.14.03
Scott, David Alexander OS.06.06
Screen4Care consortium, . PP.159
Scriba, Carolin OS.09.05, PP.148, PP.154
Scully, Tim OS.04.03
Seetam, Kumar PP.041
Seferian, Andreea M. OS.07.03, PP.002
Sehinovych, Ihor OS.07.03, PP.002
Seijka, Tim PP.092
Selvatici, Rita OS.08.04
Semeryak, Orest PP.039
Seo, Incheol OS.02.02
Seok, Hung Youl OS.02.02
Serra, Elizabeth PP.063
Serrano-Pons, Jordi PP.032
Servais, Laurent OS.06.04, OS.06.07, PP.002, PP.135
Servais, Laurent OS.06.08
Sevilla, Teresa OS.07.08
Shahrizaila, Nortina PP.081, SS.22.04
Sharma, Alok OS.09.08
Sharma, MC PP.040
Sheehan, Gerard OS.04.05
Sheehy, Tim PP.103
Shelly, Shahar OS.03.07, OS.05.06
Shepheard, Stephanie SS.14.03
Sheremeta, Chynna-Loren PP.018
Shichijo, Chika PP.069
Shieh, Perry OS.03.03, PP.135
Shieh, Perry B. PP.057
Shilling, Rebecca PP.049, PP.119
Shin, Ha Young SS.35.02
Shin, Ha Young PP.054
Shin, Jin-Hong OS.01.07
Shin, Ha-young PP.082
Shouman, Kamal OS.09.03, SS.24.04
Shtamler, Anna PP.139
Sia, Trinh PP.103
Siciliano, Gabriele PP.003, PP.019
Siddiqi, Zaeem OS.07.05
Sideris, Eleftherios OS.06.06
Sidiropoulos, Paris OS.01.08
Silvestri, Nicholas J PP.063
Simmonds, Jacob PP.037
Simons, Cas PP.080
Simu, Mihaela-Adriana OS.05.04
Singh, Bikram OS.04.03
Singh, Puneet PP.057
Singh, Rajesh Kumar PP.117
Singh, Teji OS.01.04
Sinthuwong, Chaichana PP.101
Sitaraman Das, Sheela OS.01.03
Sivakumar, Kumaraswamy OS.07.08
Skolka, Michael P. OS.05.07
Sleutjes, Boudewijn T.H.M. PP.119
Smilowski, Marek OS.07.08
Smith, N.J.C. PP.145
Smith, Robert OS.02.05
Smithuis, Frank PP.028
Smythe, Mark PP.018
Soliven, Betty PP.060
Sommer, Claudia OS.05.08
Sonoo, Masahiro SS.22.03
Soontrapa, Pannathat OS.09.03
Sparks, Susan OS.04.07
Spiliopoulos, Kanellos C. PP.160
Spinner, Robert J. OS.05.06
Spinner, Robert J. OS.05.07
Sproule, Douglas PP.004
Sreeram, Krishna Mythili PP.046
Stark, Richard PP.092
Statland, Jeffrey PP.020
Stavenhagen, Jeffrey PP.055, PP.102
Steeland, Sophie OS.07.06, PP.056
Stefanov, Rumen PP.159
Stepniak, Iwona PP.165
Sterky, Fredrik H PP.150
Stevanovski, Igor PP.080, PP.154
Stevenson, Herb OS.04.05
Steyn, Frederik SS.18.02
Stojkovic, Tanya OS.01.07
Stoll, Marion PP.155
Strathdee, James PP.092
Straub, Volker OS.01.07, PP.002, PP.015, SS.12.04, TC.09.04
Strauss, Kevin OS.06.03
Strijbos, Paul OS.06.08
Strober, Jonathan PP.061
Sturtz, Franck OS.09.07
Su, Mao-Yuan OS.05.03
Suanprasert, Narupat OS.05.07, PP.101
SUBRAHMANYAN1, V. R. PP.126
Sumbul, Anne PP.049
Sumbul, Anne PP.053
Summer, Dave OS.06.04
Sun, Hong OS.07.08
Sun, Hong PP.061
Sung, Jung-Joon PP.086, PP.087, PP.088, PP.106, PP.115
Sung, Jung-joon PP.082
Suresh, Niraja OS.05.04
Sutherland, C. Simone OS.06.06
Suzuyama, Kohei PP.069
Swangkaew, Saowanee PP.001
Swart, Grace OS.05.06
Szabo, Lena OS.06.08
Szczesniak, Dominika PP.165
Szmyd, Gabriela PP.093
T’joen, Caroline PP.065
Takahashi, Ryosuke OS.02.07
Takashima, Hiroshi PP.069, PP.079
Talaga, Anna PP.004
Talakad, Sathyaprabha PP.126
Talbot, Kevin SS.16.02
Talloen, Willem PP.119
Tan, Binghe PP.029
Tan, Cheng Yin PP.081
Tanaka, Koya PP.069
Tanboon, Jantima PP.023
Tang, Fengming OS.08.07
Tarancón, Thaïs OS.07.04
Tawil, Rabi PP.020
Taylor, C. PP.145
Taylor, Rachael OS.09.05
Taylor, Rhonda OS.01.06, OS.04.04
Taylor, Sean OS.09.03
Termglinjan, Thanes PP.023
Terrill, Jessica PP.011, PP.016
Teshima, Rie OS.06.05
Thaler, L PP.005
Theunissen, Frances PP.111, PP.124
Theunissen, Francess SS.16.03
Tierney, Anna PP.025
Tiong, Chrystal PP.007
Tizzano, Eduardo OS.06.07
Tomas, Doris OS.02.05
Tonacci, Alessandro PP.003, PP.019
Topf, Ana OS.08.05
Torrente, Yvan PP.038, PP.039
Torri, Francesca PP.003
Toscano, Antonio OS.01.03, OS.04.07
Treanor, Darran OS.04.03
Tripathi, Manjari PP.117
Triplett, James SS.28.02
Trivedi, Jaya SS.11.04
Truby, Helen PP.009
Tseng, Ping-Huei OS.05.03
Tsioutsias, Irene PP.011, PP.016
Tsuchida, Ken OS.06.05
Tsumura, Keisuke PP.069
Tu, Sicong PP.104
Tulinius, Már PP.017
Tulinius, Már OS.04.06
Turcq, Béatrice OS.09.07
Turner, Bradley OS.02.05, PP.124
Turner, Christopher OS.01.05
Udd, Bjarne TC.02.04
Udwani, Sachin PP.163
Uemoto, Mika PP.113
Ulrichts, Peter PP.085
Ulrichts, Peter PP.065
Urbano Martín, Mario PP.130
Utsugisawa, Kimiaki OS.03.05, OS.03.06, OS.08.07, PP.062
Utsugisawa, Kimiaki OS.07.06, PP.065
Uzawa, Akiyuki PP.061
Van Bragt, Tonke PP.049, PP.119
Van de Steen, Oliver PP.075, PP.093
Van de Steen, Olivier PP.094
Van de Walle, Inge PP.075, PP.093, PP.094
Van der Walt, Anneke SS.43.03
Van Doorn, Pieter OS.05.08, PP.074
Van Doorn, Pieter A. OS.05.08
Van Hoomissen, Iris PP.075, PP.093, PP.094
Van Hoorick, Benjamin OS.09.06, PP.070, PP.071, PP.072
Van Hoorick, Benjamin OS.05.05, OS.07.06, PP.065
Vandenborne, Krista PP.015
Vanderkelen, Mark OS.03.06, PP.062
Vanhauwaert, Roeland PP.049, PP.119
Vaquero, Fernando PP.032
Varghese, Anu Mary PP.112
Varin, Luc OS.04.08
Varon, Matthew PP.022
Vaziri, Ashkan PP.022
Vedartham, Veena PP.090
Veedu, Rakesh PP.122
Veerapandiyan, Aravindhan OS.01.04
Veiga, Andreia PP.048
Veltsista, Dimitra PP.160
Vengalil, Seena OS.09.08, PP.027, PP.041, PP.108, PP.109, PP.153
VENGALIL, SEENA PP.026, PP.126
Verhamme, Camiel PP.028
Verma, Kunal PP.147
Vibha, Deepti PP.117
Vicente, Manuel M PP.066
Vidmar, Suzanna OS.04.03
Vijayalakshmi, K PP.112
Vijayalakshmi, K. PP.108
VIJAYALAKSHMI, K PP.126
Vijayan, Joy SS.39.03
Villa, Luisa OS.01.08
Vincent, Angela SS.44.04
Vissing, John OS.01.07, OS.03.05, OS.07.04, OS.08.06, OS.08.07, PP.055
Vlodavets, Dmitry OS.06.04
Vo, Christina OS.01.06
Vola, Beatrice PP.038
Vola, Beatrice PP.039
Vrščaj, Eva OS.06.08
Vu, Tuan OS.03.05, OS.03.06, OS.07.04, PP.062, PP.085
Vu, Tuan OS.03.04, OS.07.06, PP.061, PP.065
Vu, Tuan OS.07.08
Vucic, Steve PP.080, SS.26.03
Vucic, Steven SS.43.04
Vujcic, Miodrag PP.075, PP.093, PP.094
van den Berg, Leonard H PP.119
van der Kooi, Anneke PP.028
van der Pol, W. Ludo PP.075, PP.094
van Doorn, Pieter A. OS.05.05, PP.074
van Eijk, Ruben P.A. PL.02.04, PP.119, SS.14.02
van Engelen, Baziel OS.01.05
van Leeuwen, Ester PP.028
van Schaik, Ivo PP.028
Waldrop, Megan PP.135
Walker, Adam SS.14.03
Walker, Michaela PP.020
Wallis, Mathew PP.080
Wallstroem, Erik OS.05.08, PP.074
Walter, Glenn PP.015
Walter, Hannah PP.028
Wandel, Christoph OS.01.04
Wang, Dazhe OS.03.03
Wang, Nasha PP.131
Wang, Stella OS.07.07
Wang, Xiaobing PP.029
Wang, Xiaochen PP.029
Warrington, Kenneth OS.09.03
Watanabe, Osamu PP.079
Watanabe, Yasuhiro PP.113
Weatherley, Emma OS.08.03
Webb, Thomas PP.067, PP.068
Wei, Cheng-Yu PP.033
Weisburd, Ben PP.154
Weiss, Michael D. OS.03.06, PP.057
Weizman, Anat PP.139
Wen, Bing SS.10.04
Weng, Wen-Chin PP.152
Werner, Christian OS.04.06, PP.017
Weyand, Cornelia OS.09.03
Wiendl, Heinz OS.07.06
Wilk-Durakiewicz, Ewa PP.002
Wilkins, H. Jeffrey PP.022
Willcocks, Rebecca PP.015
Wilton, Steve PP.124, SS.02.03
Wilton, Steve D OS.02.05
Wilton, Steve D. PP.001
Wing, J PP.005
Winkel, Antony PP.154, SS.45.04
Wiratman, Winnugroho PP.089
Wittig, Ilka PP.030
Wolf, Daniel OS.01.05
Wolff, David PP.135
Woltering, Franz OS.03.05
Wong, Ivy KK PP.138
Wong, Judith PP.155
Wong, Siaw Cheng PP.081
Wong, Sui OS.08.08
Wood, Jamie PP.075, PP.091, PP.093, PP.094
Woodcock, Ian OS.04.03, PP.009, PP.013, PP.020, PP.021, PP.145, PP.161
Woodcock, Ian PP.143
Woodcock, Ian R PP.142
Wools, C PP.107
Wools, Christine PP.103
Wright, Melissa SS.07.04
Wu, Huaxiang PP.029
Wu, Wen-Chau OS.02.06
Wu, Xin PP.029
Wu, Yanping OS.08.07
Wuu, Joanne SS.14.03
Xu, Huji PP.029
Yadavaraj, Satish PP.095, PP.118
Yan, Chuanzhu SS.10.04
Yang, Chih-Chao PP.152
Yang, Huan OS.07.08
Yang, Li Yu PP.033
Yanuar, Ahmad PP.089
Yarlagadda, Sai PP.018
Yiu, E.M. PP.145
Yiu, Eppie OS.04.03, PP.009, PP.010
Yiu, Eppie PP.143, SS.26.03
Yiu, Eppie M PP.013, PP.142
Yong, Vivien OS.09.05
Yoshikawa, Masaaki PP.069
Yoshimura, Madoka OS.01.07
Youn, Bora PP.131
Youssef, Eriene OS.07.08
Yu, Alan SS.17.03
Yu, DaeYoung PP.067, PP.068
Yu, Lixi OS.04.05
Yui-Eto, Karina OS.02.04
Yukitake, Motohiro PP.069
Zafeiriou, Dimitrios PP.047
Zaidman, Craig M. OS.01.04
Zanoteli, Edmar OS.06.04
Zekanovic, Stipe PP.155
Zhang, Wen Wen PP.147, SS.43.03
Zhang, Wen Wen PP.092
Zhang, Wenfei OS.07.03
Zhang, Wenfei OS.01.04, PP.015
Zheng, Biao PP.029
Zhou, Allen OS.07.05
Zhu, Danqing PP.155
Zhu, Liang PP.029
Zhu, Yaowei OS.07.08, PP.061
Zichlin, Miriam L PP.091
Ziora-Jakutowicz, Karolina PP.165
Zschuentzsch, Jana OS.08.04
Zschüntzsch, Jana PP.159
Zsurka, Gabor PP.030
Zuccolo, Michela OS.08.04
Züchner, Stephan TC.04.04
Zygmut, Aldona PP.159