
Abstract
Neuroinflammation plays a crucial role in the progression of Alzheimer's disease (AD), contributing to both cognitive decline and motor impairment. This review examines the dual impact of neuroinflammatory processes, particularly the activation of microglia and astrocytes, and the dysregulated release of pro-inflammatory cytokines and chemokines, on cognition and balance in AD. Neuroinflammation leads to synaptic dysfunction, oxidative stress, and mitochondrial impairment, which accelerate neuronal damage and disrupt motor circuits, such as those in the motor cortex, basal ganglia, and cerebellum. These disruptions result in motor symptoms like gait disruption and balance issues, reflecting the close interplay between cognitive and motor functions. In addition, we highlighted the role of blood-brain barrier disruption and peripheral neuropathy in increasing motor dysfunction. Despite the promise of anti-inflammatory treatments, clinical trials with non-steroidal anti-inflammatory drugs have yielded mixed results, underscoring the need for early intervention and more targeted therapies. Therefore, understanding the mechanisms linking neuroinflammation to cognitive and motor impairments may guide the development of new therapeutic approaches aimed at mitigating the complex pathology of AD.
Introduction
Alzheimer's disease (AD) is the most prevalent neurodegenerative disorder worldwide, primarily affecting older adults. 1 In the United States, around 5.8 million individuals currently live with AD, and this number is expected to rise to nearly 14 million by 2060. 2 AD is marked by progressive cognitive decline, primarily affecting the cerebral cortex, which is crucial for memory, reasoning, and language. The occurrence of AD varies on factors such as age, genetic make-up, lifestyle, and social behaviors. Early symptoms typically include mild memory loss but can worsen over time, leading to severe impairments in communication and difficulty in physical activities like walking. 2 The disease typically progresses from the preclinical phase, through mild cognitive impairment, and ultimately advancing to AD, with risk factors such as APOE4 genetic status and brain atrophy. These cognitive impairments significantly disrupt daily life activities, diminishing the quality of life of those affected.
Emerging evidence indicates a strong link between cognitive decline and motor function, especially balance and gait impairment, in individuals with AD.3–5 Slow gait speed is increasingly recognized as a consequence of cognitive decline and an early predictor of it. This connection likely arises because cognition and balance control rely on overlapping brain regions, particularly the subcortical circuits. 6 In a longitudinal study conducted by Best et al. (2016), the authors found that adults who experienced a faster decline in walking speed during the first four years of their study also exhibited more rapid cognitive deterioration in the subsequent five years. 7 Similarly, Ahn et al. (2023) found that individuals with gait and/or balance impairment are at a higher risk of incidence of AD, reinforcing the strong connection between motor and cognitive decline. 8 However, Atkinson et al. (2007) reported that cognitive decline could predict subsequent declines in motor task performance among older adults. 9 This bidirectional relationship suggests that balance and gait impairments may act as early indicators of cognitive decline, reflecting shared vulnerabilities within overlapping brain regions.
The cause of AD remains unknown despite extensive research on potential contributing factors such as oxidative stress, tau protein dysfunction, and amyloid-β (Aβ) accumulation. A central question in AD research is whether one of these factors initiates the disease process or whether they act in combination, compounding each other's effects. While numerous studies have attempted to establish a sequence or hierarchy among these pathological processes, the interrelated nature of these mechanisms complicates a clear understanding of AD causality.
Early research on neurodegenerative diseases, including AD, focused on broad anatomical changes such as protein aggregation, which is visible in the formation of neurofibrillary tangles (NFTs), Aβ plaques, etc. 10 In 1975, researchers identified immune-related proteins in the senile plaques of AD patients, suggesting a potential role for immune responses in the disease process. 11 By the 1980s, studies had further identified microglial activation as a consistent feature in neurodegenerative diseases.12–14 However, at this time, inflammation was viewed merely as a secondary effect that exacerbated AD pathology rather than a primary driver of the disease. 10 In the 1990s, however, the perspective on inflammation in AD shifted. Observational studies showed that long-term users of nonsteroidal anti-inflammatory drugs (NSAIDs) had up to a 50% reduction in AD risk, suggesting that inflammation might play a more integral role. 15 During this period, many studies investigated the origins, function, and toxicity of microglia and how they lead to neurodegenerative diseases. These findings prompted a re-evaluation of the role of neuroinflammation, leading to the growing belief that inflammation might be central to AD's development, not just a secondary effect of other pathologies.
Recent evidence has increasingly recognized neuroinflammation, particularly involving microglia and astrocytes, as a significant factor underlying cognitive and motor impairments in AD.16–18 Neuroinflammation occurs when these immune cells in the brain become persistently activated in response to accumulated pathological proteins such as Aβ plaques and tau tangles. While this immune response initially aims to protect the brain, its sustained activation leads to chronic inflammation, ultimately damaging neurons and impairing functions critical to memory, learning, and movement. 19 Studies have highlighted the dual role of microglia and astrocytes in mediating both cognitive decline and motor dysfunction in AD. Neuroinflammation disrupts not only memory-related neural networks but also motor circuits, including those in the motor cortex, basal ganglia, and cerebellum. 20 These disruptions manifest in motor symptoms such as bradykinesia, gait impairment, and balance issues, underscoring the complex interaction between cognitive and motor functions in AD.
Given the central role of neuroinflammation in these dual impairments, we conducted an extensive literature review presenting an understanding of the underlying mechanism through which neuroinflammation drives both cognitive and motor symptoms in AD. While previous research has illuminated many inflammatory mechanisms underlying cognitive dysfunction, the specific pathways through which neuroinflammation contributes to motor impairments, particularly gait Impairment, remain poorly understood. This review will explore the mechanisms by which neuroinflammation impacts both cognitive and motor functions in AD.
Overview of neuroinflammation in Alzheimer's disease
Role of microglia and astrocytes in neuroinflammation
The activation of the glial cells, primary microglia and astrocytes, leads to chronic inflammations that sequentially contribute to synaptic dysfunction, neural damage, and, ultimately, cell death in the brain (Figure 1).21–24 Microglia, the brain's resident immune cells, are the first responders to abnormal protein accumulations such as Aβ plaques. In the earliest stages of AD, Aβ formation occurs due to abnormal cleavage of amyloid-β protein precursor (AβPP) by β- and γ-secretases. Mutations in the APP or PSEN1/PSEN2 genes can further promote Aβ aggregation. Aβ monomers are intrinsically disordered and have a tendency to oligomerize and form Aβ plaques, which, when accumulated, trigger microglial activation. 10 Recognizing Aβ as a damage-associated molecular pattern (DAMP), microglia become activated via receptors like toll-like receptors (TLRs), receptors for advanced glycation end-products (RAGE), and NOD-like receptors (NLRs).25,26 Additional receptors, such as CD36, CD14, class A scavenger receptor, and α6β1 integrin, also facilitate the recognition of Aβ oligomers and fibrils, amplifying microglial activation.27–29 Initially, this activation is neuroprotective, as microglia attempt to clear Aβ deposits and limit damage. However, when this clearance fails, microglia become chronically activated, amplifying the Aβ aggregation process through upregulation of AβPP expression and activation of proteins like IFITM3, as well as releasing toxic ions such as Zn + which further fuel the feedback loop. 10 This chronic activation is further exacerbated by the release of pro-inflammatory cytokines, reactive oxygen species (ROS), and other factors like iron, which can enhance Aβ aggregation, creating a feedback loop that accelerates neurodegeneration. 26 Additionally, damage-associated molecular patterns (DAMPs) released from dying neurons, such as ATP, HMGB1, and DNA, further promote inflammation and exacerbate the ongoing neuroinflammatory cycle. These DAMPs amplify the activation of microglia and astrocytes, linking Aβ aggregation to tau pathology and perpetuating the inflammatory response. 10 Also, prolonged microglial activation results in the dysfunction of microglial phagocytic mechanisms, further impeding Aβ clearance and fueling amyloid accumulation. Microglial phagocytosis of Aβ fibrils involves pathways such as the endosomal/lysosomal degradation system; however, fibrillar Aβ is largely resistant to enzymatic breakdown, highlighting the importance of extracellular proteases like insulin-degrading enzyme (IDE) and neprilysin in soluble Aβ clearance. 30

Overview of the key neuroinflammatory pathways involving microglia and astrocytes in Alzheimer's disease. This figure illustrates the complex neuroinflammatory mechanisms that drive the pathogenesis of Alzheimer's disease (AD). Early in the disease, amyloid-β (Aβ) is produced due to the abnormal cleavage of amyloid-β protein precursor (APP) by β- and γ-secretases. These Aβ monomers, due to their disordered structure, readily aggregate into toxic oligomers and plaques. Genetic mutations in APP, PSEN1, or PSEN2 genes further exacerbate Aβ production, accelerating plaque formation. The deposition of Aβ plaques triggers the activation of microglia, the resident immune cells of the brain, which attempt to clear the accumulated Aβ through phagocytosis. However, when this clearance process fails, Feedback Loop 1 is initiated: microglia become chronically activated, enhancing the expression of IFITM3 and APP, and releasing pro-inflammatory molecules and ions such as zinc (Zn+), which promote further Aβ aggregation. This creates a positive feedback loop, sustaining neuroinflammation and amplifying Aβ deposition. Alongside microglial activation, astrocytes are also recruited and activated by the inflammatory signals. These reactive astrocytes exacerbate the inflammatory response by releasing additional pro-inflammatory cytokines, such as TNF-α, IL-6, and IL-1β, which further amplify the neuroinflammatory cascade. This persistent inflammation leads to Feedback Loop 2, in which damage-associated molecular patterns (DAMPs) such as ATP, HMGB1, and DNA are released from dying neurons. These DAMPs activate both microglia and astrocytes, perpetuating the cycle of neuroinflammation. This prolonged inflammatory state activates various kinases, leading to tau hyperphosphorylation, which causes tau to dissociate from microtubules and form neurofibrillary tangles (NFTs) in the cytosol. Therefore, neuroinflammation plays a crucial role in linking the accumulation of Aβ plaques to tau pathology, ultimately contributing to the cognitive and motor decline observed in AD.
This sustained activation of microglia also links Aβ and tau pathology. Prolonged inflammation, along with cytokine release, activates several kinases, such as glycogen synthase kinase-3β (GSK-3β), which leads to tau hyperphosphorylation and subsequent formation of NFTs.
31
NFTs within axonal fibers further destabilize neuronal transport and structure, exacerbating synaptic loss and contributing to cognitive decline. These NFTs disrupt axonal transport, impairing the delivery of essential cellular materials and further contributing to neuronal dysfunction and death
Microglia exist in a continuum of activation states, rather than the traditionally defined M1 (pro-inflammatory, neurotoxic) and M2 (anti-inflammatory, neuroprotective) dichotomy.35,36 Recent evidence suggests that microglia in AD exhibit a spectrum of functional phenotypes, dynamically shifting in response to disease progression and inflammatory cues. A subset known as Disease-Associated Microglia (DAM) has been identified in AD, characterized by upregulated expression of genes such as TREM2, APOE, CST7, and CD9, which regulate lipid metabolism, phagocytosis, and inflammatory signaling. 35 Single-cell RNA sequencing studies demonstrated that DAM undergo a two-step activation process, where an initial TREM2-independent phase primes microglia by downregulating homeostatic markers, followed by a TREM2-dependent phase that enhances phagocytosis and neuroinflammatory gene expression. While DAM initially help clear Aβ, their function becomes dysregulated as AD progresses, leading to chronic neuroinflammation and neurotoxicity. 35
In AD, microglia increasingly adopt pro-inflammatory states, releasing cytokines that exacerbate oxidative stress and neuronal damage. This activation contributes to tau pathology by enhancing tau phosphorylation through kinases such as GSK-3β and CDK5, promoting tau aggregation. Rather than a simple shift toward a fixed M1-like phenotype, microglia display heterogeneous activation patterns, with subsets that attempt to clear Aβ while others drive neurotoxicity.37,38 This dysregulation impairs microglial phagocytic functions, leading to reduced clearance of Aβ and neuronal debris, further fueling the neuroinflammatory cycle. Additionally, genetic mutations in microglial receptors, such as TREM2 and CD33, impair microglial Aβ clearance and exacerbate inflammatory responses. TREM2 mutations reduce microglial capacity to phagocytose Aβ and neuronal debris, while CD33 mutations interfere with Aβ fibril degradation, increasing toxic aggregate accumulation and accelerating neurodegeneration.39–41
Astrocytes, essential for maintaining neuronal health and synaptic support, also become reactive in response to Aβ and tau pathology. Upon activation, astrocytes release pro-inflammatory cytokines and chemokines that reinforce microglial activation, sustaining a pro-inflammatory environment. Emerging evidence now suggests that astrocytes also contribute directly to Aβ production, as reactive astrocytes express the necessary components for amyloid production, such as AβPP, β-secretase (BACE1), and γ-secretase. This makes astrocytes significant contributors to the overall amyloid burden in the brain. 42 Phillips et al. emphasized the significance of astrocyte-mediated inflammation in synaptic damage. 43 This reactive state of astrocytes causes neural stress that contributes to the development of AD. Astrocytic activation in AD also includes a stage of atrophy, which disrupts synaptic connectivity and contributes to cognitive decline. Early astrocyte atrophy, first observed in the entorhinal cortex, progresses to affect more distant areas as the disease advances. 44 In addition to interacting with microglia, astrocytes impact other central nervous system (CNS) cells, such as neurons, oligodendrocytes, and endothelial cells, further amplifying the inflammatory response within the brain.45–47 Reactive astrocytes also secrete pro-inflammatory cytokines such as interleukin-1β (IL-1β), interleukin-6 (IL-6), and tumor necrosis factor-alpha (TNF-α), which exacerbate neuroinflammation and contribute to neuronal damage. 42 Astrocytes also contribute to Aβ clearance through internalization and degradation pathways, mediated by proteases like endothelin-converting enzyme-2, neprilysin, and IDE. Moreover, astrocyte-driven paravenous drainage of soluble Aβ, facilitated by aquaporin-4, plays a critical role in mitigating Aβ deposition, although these mechanisms are impaired in AD. 48
The combined actions of microglia and astrocytes in AD create a continuous cycle of neuroinflammation. The persistent release of pro-inflammatory cytokines, such as IL-1β, TNF-α, and IL-6, contributes to synaptic dysfunction and blood-brain barrier (BBB) disruption, allowing peripheral immune cells to infiltrate the brain and exacerbate inflammation. While reactive astrocytes attempt to repair the brain, they can also contribute to neurotoxicity through the production of ROS and inflammatory cytokines, which further damage neuronal networks. 42 This inflammatory cascade damages neuronal networks and drives the disease's core pathological features, accelerating cognitive decline and motor dysfunction.37,38
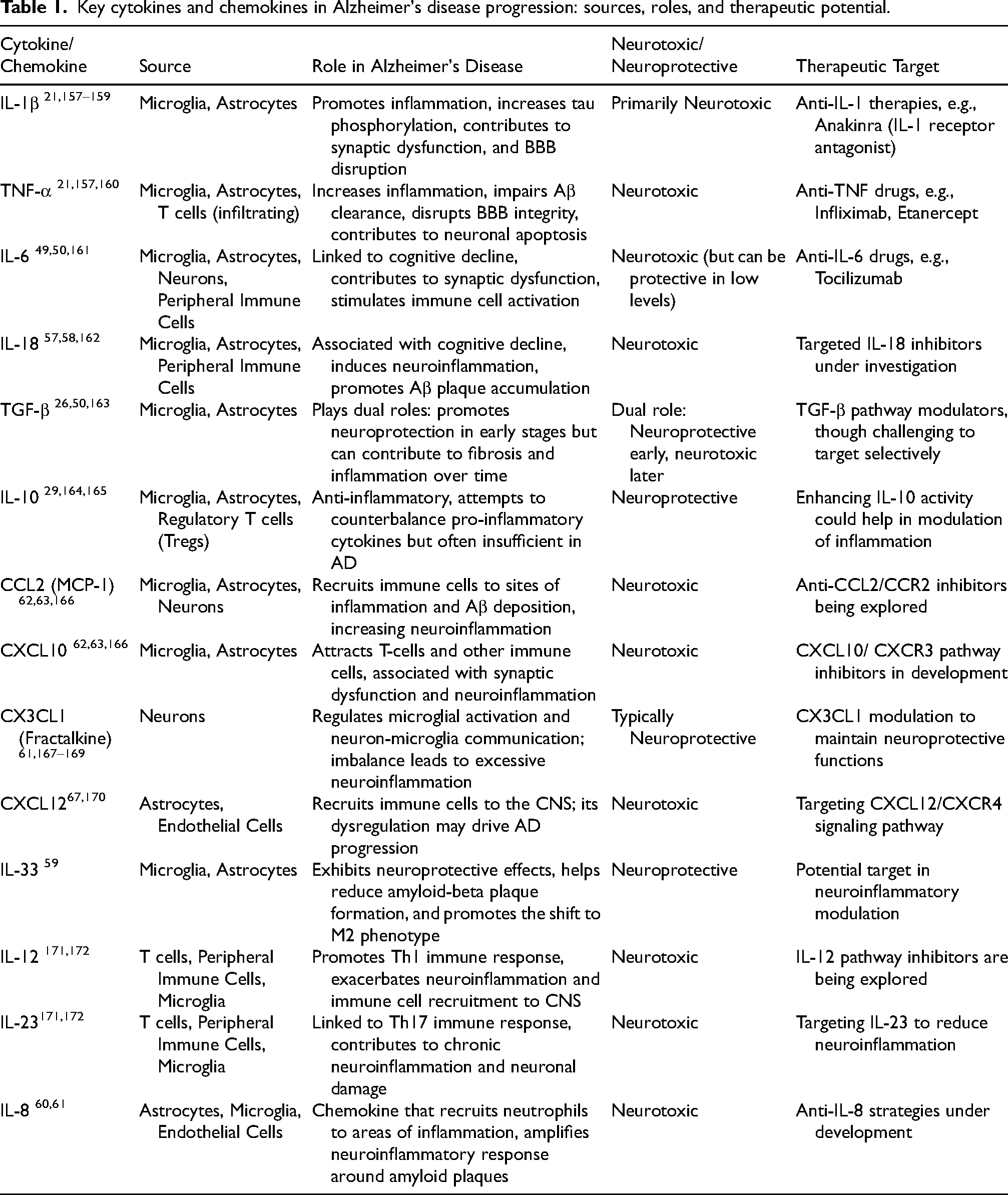
Cytokines and chemokines in AD
Cytokines and chemokines are essential regulators of immune responses in the brain, coordinating communication between cells and modulating inflammation. In AD, an imbalance in these signaling molecules fosters a chronic neuroinflammatory environment that accelerates neuronal damage and worsens disease progression. Cytokines can promote or inhibit inflammation depending on their type and concentration, while chemokines guide immune cells to sites of injury or inflammation (Figure 2).26,49,50 In AD, pro-inflammatory cytokines such as IL-1β, IL-6, TNF-α, and transforming growth factor-beta (TGF-β) are elevated, particularly near Aβ plaques, a hallmark of AD pathology. These cytokines, including microglia and astrocytes, activate immune cells to respond to Aβ accumulation. Though initially protective, prolonged cytokine activity leads to a chronic inflammatory state, causing synaptic dysfunction, neuronal damage, and disease progression.26,49,50 Increased levels of these cytokines, such as IL-1β, IL-6, TNF-α, and granulocyte macrophage-colony stimulating factor, have been associated with Aβ pathology in transgenic mouse models of AD, further supporting their role as key drivers of neuroinflammation. 51

Dynamic balance between pro-inflammatory and anti-inflammatory microglia in Alzheimer's disease. This figure illustrates the interplay between pro-inflammatory and anti-inflammatory cytokines in Alzheimer's disease (AD) and their impact on microglial activation. In response to pathological stimuli, such as amyloid-β (Aβ) plaques and tau aggregates, homeostatic microglia become activated and release pro-inflammatory cytokines (IL-1β, TNF-α, IL-6) and chemokines (CCL2, CXCL10). While this initial immune response aims to contain and clear toxic proteins, prolonged cytokine release exacerbates neuroinflammation, leading to neuronal damage and disease progression. As neurons degenerate, the inflammatory cycle perpetuates further cytokine production, reinforcing a harmful feedback loop. In contrast, anti-inflammatory cytokines, including IL-10 and TGF-β, are associated with a compensatory microglial response that emerges in later stages of the disease. These cytokines promote waste clearance, tissue repair, and neuroprotection, helping to counterbalance the detrimental effects of chronic inflammation. However, this shift to an anti-inflammatory state may become ineffective in AD, as sustained neurodegeneration and immune exhaustion impair microglial function.
IL-1β, IL-6, and TNF-α are prominent drivers of neuroinflammation in AD. TNF-α, for example, disrupts the BBB, allowing peripheral immune cells to infiltrate the brain and further intensify local inflammation. It also impairs Aβ clearance, accelerating its accumulation. 52 Additionally, IL-1β and TNF-α stimulate NFT formation from hyperphosphorylated tau, damaging neuronal signaling and synaptic stability. IL-6 is associated with synaptic dysfunction and cognitive decline due to its disruptive effects on neuronal communication. 53 As the disease progresses, a subset of cytokines, including IL-10 and TGF-β, becomes more prominent, reflecting a potential shift in the inflammatory response. This shift may serve as a compensatory mechanism aimed at mitigating excessive neurotoxicity, as chronic neurodegeneration can drive anti-inflammatory signaling pathways. 54 Anti-inflammatory cytokines like IL-10 counterbalance the inflammatory response, but in AD, their effects are often insufficient to prevent the progression of inflammation. TGF-β, which plays dual roles depending on the disease stage, can act as both a protective and a harmful factor, reflecting the complex regulation of inflammation in AD. 26 Interestingly, while IL-1β is typically associated with neurotoxicity, studies in transgenic mouse models of AD suggest it may also promote beneficial neuroinflammation, reducing Aβ burden under specific conditions.55,56
Other cytokines also influence AD pathology. For example, IL-18, is elevated in AD and worsens neuroinflammation by activating microglia, leading to further cytokine release. Increased IL-18 correlates with Aβ accumulation and cognitive decline.57,58 IL-33, however, has neuroprotective effects, reducing Aβ plaque formation and promoting the microglial shift to the anti-inflammatory M2 phenotype, which aids in Aβ clearance. 59 Cytokines like IL-12 and IL-23, associated with T-helper immune responses, recruit T-cells to the CNS, increasing inflammation and contributing to neuronal damage. IL-8, primarily a chemokine, is elevated in AD cerebrospinal fluid (CSF) and recruits immune cells to Aβ plaques, promoting neuroinflammatory responses.60,61
Chemokines are central to AD by directing immune cell migration to degenerating brain areas and maintaining chronic inflammation. CCL2 (MCP-1) is elevated in AD and recruit monocytes and microglia to Aβ plaques, amplifying local inflammation, worsening neuronal damage, and contributing to cognitive decline.62–64 CXCL10, elevated in response to inflammation, attracts T-cells and microglia to degenerative areas, exacerbating synaptic dysfunction and neurodegeneration.26,63,65 CX3CL1 (fractalkine) typically regulates neuron-microglia communication, acting as an inhibitory signal to modulate microglial activity. 66 In AD, its dysregulation leads to unchecked microglial activation, worsening inflammation, and neuronal damage. Another chemokine, CXCL12 (SDF-1), promotes immune cell recruitment to the CNS, driving neuroinflammation. 67 Dysregulation of the CXCL12-CXCR4 pathway may further propel AD progression, highlighting the importance of chemokines like CCL2, CXCL10, CX3CL1, and CXCL12 in AD pathogenesis.
Table 1 provides an overview of key cytokines and chemokines involved in AD progression, detailing their sources, roles, neurotoxic or neuroprotective effects, and potential therapeutic targets.
Key cytokines and chemokines in Alzheimer's disease progression: sources, roles, and therapeutic potential.
Role of neuroinflammation in BBB disruption and cerebral blood flow alterations
The BBB is a specialized multicellular vascular structure that plays a critical role in maintaining brain homeostasis by regulating the exchange of substances between the bloodstream and the brain parenchyma. It consists of brain endothelial cells (BECs), astrocytic end-feet, pericytes, and a basement membrane, forming a semi-permeable membrane that separates the central nervous system from the peripheral blood circulation. 68 BECs are tightly connected by junctional proteins such as claudins, occludins, and zonula occludens (ZO-1, ZO-2, ZO-3), which ensure the integrity of the barrier. 69 In a healthy brain, the BBB actively facilitates the clearance of Aβ from the brain into the peripheral circulation. This process is mediated by transporters such as low-density lipoprotein receptor-related protein 1 and P-glycoprotein, which play critical roles in Aβ efflux across the BBB. 70 Moreover, the glymphatic system, which helps remove waste products from the brain, works in coordination with the BBB to maintain Aβ homeostasis. 71
However, neuroinflammation impairs this process, allowing Aβ to accumulate, promoting the formation of plaques and compromising BBB integrity. 72 Aβ itself has been implicated in damaging the BBB, exacerbating its permeability and facilitating the entry of pro-inflammatory substances and immune cells into the brain. 73 Furthermore, studies have shown that glial cells, such as microglia and astrocytes, are activated around these plaques, releasing inflammatory cytokines and chemokines that contribute to the recruitment of immune cells, including macrophages, thereby perpetuating neuroinflammation. Importantly, rather than being a mere consequence of glial activation, BBB dysfunction plays an active role in amplifying neuroinflammation by increasing the brain's exposure to peripheral immune cells and toxins.
Once the BBB is compromised (Figure 3), neurotoxic substances from the bloodstream infiltrate the brain, triggering additional inflammatory responses from microglia and astrocytes. This influx exacerbates local inflammation and disrupts the clearance of toxic proteins, including Aβ, promoting a vicious cycle of neurodegeneration and inflammation. Vascular dysfunction thereby impairs the efflux of Aβ, further contributing to plaque accumulation and cognitive decline in AD.

Role of neuroinflammation in Alzheimer's disease, highlighting blood-brain barrier disruption and subsequent neurodegeneration. This figure illustrates the interplay between neuroinflammation, blood-brain barrier (BBB) dysfunction, cerebral blood flow (CBF) impairment, and neuronal degeneration in Alzheimer's disease (AD). The BBB, composed of endothelial cells and tight junction proteins (claudin-5, occludins, ZO-1), regulates molecular exchange between the bloodstream and the brain. In AD, this barrier becomes compromised due to Aβ accumulation and inflammation, leading to increased permeability. Aβ clearance is impaired by the reduced function of low-density lipoprotein receptor-related protein 1 (LRP1), while receptor for advanced glycation end products (RAGE) facilitates Aβ influx, further exacerbating its deposition. This accumulation promotes the formation of extracellular plaques, which activate microglia and astrocytes, triggering the release of pro-inflammatory cytokines (TNF-α, IL-1β, IL-6, IL-12, IL-23). The inflammatory response weakens the BBB by degrading tight junction proteins, allowing neutrophils and leukocytes to infiltrate the brain and amplify neuroinflammation. Additionally, reduced CBF, an early pathological event in AD, exacerbates Aβ accumulation by impairing its clearance and reducing oxygen supply, further promoting oxidative stress and vascular damage. Sustained inflammation and oxidative stress contribute to NMDA receptor overactivation, leading to excessive calcium influx and activation of apoptotic pathways (p65, p53), ultimately causing neuronal degeneration. This figure highlights how BBB disruption, neuroinflammation, impaired Aβ clearance, and excitotoxicity form a pathological feedback loop that accelerates neurodegeneration in AD.
Key inflammatory cytokines, particularly IL-1β, TNF-α, and IL-6, play significant roles in disrupting BBB integrity. These cytokines enhance the activity of matrix metalloproteinase 9 (MMP-9), leading to degradation of the endothelial barrier. 74 IL-1β contributes to BBB leakage by activating the activin receptor-like kinase 1 (ALK1) and the SMAD signaling pathway, resulting in vascular leakage and tissue damage 75 Similarly, TNF-α activates NF-κB signaling, reducing the expression of claudin-5, a key tight junction protein, weakening the BBB, and allowing immune cells to infiltrate the brain. 76 The infiltration of peripheral immune cells drives chronic neuroinflammation, accelerating neurodegeneration and contributing to both cognitive and motor decline in AD.
Evidence suggests that a reduction in cerebral blood flow (CBF) is one of the earliest events in AD pathogenesis, occurring even before cognitive symptoms manifest. 77 Reduced CBF impairs the clearance of Aβ via endothelial receptors, such as Low-Density Lipoprotein Receptor-Related Protein 1, leading to its accumulation within the brain.77,78 At the same time, RAGE expression is upregulated under inflammatory conditions, further promoting Aβ influx into the brain and exacerbating its deposition. Additionally, lower perfusion compromises the delivery of oxygen and nutrients, further exacerbating oxidative stress and neuronal dysfunction.77,78 The resulting decline in CBF also weakens the BBB,79,80 making it more susceptible to inflammatory damage and immune cell infiltration, 79 reinforcing the feedback loop that accelerates neurodegeneration and disease progression. 81
Moreover, sustained neuroinflammation promotes excitotoxicity through the excessive activation of NMDA receptors, a mechanism directly linked to neuronal injury in AD. 82 As illustrated in Figure 3, inflammatory cytokines such as TNF-α and IL-1β contribute to NMDA receptor overactivation, which leads to increased calcium influx, oxidative stress, and activation of pro-apoptotic pathways (p65 and p53). This cascade results in synaptic dysfunction and neuronal degeneration, a hallmark of AD progression. Despite being a physiological receptor involved in synaptic plasticity, NMDA receptor dysregulation in the context of chronic inflammation plays a detrimental role in neuronal survival, ultimately driving neurodegeneration. 82
Neuroinflammation's impact on cognitive decline in Alzheimer's disease
Role of oxidative stress in synaptic dysfunction
Synapses are critical junctions between neurons that enable communication through neurotransmitters, which are essential for cognitive and motor functions such as learning and memory. In AD, synaptic dysfunction, characterized by synapse loss or impaired signaling, disrupts neuronal communication and contributes to cognitive decline. This dysfunction often precedes neuronal loss in vulnerable brain regions, correlating strongly with cognitive deficits in AD. Studies indicate that synaptic loss is a key early event in AD, and its association with cognitive impairment is well-documented.83–85 Oxidative stress, primarily induced by ROS, plays a central role in synaptic dysfunction in AD.86,87 While ROS are typically involved in cellular signaling, excessive ROS production contributes to neurodegenerative processes in AD. This oxidative stress is exacerbated by neuroinflammatory processes, where glial activation (e.g., microglia and astrocytes) enhances ROS production.88,89 Microglial activation, triggered by Aβ plaques, and the subsequent inflammatory response can amplify oxidative damage, creating a vicious cycle that exacerbates synaptic impairment. The production of ROS within this neuroinflammatory context further destabilizes synaptic structures and promotes the accumulation of harmful Aβ oligomers and tau. 90
Studies suggest that the accumulation of Aβ oligomers, particularly high molecular weight oligomers, contributes to synaptic dysfunction in AD. While these larger Aβ aggregates show limited cytotoxicity, they dissociate into smaller, more toxic forms that impair hippocampal long-term potentiation (LTP), a process essential for memory consolidation.91–93 However, oxidative stress, through ROS production, can modulate synaptic activity, affecting N-methyl-D-aspartate (NMDA) receptor signaling, which is critical for LTP.91–93 Excessive ROS, in combination with Aβ and tau, can lead to NMDA receptor dysfunction, promoting receptor internalization and impairing synaptic plasticity, which further contributes to cognitive decline.94–96
Furthermore, mitochondrial dysfunction, another result of neuroinflammation, contributes to ROS production, destabilizing NMDA receptors and causing excitotoxicity. 86 This excess ROS not only impairs synaptic plasticity but also accelerates neurodegeneration. Moreover, ROS and reactive nitrogen species trigger pathways that lead to tau hyperphosphorylation, exacerbating synaptic dysfunction. This oxidative stress-induced tau phosphorylation has been linked to NFT formation, which disrupts neuronal signaling and accelerates cognitive decline.
The relationship between oxidative stress, Aβ, tau, and synaptic dysfunction is further amplified by the activation of stress-related kinases, such as p38 mitogen-activated protein kinase (MAPK). 85 These kinases amplify oxidative stress and activate apoptotic pathways, contributing to NFT formation and neuronal loss. This cycle of oxidative damage and neuroinflammation further exacerbates synaptic dysfunction, a key driver of cognitive decline in AD.
Role of mitochondrial dysfunction and its impact on the hippocampus
Mitochondrial dysfunction is a hallmark of AD, particularly affecting the hippocampus, a brain region essential for memory formation and consolidation. This dysfunction impairs several cellular processes, including synaptic transmission, energy metabolism, and oxidative stress regulation. Mitochondria maintain their function through continuous fission and fusion processes, which ensure structural integrity and adaptability. Key regulators of this dynamic balance include Dynamin-related protein 1 (Drp-1), mitochondrial fission factor (Fis-1), and fusion proteins such as Mitofusin 1, Mitofusin 2, and Optic atrophy protein 1. 97 In AD, neuroinflammation drives mitochondrial dysfunction by increasing ROS production. This disrupts energy metabolism and promotes the abnormal processing of AβPP. This leads to the accumulation of Aβ, further impairing mitochondrial function and accelerating disease progression. 98
Recent studies highlight the genetic underpinnings of mitochondrial dysfunction in AD. Tian et al. identified several genes associated with AD susceptibility in the mitochondrial solute carrier family (SLC25). 99 A meta-analysis of hippocampal transcriptome-wide association studies (TWAS) identified SLC25A10, SLC25A17, and SLC25A22 as key genes linked to hippocampal atrophy. SLC25A10 encodes a mitochondrial dicarboxylate carrier involved in metabolic cycles, SLC25A17 is a peroxisomal membrane transporter facilitating fatty acid metabolism, and SLC25A22 encodes a glutamate transporter essential for neuronal excitability and metabolism. Among them, SLC25A22 exhibited a strong inverse correlation with hippocampal atrophy, with its downregulation closely associated with AD onset, highlighting its potential role in disease progression. 99 These findings align with the mitochondrial cascade hypothesis, suggesting that mitochondrial dysfunction plays a critical role in AD pathogenesis and represents a promising target for early diagnosis and intervention. 99
Manczak et al. explored the interaction between voltage-dependent anion channel 1 (VDAC1), Aβ, and phosphorylated tau in AD brains and transgenic mouse models (APP, APP/PS1, and 3xTg.AD). 100 Their research revealed that VDAC1 interacts with Aβ and tau, leading to mitochondrial pore dysfunction and impaired ATP production. In transgenic mice, increased VDAC1 expression exacerbated mitochondrial dysfunction with age, underscoring the link between mitochondrial dysfunction and AD progression. 100 These findings suggest that reducing VDAC1 expression and mitigating Aβ and tau accumulation may restore mitochondrial function and slow cognitive decline. In summary, mitochondrial dysfunction, driven by neuroinflammation and oxidative stress, contributes to hippocampal vulnerability, synaptic dysfunction, and cognitive decline in AD. Therapeutic strategies targeting mitochondrial genes such as SLC25A22 and reducing VDAC1 expression offer promising avenues for improving mitochondrial health and preventing cognitive deterioration.
Neuroinflammation's impact on balance and motor impairment in Alzheimer's disease
Neuroinflammation plays a crucial role in the pathophysiology of AD, driving cognitive decline and contributing to motor impairments. The activation of microglia and astrocytes, hallmarks of neuroinflammation, leads to the release of pro-inflammatory cytokines and chemokines, which disrupt neural signaling and contribute to neurodegeneration.29,101,102 This inflammatory process extends beyond the brain, impairing motor coordination and balance. Motor symptoms such as disequilibrium (balance disorders) and dysplasia (impaired gait) can be directly linked to these neuroinflammatory processes, which impair the ability of the CNS to integrate sensory input and coordinate movement.103,104
Motor cortex and basal ganglia dysfunction
The motor cortex and basal ganglia are integral components of the neural network responsible for planning, initiating, and executing voluntary movements. The motor cortex—comprising the primary motor cortex, premotor cortex, and supplementary motor area—orchestrates muscle contractions and coordinates complex motor tasks. The basal ganglia, particularly the striatum, serve as the principal input center for glutamatergic and dopaminergic signals essential for modulating motor control and coordination. The striatum includes the caudate nucleus and putamen in the dorsal striatum, and the nucleus accumbens and olfactory tubercle in the ventral striatum. 105 Given the well-documented impact of neuroinflammation on motor regions in other neurodegenerative diseases,106–108 it is plausible that similar processes influence the motor cortex and basal ganglia in AD. Activation of microglia and astrocytes in response to increased Aβ plaques and tau tangles leads to the release of pro-inflammatory cytokines such as IL-1β and TNF-α within these motor regions. 29 These cytokines disrupt neuronal function by altering synaptic transmission and plasticity, ultimately impairing motor control and contributing to motor deficits observed in AD patients.
Oxidative stress further exacerbates dysfunction in the motor cortex and basal ganglia. Chronic neuroinflammation leads to the excessive production of ROS, causing oxidative damage to neuronal cells through mechanisms such as lipid peroxidation, DNA fragmentation, and protein oxidation. 109 In the motor cortex, oxidative damage compromises neuronal integrity and viability. In Huntington's disease, oxidative stress in the striatum impairs mitochondrial function in medium spiny neurons (MSN), leading to energy deficits that compromise neuronal survival and synaptic activity essential for motor control. 108 A similar mechanism may also be involved in AD. Li et al. (2022) identified increased oxidative damage in the caudate and putamen of AD patients, correlating with disease progression and neurodegeneration. 110 Although the study did not directly link oxidative damage to motor impairments, these brain regions are critical for motor integration, as the caudate and putamen are key components of the basal ganglia, which help regulate voluntary movement.111,112 The interplay between neuroinflammation and oxidative stress creates a detrimental cycle in both the motor cortex and basal ganglia. Neuroinflammation increases ROS production, while oxidative stress further activates inflammatory pathways, accelerating neuronal degeneration in these motor regions. 113
Striatal neurotransmitter imbalance due to neuroinflammation
Neuroinflammation in AD significantly disrupts neurotransmitter systems within the striatum, particularly affecting dopamine and gamma-aminobutyric acid (GABA) signaling, which are crucial for motor control. Dopaminergic neurons originating from the substantia nigra pars compacta project to the striatum and modulate the activity of MSNs via dopamine receptors D1 (DRD1) and D2 (DRD2).114–116 DRD1-expressing neurons enhance the direct pathway to facilitate movement, while DRD2-expressing neurons modulate the indirect pathway to inhibit excessive movements. 117 Chronic neuroinflammation leads to alterations in dopaminergic signaling by decreasing dopamine synthesis and release, as well as impairing receptor function. Pro-inflammatory cytokines like TNF-α and IL-1β can downregulate dopamine receptor expression and interfere with dopamine transporter function, resulting in diminished dopaminergic neurotransmission. 113 Recent studies, such as those by Tournier et al. (2024), have shown that hippocampal neuroinflammation in AD can further exacerbate these disruptions by influencing dopaminergic signaling in the striatum. 116 Additionally, dopaminergic dysfunction in AD is not limited to the striatum. Nobili et al. (2017) highlighted that degeneration of dopaminergic neurons in regions such as the ventral tegmental area contributes to reduced dopamine availability, further impacting brain regions like the hippocampus and striatum. 115 This reduction in dopamine contributes to motor deficits such as bradykinesia observed in AD patients. 118
Similarly, neuroinflammation affects GABAergic signaling within the striatum. GABA is the primary inhibitory neurotransmitter in the CNS, essential for regulating neuronal excitability and motor coordination. Inflammatory cytokines can alter GABA receptor expression and function, disrupting inhibitory control over motor circuits. Elevated levels of pro-inflammatory mediators have been associated with changes in GABA_A receptor subunit composition, leading to impaired GABAergic neurotransmission. 119 This imbalance may result in hyperexcitability of motor pathways and contribute to motor dysfunction in AD. Empirical studies have established a link between neuroinflammation-induced neurotransmitter imbalance and motor impairments in AD. Interventions that reduce neuroinflammation have been shown to restore dopaminergic signaling and improve motor performance. 120 These findings underscore the critical role of neuroinflammation in disrupting neurotransmitter systems within the striatum. By altering dopamine and GABA signaling, neuroinflammation contributes significantly to the motor impairments observed in AD.
Toll-like receptor 4 signaling pathways
Toll-like receptors (TLRs) are a family of pattern recognition receptors crucial for initiating innate immune responses by detecting pathogenic molecules. Among them, Toll-like receptor 4 (TLR4) has gained significant attention for its role in neuroinflammation associated with neurodegenerative diseases like AD. 121 TLR4 is predominantly expressed on microglia, the resident immune cells of the CNS, and its activation leads to the production of pro-inflammatory mediators that can contribute to neuronal damage and motor dysfunction. Activation of TLR4 occurs upon recognition of specific ligands, such as lipopolysaccharide (LPS) found in Gram-negative bacteria. In AD, Aβ peptides and damaged neuronal components can also activate TLR4. 122 Upon activation, TLR4 initiates two primary intracellular signaling pathways: the MyD88-dependent pathway and the MyD88-independent (TRIF-dependent) pathway. Both pathways culminate in the activation of transcription factors like NF-κB and interferon regulatory factors, leading to the transcription of pro-inflammatory cytokines such as TNF-α, IL-1β, and IL-6. 123
Empirical studies have demonstrated the impact of TLR4-mediated neuroinflammation on motor behavior. Qin et al. (2007) showed that systemic administration of LPS in animal models led to chronic neuroinflammation and progressive neurodegeneration, resulting in significant motor deficits. Behavioral changes included reduced locomotor activity, impaired motor coordination, and decreased exploratory behavior. 124 These findings highlight how TLR4 activation can adversely affect motor function. However, the inhibition of TLR4 signaling has been shown to mitigate neuroinflammation and improve motor outcomes. Hutchinson et al. (2008) reported that blocking TLR4 with specific antagonists suppressed sickness behavior and prevented LPS-induced motor deficits in rodents. 125 These findings highlight how TLR4 activation can adversely affect motor function.
Given the significant role of TLR4-mediated neuroinflammation in motor impairments, targeting TLR4 signaling pathways presents a promising therapeutic avenue. Potential interventions include the development of TLR4 antagonists or inhibitors that can cross the BBB and selectively inhibit TLR4 signaling in the CNS. Additionally, modulating downstream signaling components, such as NF-κB inhibitors or agents that enhance anti-inflammatory cytokine production like interleukin-10 (IL-10), may offer neuroprotective benefits and improve motor function in AD patients.
Peripheral neuropathy and BBB disruption
Systemic inflammation extends to the peripheral nervous system (PNS), further contributing to motor dysfunction in AD. Neurogenic inflammation in the PNS is characterized by the release of pro-inflammatory cytokines that sensitize sensory neurons, particularly nociceptors, which transmit pain and sensory signals.126,127 As a result, individuals with AD may experience hyperalgesia (increased pain sensitivity) or hypoesthesia (diminished sensory perception), both of which disrupt sensory processing and impair proprioception, the body's ability to sense its position in space—a relationship observed in individuals with osteoarthritis and stroke,128,129 though direct evidence in AD populations remains lacking. Proprioceptive deficits arise from impaired mechanoreceptors in muscles and joints, causing abnormal neural discharges and reduced γ-motor neuron excitability. These impairments lead to muscle weakness and balance issues as the CNS struggles to process inaccurate sensory input, further compounding motor dysfunction in individuals with osteoarthritis and stroke.128,129 A similar mechanism may underlie the observed balance deficit in AD, though further research is needed to confirm this connection.
The BBB, which generally protects the CNS from peripheral inflammatory factors, becomes compromised during systemic inflammation, allowing immune cells and cytokines to infiltrate the brain. 130 This infiltration exacerbates neuroinflammation, disrupting neural circuits essential for motor coordination. Inflammatory mediators within the CNS also increase oxidative stress, damaging neurons in motor-related areas such as the cerebellum and motor cortex, further impairing balance and coordination. 131
The disruption of the BBB creates a feedback loop where systemic inflammation intensifies neuroinflammation, leading to further motor impairments. This interaction between different components of the peripheral and central nervous system inflammatory cascades highlights the need to target neuroinflammatory pathways to mitigate motor dysfunction and cognitive decline in AD.
Neuroinflammation in Alzheimer's disease: diagnostic and therapeutic perspectives
Neuroinflammatory biomarkers: a potential option to diagnose and monitor AD progression
Biomarkers reflecting neuroinflammation are emerging as critical tools for diagnosing and monitoring AD. Findings from a recent study by Foley et al. (2023) involving 837 AD participants emphasize the role of neuroinflammatory biomarkers in tracking the disease progression. 132 The study demonstrated significant correlations between hallmark AD plasma biomarkers (e.g., p-tau181, Aβ42/Aβ40 ratio, and neurofilament light chain (NfL)) and inflammatory biomarkers, including IL-6, IL-8, IL-10, and GFAP. The strongest associations were observed between NfL and inflammatory markers, indicating a close link between systemic inflammation and axonal degeneration. 132 These results highlight the utility of inflammatory biomarkers in capturing immune system activation during AD progression and suggest their potential for monitoring the interplay between amyloid, tau, and inflammation in real-time.
Similarly, elevated levels of cytokines in CSF and blood have been linked to progressive neuronal dysfunction and synaptic loss, correlating strongly with cognitive decline and motor impairments.133,134 These cytokines provide a non-invasive means of assessing the inflammatory burden in the brain, enabling clinicians and researchers to track how inflammation exacerbates amyloid and tau pathology over time. Emerging biomarkers of glial activation, such as soluble TREM2 (sTREM2), offer additional precision in monitoring neuroinflammation.135,136 Detectable in plasma and CSF, sTREM2 correlates with amyloid plaque burden and tau pathology, reflecting the extent of microglial activation. 136 Astrocytic markers like glial fibrillary acidic protein (GFAP) and YKL-40 may further enhance the capacity to monitor inflammation in AD, 137 as elevated levels of these markers have been associated with astrocytosis, neuronal injury, and disease progression, emphasizing their relevance in assessing both cognitive and motor decline. 137
In addition, NfL, a marker of axonal damage, is released into CSF and blood during neuronal injury and correlates with both cognitive decline in AD. 138 NfL levels in CSF and blood rise significantly in response to neuronal injury. With plasma, NfL increases are detectable up to 22 years before the clinical onset of familial AD, highlighting its potential as an early diagnostic tool. 139 Elevated levels of GFAP and NfL provide a clear indication of the extent of astrocytic reactivity and axonal degeneration, respectively, offering a comprehensive picture of the disease's progression.140,141 These biomarkers may be particularly useful in identifying early pathological changes and tracking the impact of therapeutic interventions to reduce inflammation. Imaging biomarkers also provide an additional layer of precision by enabling the visualization of neuroinflammatory processes and structural brain changes in vivo. Positron emission tomography (PET) imaging with translocator protein (TSPO) tracers identifies microglial activation, while magnetic resonance imaging detects brain atrophy and vascular dysfunction associated with AD. 142 PET tracers for amyloid and tau allow clinicians to map the spatial distribution of pathological deposits, while advanced modalities such as 11C-DED PET and magnetic resonance spectroscopy measure astrocytic reactivity. 142 These imaging tools complement fluid biomarkers, linking molecular changes to localized brain dysfunction, and provide a robust framework for diagnosing and monitoring AD progression.
Neuroinflammatory biomarkers hold immense promise for diagnosing and monitoring AD progression. Their ability to capture the interplay between glial activation, neuroinflammation, and neurodegeneration provides critical insights into disease mechanisms. By reflecting early pathological changes and correlating with cognitive and motor impairments, these biomarkers could revolutionize how AD is diagnosed and managed, paving the way for more effective interventions and personalized treatment strategies.
Clinical implications and therapeutic interventions
The possibility that neuroinflammation drives both cognitive decline and motor dysfunction in AD has led to numerous studies exploring the efficacy of anti-inflammatory treatments. Parkinson's disease (PD), which also involves chronic neuroinflammation, provides valuable insights into the potential of these treatments. Observational studies in PD have shown that non-aspirin NSAIDs, such as ibuprofen, are associated with a reduced risk of developing PD, and this has sparked further investigation into their use in AD. 143 However, the outcomes in AD trials have been less conclusive.
Early epidemiological studies suggested that long-term use of NSAIDs could reduce the risk of developing AD.144–147 These findings, supported by animal model studies, led to the hypothesis that NSAIDs might also alleviate cognitive decline and motor symptoms once AD has been diagnosed. 148 Despite this promise, clinical trials have consistently failed to show significant benefits in cognitive function for patients already suffering from AD. Drugs such as naproxen, ibuprofen, and celecoxib did not demonstrate substantial improvements in cognition or motor functions like gait.143,149 The mixed results raise important questions about the role of inflammation in cognitive and motor impairments. In PD, NSAIDs have shown some promise in delaying disease onset but have not been successful in modifying disease progression once motor symptoms have emerged. 143 This parallel with AD suggests that anti-inflammatory treatments may be more effective if administered early before substantial neurodegeneration has occurred. Neuroinflammation may contribute to both cognitive decline and gait impairment in AD. Still, the failure of NSAIDs in clinical trials suggests that reducing inflammation alone may not be sufficient to reverse these impairments once the disease has progressed. 149 Therefore, recent research has increasingly focused on alternative strategies targeting specific pathways implicated in neuroinflammation. For example, modulating insulin resistance is recognized as a promising approach, given the link between metabolic dysfunction and increased neuroinflammatory processes.150,151 Therapies aimed at improving insulin sensitivity, such as AMPK activators like metformin, have demonstrated potential in reducing inflammatory profiles and cognitive impairment in patients with mild cognitive impairment. 151 Future therapeutic strategies may thus benefit from focusing on metabolic modulation as a means to interrupt the cycle of insulin resistance and neuroinflammation.
Furthermore, in both PD and AD, the role of neuroinflammation is complex. In AD, neuroinflammation affects multiple brain regions, including those responsible for memory, executive function, and motor control, such as the hippocampus and basal ganglia. This suggests that inflammation might be a common cognitive and motor dysfunction driver. However, clinical trials have indicated that the broad anti-inflammatory effects of NSAIDs might not adequately target the specific inflammatory pathways involved in AD. Additionally, certain NSAIDs, such as ibuprofen and indomethacin, have been found to lower Aβ levels, especially the more toxic Aβ42 variant, through γ-secretase modulation, however, these effects are independent of their anti-inflammatory actions. 149 This complexity may help explain the lack of clinical efficacy seen in AD trials (Table 2). Drawing from PD research, it is possible that more targeted anti-inflammatory therapies could yield better results if they are administered early and tailored to specific neuroinflammatory pathways. The failure of COX-2 inhibitors, such as rofecoxib and celecoxib, in AD trials, despite their success in reducing peripheral inflammation, further highlights the need for treatments that more precisely address central neuroinflammation. 143 149
List of anti-inflammatory drugs accepted for clinical trials at ClinicalTrials.gov.

In addition to pharmacological interventions, dietary and microbiome-based approaches have gained attention as alternative strategies for managing neuroinflammation. Nutraceutical compounds such as lactoferrin, polyphenols (e.g., resveratrol, curcumin), and B vitamins have been investigated for their ability to modulate immune responses, reduce oxidative stress, and support neuronal function. 151 Studies suggest that resveratrol activates SIRT1, a protein involved in inflammation control, while curcumin exhibits anti-amyloid and neuroprotective properties.151,152 Moreover, gut microbiome interventions, including probiotics and fecal microbiota transplantation, have demonstrated potential in reducing neuroinflammation and improving cognitive function in preclinical AD models. 153
Moreover, some evidence suggests that neuroinflammation may exacerbate the disease and play a dual role by engaging protective immune responses. The challenge for future treatment lies in striking the right balance between reducing harmful inflammation and preserving these neuroprotective effects.
Recommendations for future research and therapeutic strategies in Alzheimer's disease
This review has emphasized the pivotal role of neuroinflammation in the progression of AD, particularly in its contributions to BBB dysfunction, Aβ accumulation, and subsequent neuronal degeneration. Given that neuroinflammation initially serves a protective function but can transition into a chronic, neurotoxic state, therapeutic interventions must be both targeted and carefully timed. Here, we outline key recommendations to guide future research and clinical strategies for addressing neuroinflammation in AD.
In summary, a multifaceted approach that combines early, targeted, and holistic interventions is essential to effectively address neuroinflammation in AD. Future treatments can more accurately target the underlying mechanisms driving AD progression by focusing on specific inflammatory pathways, protecting BBB integrity, and utilizing personalized biomarker-based diagnostics. Additionally, longitudinal studies on neuroinflammation's impact on motor symptoms and the integration of lifestyle changes could enhance preventive and therapeutic strategies, ultimately improving outcomes for individuals affected by this complex and debilitating disease.
Conclusion
In conclusion, neuroinflammation is a pivotal factor in both cognitive decline and motor impairments observed in AD. The intricate roles of microglia, astrocytes, and the dysregulated release of pro-inflammatory cytokines and chemokines highlight the dual impact of neuroinflammation on cognitive and motor circuits. As inflammation exacerbates synaptic dysfunction, oxidative stress, mitochondrial impairment, and BBB breakdown, the convergence of these factors accelerates neurodegeneration, impacting cognition, balance, and motor coordination. Future research should focus on developing targeted anti-inflammatory treatments that can mitigate both cognitive and motor dysfunction early in AD progression, potentially offering new therapeutic strategies that address the multifaceted effects of neuroinflammation.
Footnotes
Acknowledgements
We would like to thank all the researchers whose work contributed to the understanding of neuroinflammation in Alzheimer's disease.
Author contributions
Funding
The author(s) received no financial support for the research, authorship, and/or publication of this article.
Declaration of conflicting interests
The author(s) declared no potential conflicts of interest with respect to the research, authorship, and/or publication of this article.