
Abstract
Background:
Phage therapy has demonstrated remarkable efficacy in treating multidrug-resistant (MDR) Klebsiella pneumoniae-related infections. However, information on the physicochemical stability and molecular characteristics of Klebsiella phages is limited.
Materials and Methods:
Phages targeting MDR K. pneumoniae subsp. pneumoniae clinical isolates were isolated from wastewater through the enrichment method. Host range analysis, efficiency of plating, and genomic studies were conducted to identify the most effective phage for further growth kinetics, physicochemical studies, and molecular characterization, respectively.
Results:
Klebsiella phage vB_Kpn_001_Koku targeting MDR K. pneumoniae subsp. pneumoniae isolated from market sewage exhibited optimal viability at 4°C and at pH 7.5. Its genome has a size of 38,232 bp of double-stranded linear DNA with a G + C content of 51% and 29 functional genes (54.7%), with no deleterious genes.
Conclusion:
Klebsiella phage vB_Kpn_001_Koku is the best candidate for phage therapy against MDR K. pneumoniae subsp. pneumoniae owing to its broader host range, stability across a wide range of temperatures and pH levels, and notable molecular characteristics.
Keywords
Introduction
About one-third of all gram-negative infections are caused by the lactose-fermenting, encapsulated, nonmotile bacterium Klebsiella pneumoniae. 1 K. pneumoniae accounts for ∼85% of clinical isolates within the K. pneumoniae species complex (KpSC), making it of significant clinical importance. 2 There are two known K. pneumoniae pathotypes: classical K. pneumoniae (opportunistic strains) and hypervirulent K. pneumoniae, hvKp (strains that infect both healthy and immunocompromised individuals with primary liver abscess, a hallmark infection in the absence of hepatobiliary disease). 3 The rmpA and rmpA2 genes (capsular production regulatory genes) are hvKp-specific, making molecular markers the most accurate way to distinguish between the two strains. 4
K. pneumoniae colonization in the mucosal surfaces of the gastrointestinal and/or oropharynx of humans is a major contributing factor to the establishment of infection, but not necessarily a prerequisite condition. 5 Colonization, together with possession of several virulence factors by the bacterium (capsule, 6 lipopolysaccharide, 7 fimbriae, 8 and siderophores9,10), facilitates the establishment of K. pneumoniae infections such as pneumonia, 3 urinary tract infections, 11 and either primary or secondary bacteremia 12 with subsequent metastatic spread, causing meningitis, endophthalmitis, empyema, spontaneous bacterial peritonitis, and pyomyositis.13,14 K. pneumoniae harbors several antibiotic resistance genes located on either its chromosome (transposon) or the plasmid. 15 This bacterium has also developed mechanisms that hinder antibiotic effectiveness, such as biofilm formation, which contribute significantly to the emergence of antibiotic-resistant strains.16,17
Global pooled prevalence of nosocomial multidrug-resistant (MDR) K. pneumoniae infections is ∼32.8%. 18 In Kenya, the prevalence of MDR K. pneumoniae in one of the tertiary hospitals was estimated to be 23%. 19 The neonatal unit accounted for an unexpected 82.6% of these clinical isolates. There is an urgency to develop alternative, innovative therapeutic measures, such as bacteriophages, to eradicate these MDR strains. Phage therapy uses the bacterial viruses (bacteriophages) for the management of antibiotic-resistant infections with great success. 20 Several studies have documented the efficacy of phage therapy in controlling MDR K. pneumoniae-related infections, such as pneumonia, 21 chronic urinary tract infections, 11 osteomyelitis, 22 burn wound infections, 23 prosthetic joint infections, 24 and bacteraemia.21,25
There are limited data concerning the physicochemical stability and molecular characteristics of MDR K. pneumoniae phages; thus, this study focused on isolating and characterizing MDR K. pneumoniae phages, especially at the molecular level, to rule out the presence of virulence-related genes, lysogenic genes, and antibiotic-resistant genes, a critical step before phage therapy application.
Materials and Methods
Host bacterial strains and antibiotic susceptibility testing
A total of 10 clinical Klebsiella-like isolates were outsourced from the National Microbiology Reference Laboratory-Nairobi, Kenya, including KPATCC, a standard bacterial culture grown on Mueller–Hinton agar. 26 The isolates were subcultured in Tryptone Soy Broth (TSB) for 24 h at 37°C to assess their viability and on MacConkey agar (HiMedia, Mumbai, India) to characterize them phenotypically. 27 These clinical isolates underwent automated identification and antibiotic susceptibility testing (AST) using the VITEK 2 system version 9.02 machine as previously described. 28 The clinical isolates that were found to be MDR strains were used as bacterial hosts for phage isolation.
Sample collection
Wastewater samples (1 L each) were collected from five locations within the Nairobi, Kilifi, and Kajiado counties from June 2023 to February 2024. The sampling sites included markets, hospitals, ferry terminals, sewage treatment plants, and residential areas. At each sampling site, wastewater was collected at three different points (upstream, middle section, and downstream for sewage and inlet, midsection, and outlet points for sewage treatment plants) as previously described. 29 The collected wastewater samples were coded as KSTW (from Nairobi), KOFFS, MH, KM (from Kilifi), and ORH (from Kajiado), while the sampling points were numbered 1, 2, and 3, thus making a total of 15 wastewater samples. The wastewater samples were collected and placed in dark containers, then transported in cooler boxes at a temperature of 4°C to the Kenya Institute of Primate Research facility, where they were processed within 2 weeks.
Phage isolation, purification, and titration
Bacteriophages were isolated from wastewater samples using an enrichment method. 30 Briefly, 45 mL of each of the wastewater samples (n = 15) was centrifuged at 7,871 g at 21°C for 10 min, then filtered through a 0.22 µm syringe filter. Approximately 2.5 g of the TSB powder and 100 µL of host bacteria were added to the filtrate and then incubated in a shaker incubator at 2 g for 18 h at 37°C. Subsequently, centrifugation was done at 7,871 g for 10 min at 4°C. The mixture was then filtered through a 0.22 µm microfilter, and the obtained phage lysate was stored at 4°C in sterile 1.5 mL Eppendorf tubes for phage screening.
The susceptibility of each bacterial isolate to the isolated phage was obtained through a spot test as described previously. 31 In summary, the phage lysate was first serially diluted using saline magnesium buffer, SM buffer (10 mM magnesium sulfate, 100 mM sodium chloride, 50 mM Tris-HCl, and 0.01% w/v gelatin, pH 7.5) in decimal dilutions (20 µL:180 µL). The top agar was prepared by dispensing 100 µL of each of the host bacterial isolates into 5 mL of warm semisolid media (TSB powder and 0.6% agar–agar powder; HiMedia, Mumbai, India), mixed gently, then laid onto respective TSA plates. Once the overlay was set after 15 min, 2 µL of the diluted phage lysate was spotted on TSA plates, set to dry for 15 min in the laminar flow, and then incubated for 3–24 h at 37°C depending on the host bacterial growth rate.32,33 The presence of plaques postincubation was recorded as positive for phage screen, and the plaques were counted to determine the plaque-forming units per milliliter (PFU/mL) per source.
Following phage screening, the titration and purification processes were achieved using the methods previously described. 31 Approximately 25, 50, or 100 µL of diluted phage lysate and 100 µL of host bacterial culture (3 h postincubation) were added to 5 mL of warm semisolid media, shaken, and overlaid on a TSA plate, and then incubated at 37°C for 3 h. A distinct plaque was picked using a sterile Pasteur pipette and resuspended in 200 µL of saline magnesium buffer. The process was repeated for a maximum of four cycles to obtain pure phage, and its titer was determined.
Determination of host range and efficiency of plating
Into 5 mL of warm semisolid media, 100 µL of host bacteria was added and laid on a TSA plate. Approximately 2 µL of each phage was spotted on the host bacterial lawn in duplicate, and the plates were then incubated at 37°C for 3 h. The formed plaques were classified according to the validated scoring system, whereby score +4: complete clear zone (complete lysis); score +3: mostly clear zone with faint hazy background (lysis with minimal bacterial resistance); score +2: pronounced turbidity throughout a clear lytic zone (lysis with marked bacterial resistance); score +1: individually opaque zone (lysis with obvious bacterial resistance); and score 0: no lytic zone. 34
Each isolated phage was spotted on the K. oxytoca bacterial lawn to determine phage species-specific characteristics using the spot test method described above. The plates were observed for lytic zones and recorded.
The isolated phages that lysed two or more of the six MDR host bacterial strains with a score of +4 were selected for efficiency of plating (EOP) studies; decimal dilutions and spot tests were carried out as described above, and the test strain titer was determined (PFU/mL). The EOP was given by the ratio of test strain titer to the reference titer (phage stock titer). Phage efficiency was categorized as inefficient (EOP < 0.001), low productive (0.001 > EOP < 0.1), moderately productive (0.1 > EOP < 0.5), or very productive (EOP ≥ 0.5). 35 The mean of two independent measurements was used to report the results.
Determination of multiplicity of infection
The most potent phage was further characterized to determine its growth kinetics. The optimal multiplicity of infection (MOI) was determined by the method as previously described. 36 Briefly, 500 µL of a bacterial culture (106 CFU/mL) was mixed with 500 µL of phage at different titers (102, 103, 104, 105, 106, 107, and 108 PFU/mL) and incubated at room temperature for 15 min. Into each 1 mL of bacteria–phage mixture, 9 mL of TSB was added and cultured in a shaker incubator at 120 rpm for 2 h at 37°C. Centrifugation at 7,871 g for 10 min at 21°C followed by serial dilution of the supernatant using SM buffer and phage titer determination by a double-layer agar method whereby the highest production indicated the optimum MOI.
Determination of a one-step growth curve
One-step growth curve determination was achieved using the method as previously described. 37 Approximately 500 µL of the most potent phage suspension was added to an equal volume of an exponentially growing bacterial host at an MOI of 0.01. The mixture was incubated at 37°C for 15 min to allow phage adsorption.34 After adsorption, the mixture was centrifuged at 12,298 g for 2 min to remove unadsorbed phages, the pellet was resuspended in 10 mL of TSB, and incubated further at 37°C for 2 h. At an interval of 10 min, the phage titer was determined using the double-layer agar method in duplicate. The burst size of the phage was determined as the ratio of phage titer (maximum phage titer minus phage titer at 0 min) to that of the initial infected host bacterial cells. The results were presented as the average of two separate measurements.
Determination of physicochemical stability
The most potent phage tolerance at different temperatures and pH was determined using the method as previously described. 36 Briefly, 1 mL of pure phage at 1010 PFU/mL was placed at 4°C (control temperature), 25°C, 37°C, 45°C, 60°C, and 70°C. The latter three temperatures were maintained using a water bath. The phages were incubated for 60 min, and every 10 min, starting from 0 min, the phage titer was determined using the double-layer agar method. The results were recorded as the mean of two independent values. Using the SM buffer adjusted to pH of 2, 4, 6, 7.5 (control pH), 8, 10, 12, and 14, 0.1 mL of pure phage at 1010 PFU/mL was serially diluted into 0.9 mL of the respective pH-adjusted buffers. The diluted phage was incubated at room temperature for 60 min before phage titer determination using the double-layer agar method. 36 Results were recorded as the mean of two independent measurements.
Phage DNA extraction
The most potent phage with the widest host range, having an EOP of >0.5, was subjected to molecular characterization. Before the phage DNA extraction process, phage sample pelleting was done to concentrate the phage solution. First, a pure phage solution was propagated using a method previously described 31 followed by filtration of the high-titer phage solution through a 0.22 µm syringe filter. At a concentration of 1010 PFU/mL, ∼20 mL of the pure phage solution was centrifuged at 17,709 g for 6 h at 4°C. After centrifugation, the supernatant was discarded, leaving behind 2 mL for pellet resuspension. The resuspended pellet was first disrupted and stored at 4°C, ready for genome extraction.
Bacterial host DNA and RNA acids were eliminated by treating the sample with DNase I and RNase A (Thermo Fisher Scientific, USA), followed by deproteinization using Proteinase K at 56°C for 1 h and 30 min. Phage DNA was then extracted using the QIAamp® DNA Mini Kit (Qiagen) according to the manufacturer’s instructions. 38 The purity of the extracted phage DNA was assessed using a Nanodrop One spectrophotometer (Thermo Fisher Scientific), and its concentration was quantified with a Qubit fluorometer (Invitrogen).
16S PCR to confirm removal of host DNA
To confirm the removal of contaminating host bacterial DNA, PCR targeting the 16S rRNA gene was performed using the universal primers 27 F and 1492 R. Reactions were set up in a 25 µL volume containing 12.5 µL of DreamTaq (Thermo Fisher Scientific), 1.0 µL each of 10 pmol primers, and 2.0 µL of template DNA. PCR conditions included an initial denaturation at 95°C for 1 min, followed by 40 cycles of 95°C for 1 min, 60°C for 30 s, and 72°C for 45 s, with a final extension at 72°C for 10 min. 38 PCR products were resolved on a 1.25% agarose gel stained with SYBR™ Gold nucleic acid gel stain (Thermo Fisher Scientific) and visualized using the iBright Vision Imaging System.
Whole-genome sequencing
Long-read sequencing libraries were constructed using NEBNext reagents and Rapid PCR Barcoding Kit 24 V14 (SQK-RPB114.24) following the manufacturer’s protocol. 39 Libraries were loaded onto R10.4.1 FLO-MIN114 flow cells and sequenced on minION using MinKNOW software version 24.06.16. Dorado v0.7.0 was utilized for base-calling (super accuracy model), trimming of adapters, and filtering low-quality reads (Q score <8). The raw reads’ quality was checked using the NanoQC pipeline (Nanoplot v.14.2). 40
De novo assembly and functional genome annotation
The Flye tool v2.9.4 was used to assemble the raw reads de novo. 41 The quality of the assembly was assessed using Quast. 42 The phage genome was annotated using Pharokka v1.7.1. 43 Coding sequences (CDSs) were identified with PHANOTATE, 44 while tRNA and tmRNA genes were predicted using tRNAscan-SE 2.0 45 and Aragorn, 46 respectively. CRISPR elements were detected with the CRISPR Recognition Tool. 47 Functional annotations were assigned by aligning CDSs to the PHROGs, 48 Virulence Factor Database, 49 and Comprehensive Antibiotic Resistance Database 50 using MMseqs2 51 and PyHMMER. 52 In addition, contigs were compared against the INPHARED database 53 using Mash 54 to determine their closest reference matches. The annotated phage genome was visualized using Circular Genome View (CG View/Proksee). 55 Finally, the phage’s life cycle was predicted using PhageLeads (https://phageleads.dk/) 56 and was taxonomically classified using the taxmyPhage program. 57
Comparative genome analysis
The phylogenetic analysis was based on the large terminase subunit, the conserved region within the phage genome. From the NCBI BLASTp database (https://blast.ncbi.nlm.nih.gov/Blast.cgi), 58 approximately 22 phages that were homologous to the most potent isolated phage, having a query sequence coverage of 100%, were first aligned using the ClustalW algorithm tool before phylogenetic tree construction. 59 To achieve comparative genomic analysis, the phylogenetic tree was constructed by the MEGA 12 tool using the neighbor-joining method and the Jones–Taylor–Thornton model with bootstrap analysis (1,000 replicates). 60 Using the Virus Intergenomic Distance Calculator tool, 61 the intergenomic comparison of the isolated whole phage genome with its 16 homologous phages from the NCBI nucleotide database having a query coverage of >90% was also determined.
Statistical analysis
The analysis was conducted using GraphPad Prism version 8.0.2 software, and the data were represented as the mean and standard deviation.
Results
Phenotypic identification of host bacterial strains
A total of 10 clinical bacterial isolates, including KPATCC, a standard bacterial culture, were purely lactose fermenters exhibiting shiny, pinkish, mucoid medium- to large-sized colonies after being incubated overnight at 37°C on MacConkey agar plates.
Automated host bacterial strain identification and AST
The 10 clinical bacterial isolates evaluated by the VITEK system version 9.02 machine were found to have a 98–99% probability of being K. pneumoniae subsp. pneumoniae. Resistance to ampicillin, tetracycline, and chloramphenicol was ∼100%, while resistance to levofloxacin, ceftriaxone, cefuroxime, cefepime, and aztreonam was ∼60%. Ampicillin/sulbactam was 57.14% resistant, whereas resistance to cefazolin was 42.86%. Nitrofurantoin, trimethoprim, and trimethoprim/sulfamethoxazole antibiotics were 28.57% resistant. The least antibiotic-resistant profiles were found in gentamicin and ciprofloxacin (14.29%), while only 10% of the isolates were resistant to meropenem (Fig. 1).
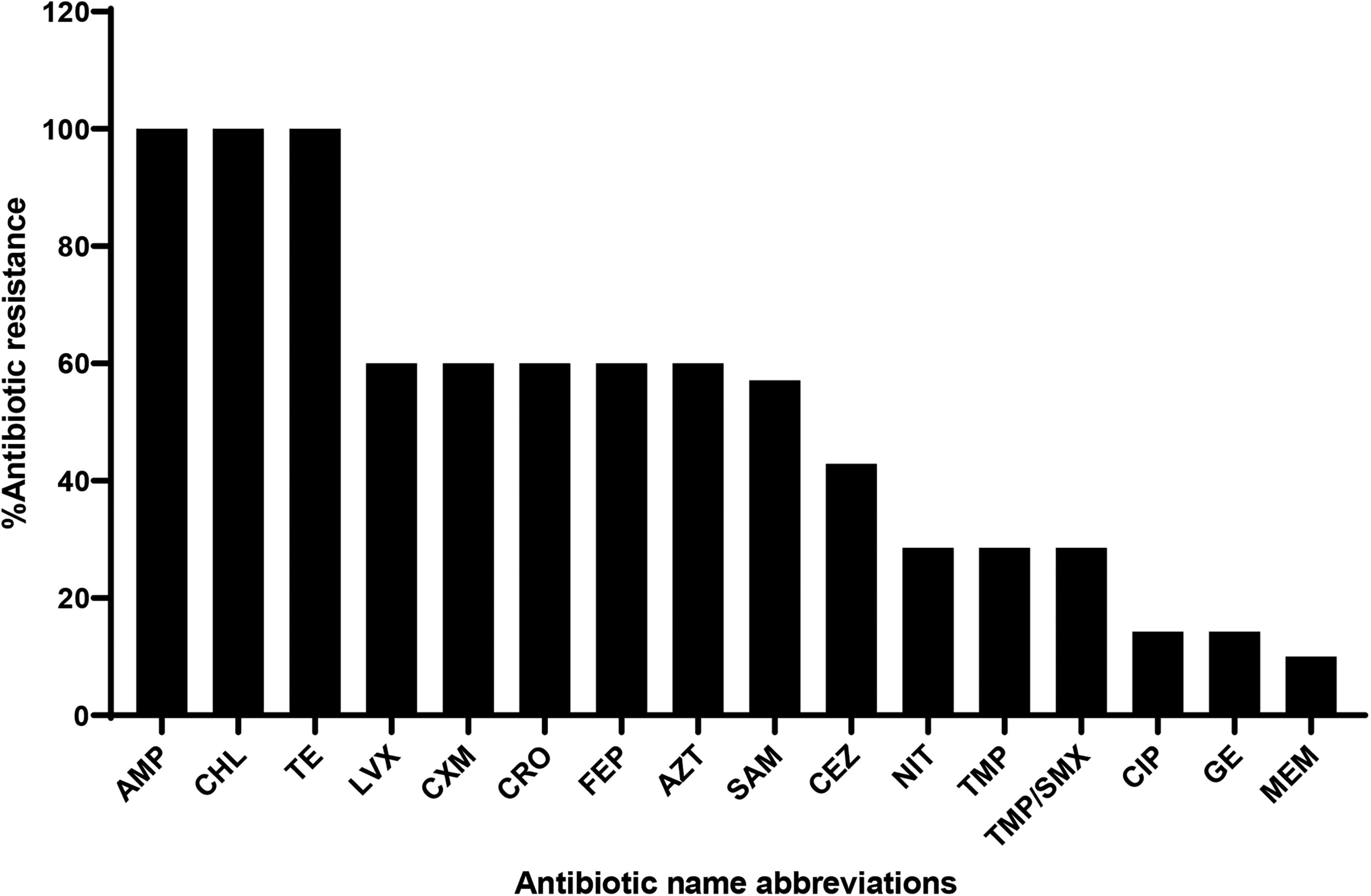
Antibiotic resistance profile of Klebsiella pneumoniae subsp. pneumoniae clinical isolates determined by the VITEK system version 9.02. The results were interpreted as being sensitive
Of the 10 K. pneumoniae subsp. pneumoniae isolates, 6 out of 10 (60%) of the isolates displayed a MDR pattern (Table 1).
Multidrug-Resistant Pattern of Klebsiella pneumoniae subsp. pneumoniae Clinical Isolates
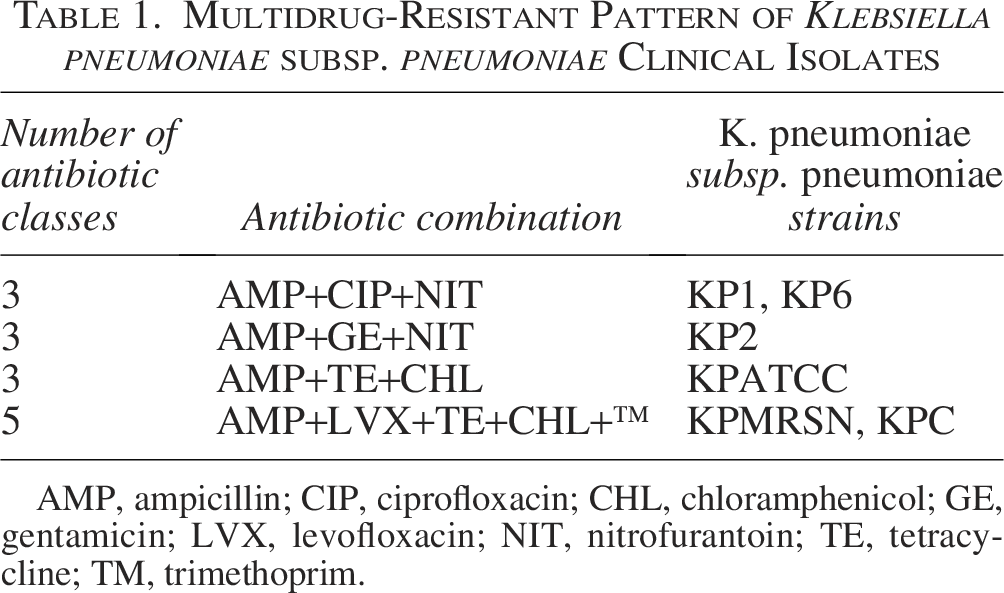
AMP, ampicillin; CIP, ciprofloxacin; CHL, chloramphenicol; GE, gentamicin; LVX, levofloxacin; NIT, nitrofurantoin; TE, tetracycline; TM, trimethoprim.
Isolation and characterization of phages targeting MDR K. pneumoniae subsp. pneumoniae
Six MDR K. pneumoniae subsp. pneumoniae clinical isolates (KP1, KP2, KP6, KPC, KPATCC, and KPMRSN) were used as bacterial hosts to isolate phages from three out of five wastewater sources (ORH, KM, and MH). A total of 16 phages against MDR K. pneumoniae subsp. pneumoniae numbered P1–P16 were isolated; ineffective phage P13 was dropped from the analysis, thereby leaving 15 phages (Table 2).
Number of Phages Against Multidrug-Resistant Klebsiella pneumoniae subsp. pneumoniae Isolated from Each Wastewater Source
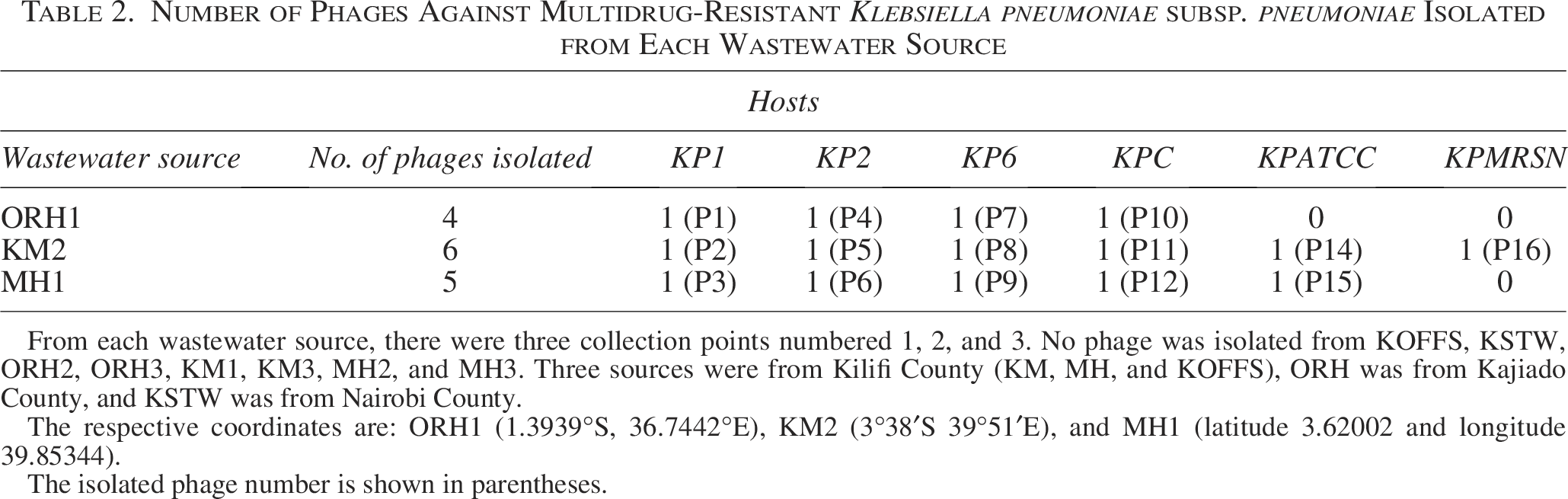
From each wastewater source, there were three collection points numbered 1, 2, and 3. No phage was isolated from KOFFS, KSTW, ORH2, ORH3, KM1, KM3, MH2, and MH3. Three sources were from Kilifi County (KM, MH, and KOFFS), ORH was from Kajiado County, and KSTW was from Nairobi County.
The respective coordinates are: ORH1 (1.3939°S, 36.7442°E), KM2 (3°38′S 39°51′E), and MH1 (latitude 3.62002 and longitude 39.85344).
The isolated phage number is shown in parentheses.
All the isolated phages against MDR K. pneumoniae subsp. pneumoniae formed clear plaques (2–5 mm) with halo nuclei signifying phage depolymerase enzyme activity on the host bacterial polysaccharide matrix (Fig. 2).
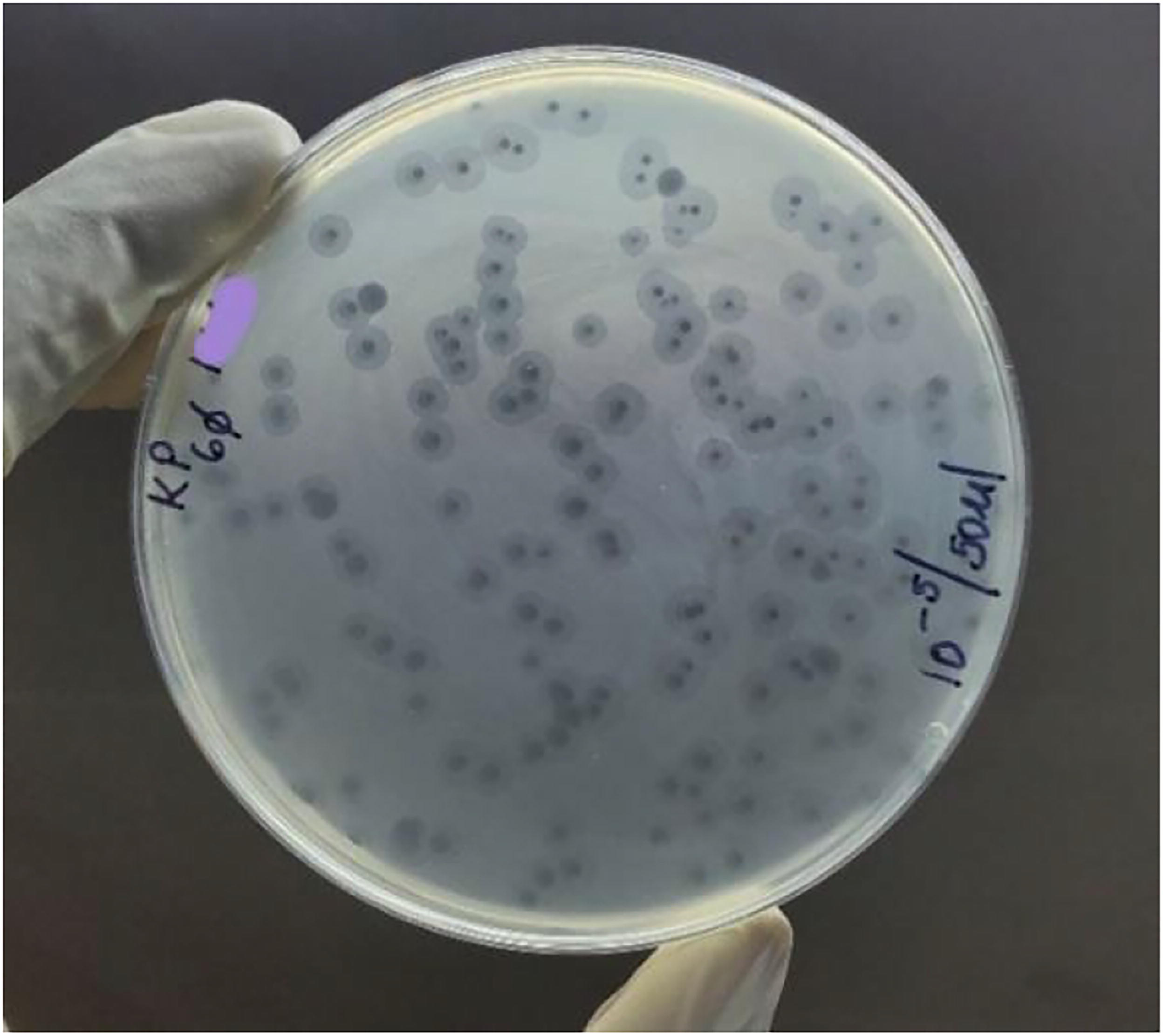
Clear phage-mediated host bacterial lysis with halo nuclei signifying the activity of phage depolymerase enzyme on the bacterial polysaccharide matrix.
Host range analysis
The most potent phage was phage number 11 (P11), while the least potent phage was phage number 6 (P6). Phage potency from highest to lowest: 100% (P11), 83.33% (P7, P14, and P16), 66.67% (P3, P10, P12, and P15), 50% (P5 and P9), 33.33% (P1, P2, P4, and P8), and 16.67% (P6). The most susceptible bacterial strain was KP2, while KPMRSN was the least susceptible bacterial strain. Bacterial susceptibility trend from most to least susceptible: 93.33% (KP2), 80% (KP1), 60% (KPC), 53.3% (KP6), 46.67% (KPATCC), and 13.33% (KPMRSN) (Fig. 3).
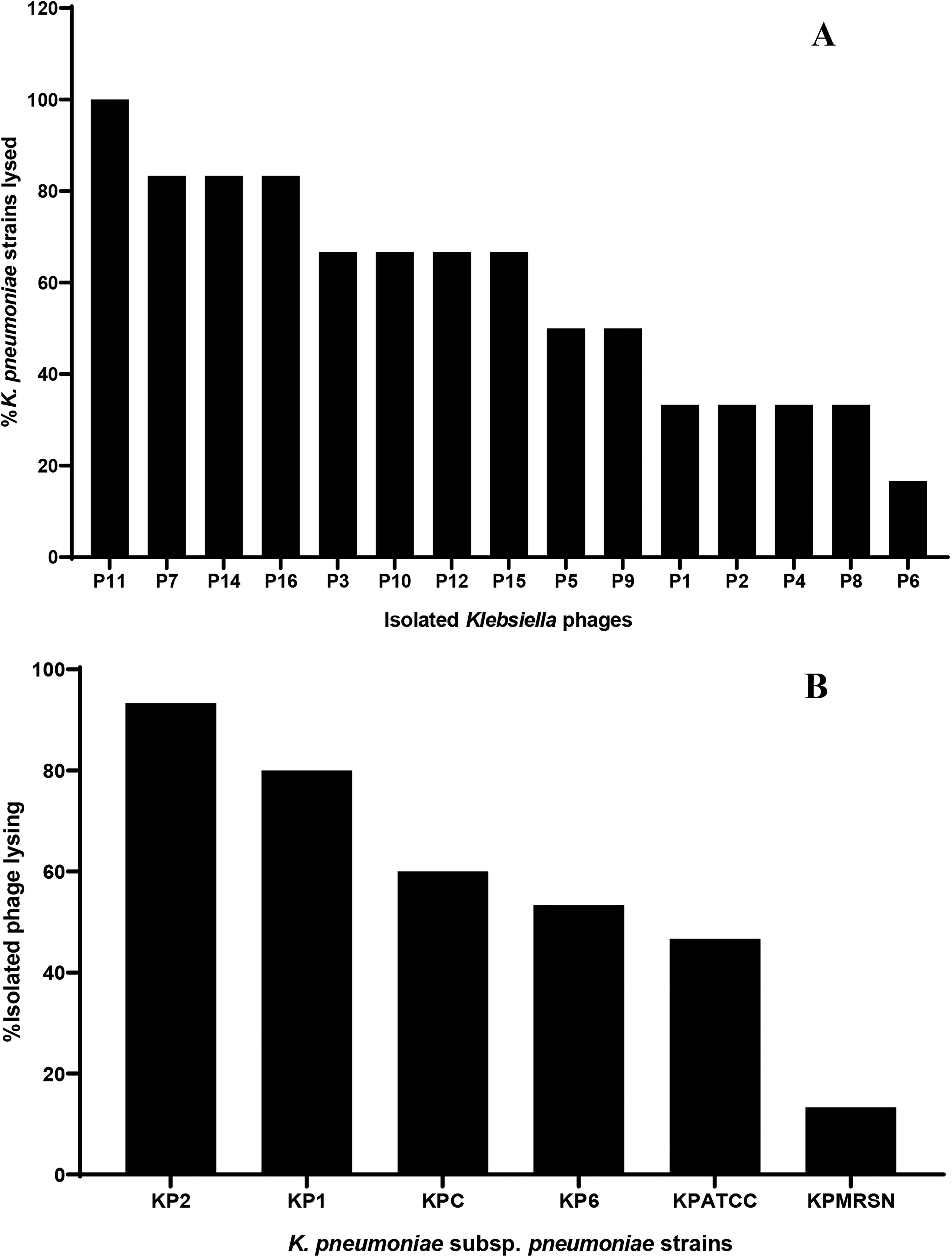
Host range analysis of 15 isolated phages against MDR Klebsiella pneumoniae subsp. pneumoniae.
Potent phage efficiency of plating
Out of the 16 phages isolated against MDR K. pneumoniae subsp. pneumoniae, 5 phages fulfilled the selection criteria for the EOP study (P10, P11, P12, P14, and P15) (Supplementary Table S1). P11 completely lysed three out of six strains of K. pneumoniae subsp. pneumoniae (KP6, KPC, and KPATCC) with a score of +4 (Fig. 4A–C).
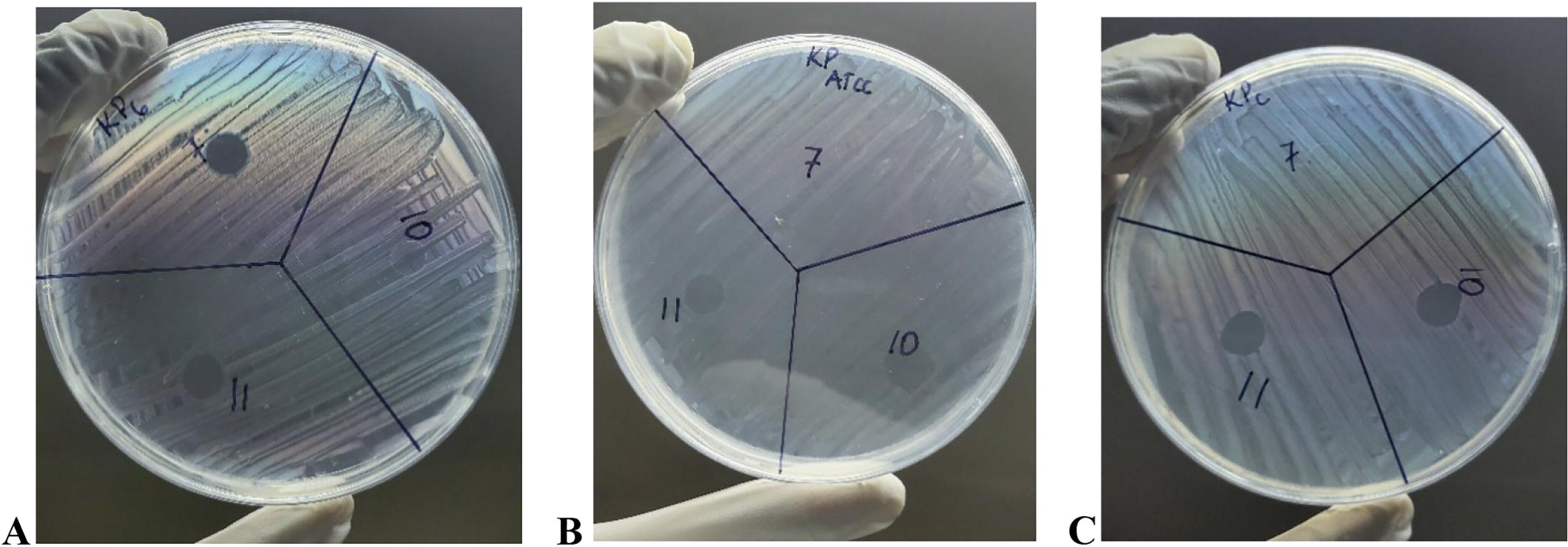
Complete lysis (score +4) of KP6, KPATCC, and KPC by phage P11.
Phage number 10 (P10) produced an EOP of >0.5 (high production) in two different MDR K. pneumoniae subsp. pneumoniae strains (KPC and KPATCC) compared with P11 and P12, which produced EOP >0.5 in a single MDR strain each (KPC). P14 and P15 produced an EOP of <0.5 (moderate production) on the KPATCC strain (Fig. 5).
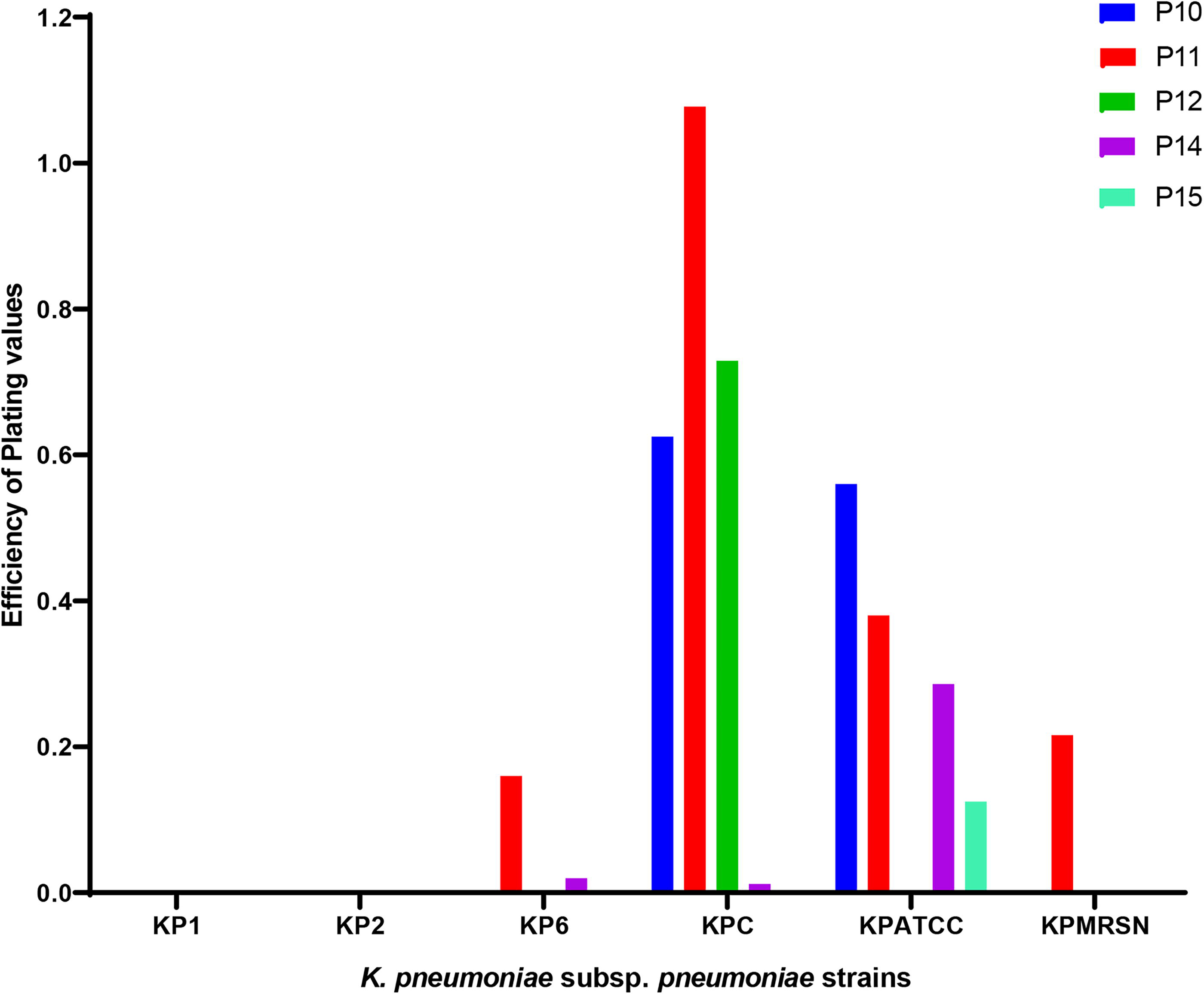
Efficiency of plating of five phages against MDR Klebsiella pneumoniae subsp. pneumoniae. Each phage was spotted on all six MDR K. pneumoniae subsp. pneumoniae bacterial lawns (18-h-old cultures) in duplicate and incubated for 3 h at 37°C. The ratio of test strain titer to reference titer (EOP) was determined.
Isolated phage species specificity
None of the isolated phages against MDR K. pneumoniae subsp. pneumoniae lysed the K. oxytoca bacterial lawn (Supplementary Fig. S1).
MOI and one-step growth curve of the most potent phage (P11)
The optimum MOI giving the highest phage titer was 0.01 when 104 PFU/mL of phage was incubated with 106 CFU/mL of the bacterial host. The one-step growth curve of P11 revealed that the phage maintained a latent period of ∼40 min, and the plateau phase was attained at 70 min. The calculated burst size at 70 min was therefore 30 PFU per host bacterial cell (Fig. 6).
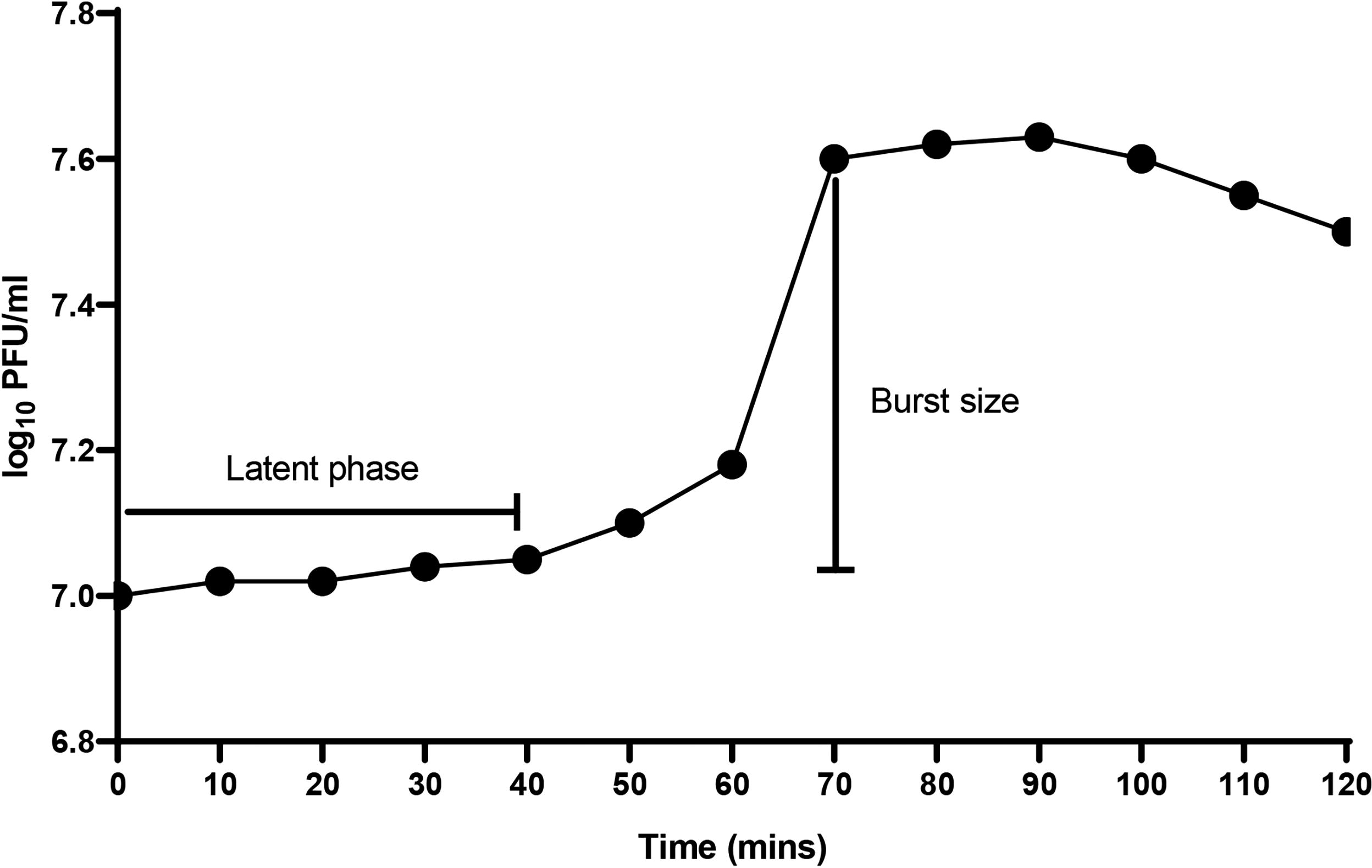
One-step growth curve of phage P11 in Klebsiella pneumoniae subsp. pneumoniae strain (KPC). Phage P11 was added at a multiplicity of infection (MOI) of 0.01 to the host bacterial culture, allowed to adsorb for 15 min, centrifuged, and the pellet with adsorbed phages resuspended in Tryptone Soy Broth (TSB) media and cultured. At regular intervals, the phage concentration in log10 PFU/mL was determined. Results are presented as the mean of two independent values. PFU/mL, plaque-forming units per milliliter.
Physicochemical stability of the most potent phage (P11)
Purified phage P11 at a concentration of 1011 PFU/mL was stable at a wide range of temperatures and pH. In a temperature range between 4°C and 45°C, the percentage phage survival rate was ∼80–100% after 60 min of incubation, with optimum survival at 4°C. At 60°C, the percentage phage survival rate dropped to 40% and went down to 0% within the first 10 min of incubation. No surviving phage was detected at 70°C. In a pH range of between 4 and 10, the phage survival rate was found to be between 50% and 96% after 60 min of incubation, with optimal survival at a pH of 7.5. At extreme acidic and basic pH conditions (pH 2 and pH 12), the phage survival rate was ≤10%. At pH 14, there were no surviving phages detected (Fig. 7).
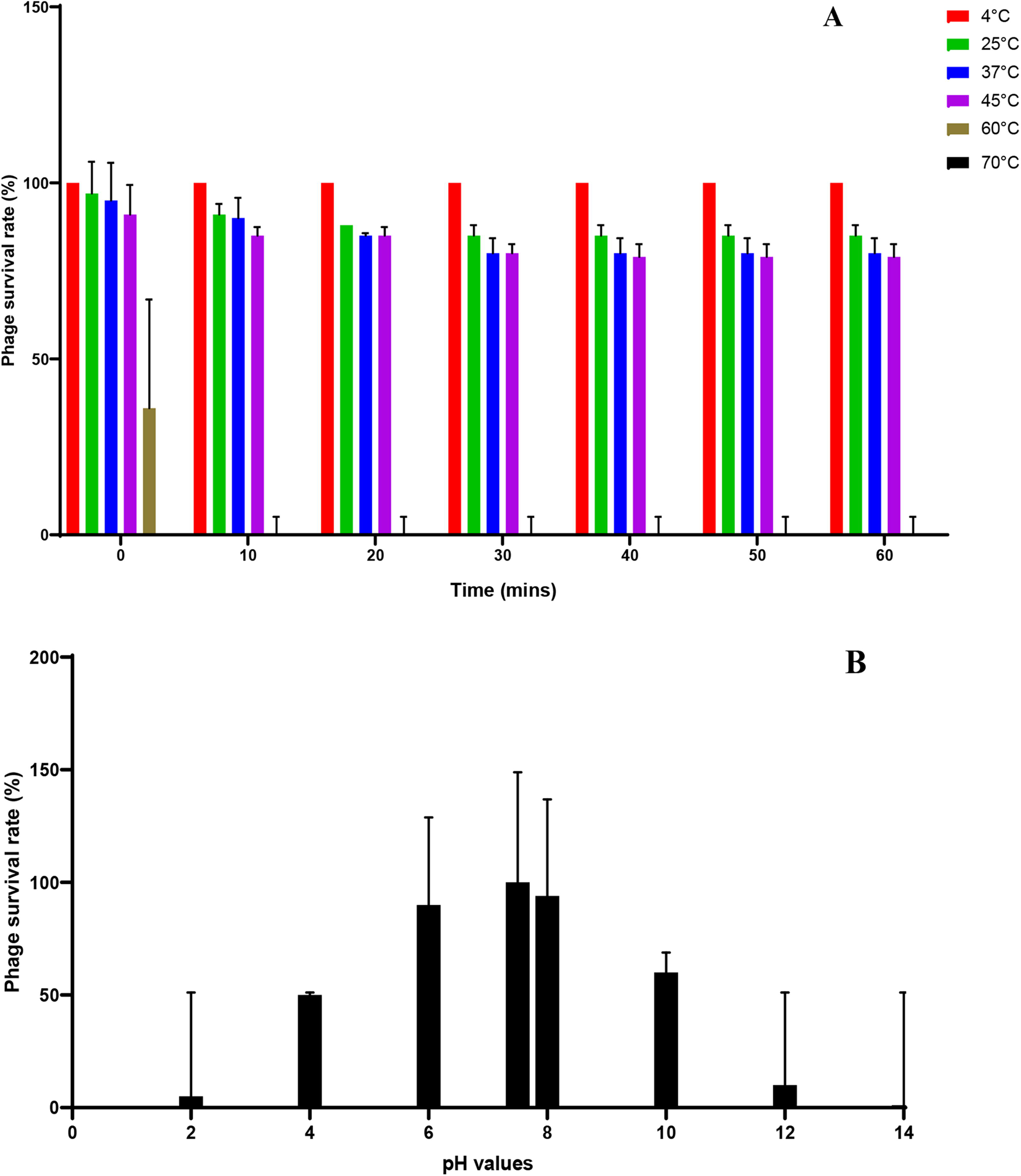
Stability of phage P11 under various conditions:
Molecular characterization of the most potent phage (P11)
The concentrated purified phage P11 at a concentration of 109 PFU/mL had its DNA extracted and found to have a concentration of 13.6 ng/µL (Nanodrop) and 11.6 ng/µl (Qubit). The protein concentration (A260/280) was 1.84, while the salt concentration (A260/230) was 1.37. The extracted phage P11 genome was found to have no contaminating host bacterial genome. The sequencing generated 214,156 reads (read N50: 4,477), which were sequenced with a coverage of 259× and assembled into a single contig with a length of 38,232 bp. The contig N50 was 38,232 bp. The phage termini could not be identified due to lack of a comprehensive reference database. Phage P11 was named Klebsiella phage vB_Kpn_001_Koku (accession number PV469479).
The genome of this bacteriophage consists of a double-stranded linear DNA molecule, 38,232 base pairs in length, with a 51% G + C content. No tRNA, tmRNA, CRISPR, virulence, antimicrobial resistance, integration, and excision genes were detected within the phage genome. This phage has a lytic mode of replication and belongs to the family Autotranscriptaviridae, genus Teetrevirus, and an unclassified Teetrevirus species. The genome of Klebsiella phage vB_Kpn_001_Koku was found to contain 53 open reading frames (ORFs), out of which 48 (90.57%) were encoded on the positive strand and the remaining 5 (9.43%) were encoded on the negative strand. Twenty-nine ORFs (54.7%) encoded for functional genes, while 24 ORFs (45.3%) were regarded as having unknown functions (Supplementary Table S2).
The functional genes of Klebsiella phage vB_Kpn_001_Koku were divided into seven modules: connector module, head and packaging module, lysis module, tail module, nucleotide metabolism module, moron, auxiliary metabolic gene, the host takeover module, and the other module. This Klebsiella phage was found to have three lytic proteins (holin, Rz-like spanin, and amidase) and three tail proteins, one being a tail fiber protein. The circular genome map of Klebsiella phage vB_Kpn_001_Koku is illustrated in Figure 8.
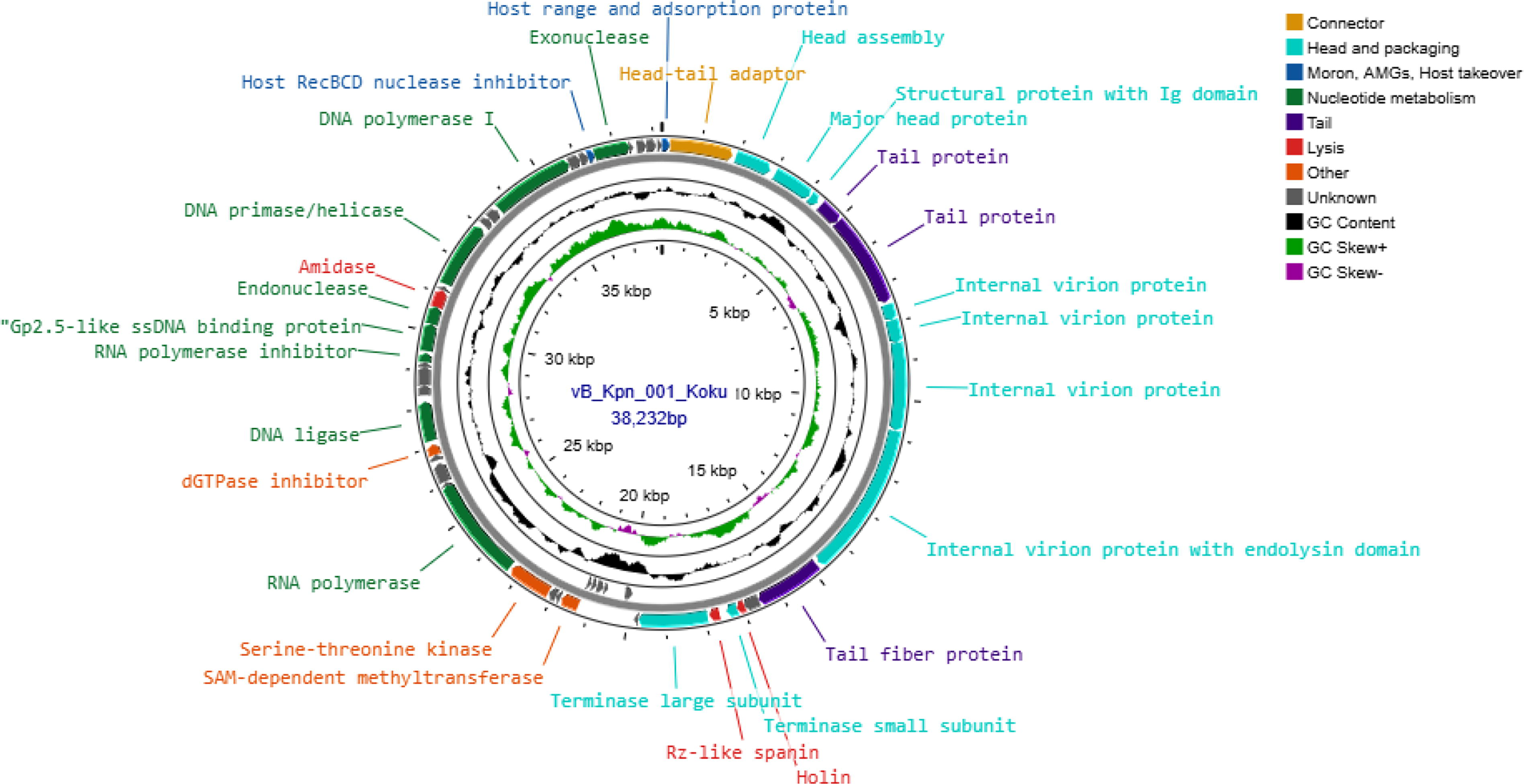
The circular genome map of Klebsiella phage vB_Kpn_001_Koku. The map is divided into four circles. From the outermost to the innermost circle: forward reading frame (positive strand), reverse reading frame (negative strand), GC content, and GC skewness. The genes are divided into eight modules represented by different colors: the connector module (light brown color), head and packaging module (turquoise color), the moron, AMGs, and host takeover module (dark blue color), nucleotide metabolism module (dark green color), tail module (dark purple color), lysis module (red color), the other module (orange color), and the genes with unknown function (gray color).
Comparative genomic analysis
The phylogenetic tree was constructed using large subunits of the terminase protein, a conserved protein within the phage genome, to extrapolate the evolutionary relationship of Klebsiella phage vB_Kpn_001_Koku with the other 22 phages having a query sequence coverage of 100% from the NCBI BLASTp database (Supplementary Table S3). Klebsiella phage vB_Kpn_001_Koku was found to form clusters with other phages, and it was more closely related to Serratia phage SM9-3Y isolated from raw hospital sewage in China than to Citrobacter phage SH1 that was isolated from a treatment plant in Tunis (Fig. 9).

Phylogenetic tree of Klebsiella phage vB_Kpn_001_Koku terminase large subunit sequence (highlighted in red) with the other 22 phages’ terminase large subunit sequences from the NCBI BLASTp database with 100% query sequence coverage. The sequences were aligned using the ClustalW algorithm, and the evolutionary history was inferred using the neighbor-joining method. The evolutionary distances were computed using the Jones–Taylor–Thornton (JTT) matrix-based method of MEGA 12 software. The percentage of replicate trees above 50% in which the associated taxa clustered together in the bootstrap test (1000 replicates) is shown next to the branches.
Upon intergenomic comparison of the Klebsiella phage vB_Kpn_001_Koku with 16 other phages from the NCBI nucleotide database having a query sequence coverage of >90%, the intergenomic similarity was found to be between 70.8% and 77.1% (Fig. 10).
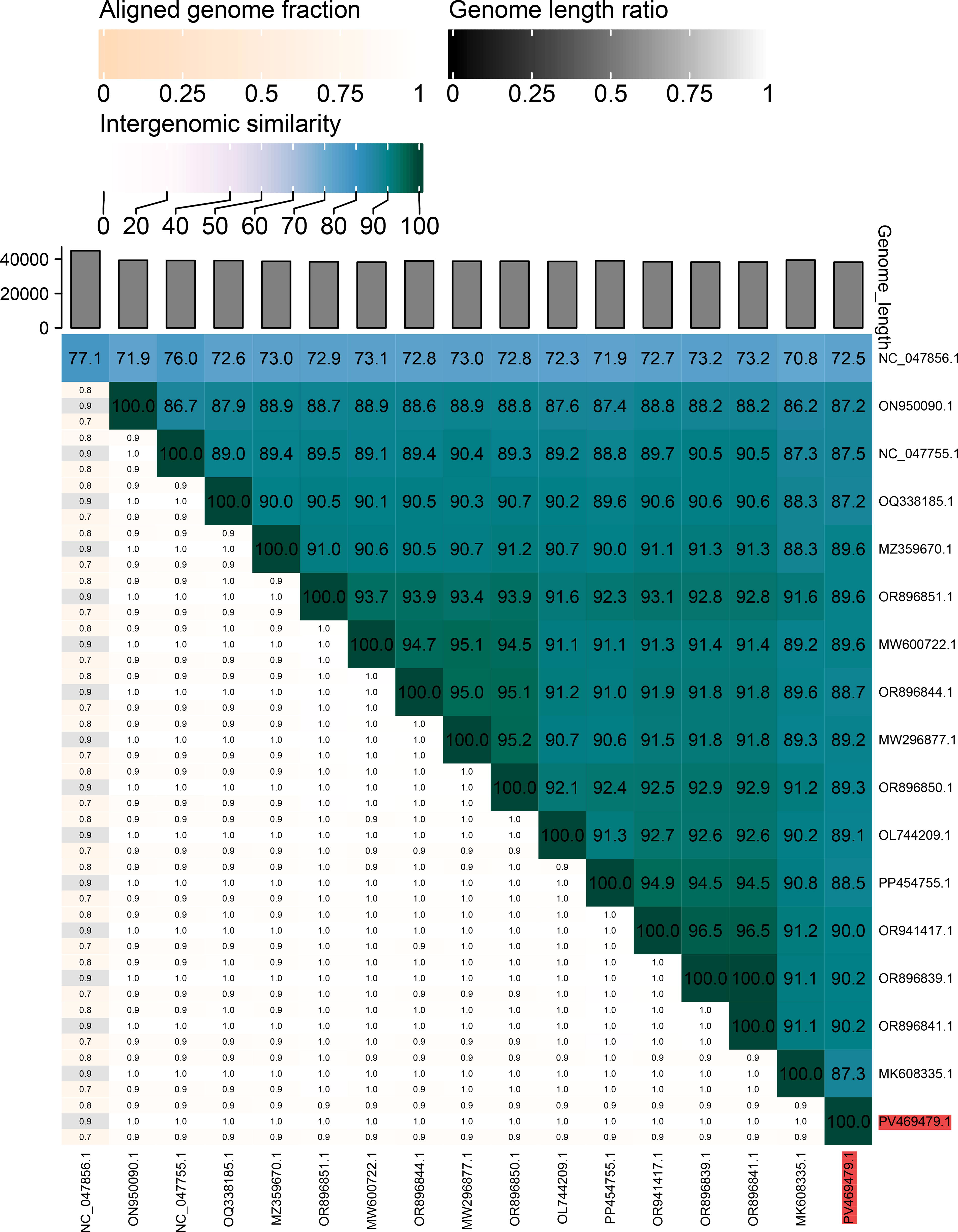
A heatmap created by the VIRIDIC analysis tool showing the intergenomic similarity of the isolated Klebsiella phage vB_Kpn_001_Koku genome (accession number highlighted in red) with other 16 phages’ nucleotide sequences from the NCBI nucleotide database, having a query sequence coverage of >90%.
Discussion
K. pneumoniae is accountable for both community-acquired and hospital-acquired infections, whose severity depends on the immune status of the infected individual and the antibiotic resistance pattern of the infecting bacterial strain.5,62 Global prevalence of MDR K. pneumoniae is alarming, necessitating a need for research and development of alternative therapeutic measures. 18 From this study, out of 10 identified K. pneumoniae subsp. pneumoniae clinical isolates, six were MDR strains exhibiting 100% resistance to ampicillin, chloramphenicol, and tetracycline, and 10% resistance to meropenem. A similar antibiotic resistance profile was reported by a study conducted in one of the tertiary hospitals in Kenya. 19
This study managed to isolate a novel Klebsiella phage, vB_Kpn_001_Koku, from the market sewage, displaying 100% potency in lysing MDR K. pneumoniae subsp. pneumoniae clinical isolates, while the least potent phage (P6) exhibited a 16.67% potency. These results are consistent with the results obtained from previous studies: vB_KshKPC-M (97.7%), 33 vB_KleS-HSE3 phage (25%), 63 and Phage IME268 (22.97%) 32 isolated from hospital sewage in China and Iran. Klebsiella phage vB_Kpn_001_Koku showed no lytic potential against K. oxytoca, thus maintaining its species-specific characteristic compared with the vB_KleS-HSE3 phage, which also lysed Yersinia pseudotuberculosis apart from the K. pneumoniae species,64 thus having a polyvalent host range characteristic.
Phage–bacterial coevolution in our environments shapes both the phenotypic and genotypic characteristics of the ecosystem, with the emergence of phage-resistant host bacterial strains and phage variants that have a wider host range with compensatory mechanisms to overcome bacterial resistance. 64 For instance, from this study, market sewage acted as a reservoir of phage variants that have undergone evolution over time, with the resultant ability to target their host bacterial closely related species, which are isolated in the hospitals. Cross-contamination of nosocomial antibiotic-resistant bacterial strains between the community and the hospital settings is significantly suggested in this study.
A total of 16 phages were isolated from environmental wastewater, forming clear plaques with halo nuclei signifying the phage-mediated depolymerase enzyme activity, an enzyme that plays a major role in destroying bacterial polysaccharides and is consequently effective in managing bacterial biofilms. 65 One out of the 16 isolated phages was dropped out of the study (P13) following the development of resistance by the host bacterium. Bacterial resistance to phages has been documented previously by several studies, which is highly contributed to by the development of bacterial protective mechanisms to phage infection, bacterial-mediated abortive infection, and bacterial-mediated receptor site modifications. 66
To counterattack bacterial defensive mechanisms, phages have also developed several techniques, as displayed by the two important functional proteins coded by the isolated Klebsiella phage vB_Kpn_001_Koku. The SAM-dependent methyltransferase is responsible for mediating the phage DNA methylation process, 67 while the dGTPase inhibitor hinders the activity of the host bacterial dGTPase, an enzyme that hydrolyzes dGTP to dGDP to deplete the levels of dGTP, a crucial DNA building block. 68
The isolated Klebsiella phage vB_Kpn_001_Koku showed a latent period of ∼40 min with a burst size of 30 PFU per host bacterial cell. These results are consistent with those of Phage IME268, which had a latent period of 30 min with a burst size of 40 PFU per bacterial cell. 32 The previously isolated Klebsiella phages vB_KshKPC-M and vB_KleS-HSE3 proved to be more potent since they had a latent period of 20 and 30 min with a burst size of 260 PFU and 277 PFU per bacterial cell, respectively.33,63 Klebsiella phage vB_Kpn_001_Koku is the best candidate for phage therapy compared with the rest due to its widest host range.
The high tolerability to varying temperatures and pH ranges of the isolated Klebsiella phage vB_Kpn_001_Koku makes it suitable for phage therapy application. The phage survived best at temperatures between 4°C and 45°C after 60 min of incubation (80–100% survival rate), thus being suitable for managing patients with severe resistant bacterial infections presenting with hyperpyrexia (body temperature above 41°C), and optimal survival at 4°C is important for storage purposes. This phage survived best at pH between 4 and 10 (≥50% survival rate), with maximum viability at a pH of 7.5. At extreme acidic and basic conditions (pH 2 and 12), the survival rate was below 10%, making this phage suitable for parenteral administration rather than the oral route of administration to evade the low pH in the stomach. No detectable phages were surviving at pH 14. Almost similar results were obtained for the previously isolated Klebsiella phages vB_KshKPC-M, 33 vB_KleS-HSE3, 63 and Phage IME26833 on their survival rate at different temperatures and pH values, although the isolated Klebsiella phage vB_Kpn_001_Koku was the most tolerant.
Klebsiella phage vB_Kpn_001_Koku has a double-stranded linear DNA with a 38,232 bp length and a 51% G + C content. Out of 53 predicted ORFs, 29 were functional (54.7%). The previously isolated Klebsiella phage vB_KshKPC-M, isolated from Iranian hospital sewage, was a linear dsDNA phage, 54,378 bp long with 51.8% G + C content. This phage genome had 84 ORFs, of which 23 were functional (27.4%). 33 Klebsiella phage vB_KleS-HSE3 isolated from a hospital sewage in China was a dsDNA phage, 46,747 bp long with 56.47% G + C content. A total of 67 ORFs were anticipated on this phage genome: 27 were functional (40.3%). 63 Klebsiella phage IME268 from the hospital sewage in China was a dsDNA phage, 49,552 bp long with 50.5% G + C content. Out of 77 ORFs, 33 were functional (42.9%). 32
Klebsiella phage vB_Kpn_001_Koku formed clusters when its terminase large (TerL) subunit sequence was phylogenetically analyzed with other TerL subunit sequences from the NCBI BLASTp database belonging to other phages, having a query sequence coverage of 100%. Klebsiella phage vB_Kpn_001_Koku was more closely related to Serratia phage SM9-3Y isolated from raw hospital sewage in China than to Citrobacter phage SH1 that was isolated from a treatment plant in Tunis. Terminase is a complex made up of two subunits: the TerL subunit and the terminase small (TerS) subunit.
The two subunits play distinct functions during the phage DNA packaging process. The TerS subunit recognizes and binds to the specific packaging sites (cos or pac sites) on the phage DNA. 69 The TerL subunit has both ATPase and endonuclease activities. The TerL subunit cleaves the phage DNA at specific sites to initiate and terminate the packaging process. The cleaved DNA sequence is then translocated into an empty phage capsid, an ATP-dependent process, as the TerL hydrolyzes ATP to produce energy. 70 Both the ATPase and nuclease domains of the TerL are highly conserved and hence used to establish evolutionary relationships between phages. 69
The Klebsiella phage vB_Kpn_001_Koku genome exhibited between 70.8% and 77.1% intergenomic similarity with the other 16 phage genomes downloaded from the NCBI nucleotide database, having a query sequence coverage of >90%. According to the International Committee on Taxonomy of Viruses, the threshold criterion for classifying bacteriophages within the same species is a nucleotide sequence identity similarity of >95%. 71 Thus, the isolated Klebsiella phage vB_Kpn_001_Koku is a novel phage.
Apart from the two distinct proteins, SAM-dependent methyltransferase and dGTPase inhibitor, present on the genome of Klebsiella phage vB_Kpn_001_Koku, amidase (ORF 40) was another distinct encoded protein found on the genome of Klebsiella phage vB_Kpn_001_Koku. Amidase is a catalytic-specific endolysin that cleaves the amide bonds of the peptidoglycan layer of the host bacterial cell wall, eventually releasing new phage particles. 72 Most dsDNA phages lyse the host bacterial cells at the end of each lytic cycle through the holin–endolysin system from within. Once holin accumulates in the cytoplasm, it forms holes in the cytoplasmic membrane, enabling endolysin to escape from the cytoplasm toward the peptidoglycan layer with its subsequent degradation and host bacterial lysis, hence releasing new phages extracellularly. 73
Gram-negative bacterium cell walls are very different from gram-positive bacterium’s; hence, endolysins that target them have different structures. The endolysins targeting the gram-negative bacteria are mainly globular in shape, with a molecular weight of 15–20 kDa, and are composed of an enzymatically active domain (EAD). 74 Few of these endolysins are known to have a modular structure with two endolysin domains, an EAD and a cell wall binding domain (CBD). 75 The former endolysins depend on the positive charge and hydrophobic nature of the EAD to cross the host bacterial outer membrane, hence accessing the peptidoglycan layer. 76 The modular-structured endolysins first recognize special molecules on the host bacterial cell wall using the CBD, followed by enzymatic degradation of the peptidoglycan layer by the EAD. The CBD-lacking endolysins have a low lytic potential compared with endolysins with CBD. 77
Son et al. (2018) showed an auxiliary role of an amidase domain in cell wall binding and exolytic activity of Staphylococcal phage endolysins. It was found that this phage endolysin had a central amidase domain apart from the CBD and EAD. The CBD was found to direct the endolysins to the peptidoglycan layer, increasing the chances of bacterial host cell lysis. The lytic effect was significantly potentiated by the presence of a central amidase domain, which enhanced the binding activity of the CBD to the bacterial peptidoglycan. 78
Conclusion
In conclusion, the isolated novel Klebsiella phage vB_Kpn_001_Koku from market sewage in Kenya, belonging to an unclassified Teetrevirus species of the family Autotranscriptaviridae, is a potent phage for phage therapy application due to its broader host range, high production characteristic, and distinctive functional proteins. Hospital sewage has been documented by several studies as the main source of phages targeting MDR strains of K. pneumoniae, with none from market sewage to the best of our knowledge. Sewage systems located outside the hospitals should be used in the research and development of novel phages with unique properties that are beneficial for the success of phage therapy, unless cross-contamination of MDR bacterial strains between hospital and community settings is effectively controlled. Engineering of phages with functional proteins capable of escaping bacterial defensive mechanisms is another area of research to be emphasized, consequently leading to the production of phages with significant antibacterial activity. Further study of Klebsiella phage vB_Kpn_001_Koku tail fiber proteins will provide an understanding of its 100% potency in lysing MDR K. pneumoniae clinical isolates, thus engineering phages with the widest host range.
Ethical Considerations
The National Commission for Science and Technology issued the research permit for this study (NACOSTI/P/24/414512). The Nairobi Water and Sewerage Company provided authorization for this study for the collection of wastewater samples (NCWSC/HR/TRG.27/VOL 8/22/MMM/ak). The National Microbiology Reference Laboratory-Nairobi, Kenya, issued the clinical isolates (MOH/DLS/NPHL/NMRL/T/02). The Institutional Scientific Ethics Review Committee (ISERC) of the Kenya Institute of Primate Research issued an ethical clearance certificate (REF: ISERC/04/2023).
Authors’ Contributions
I.K.L.: Conceptualization (lead), data curation (lead), formal analysis (lead), investigation (lead), methodology (lead), writing—original draft (lead), and writing—review and editing (equal). I.J.M.: Data curation (equal), formal analysis (lead), methodology (equal), visualization (lead), and writing—review and editing (lead). J.N.: Investigation (equal) and methodology (equal). A.A.J.: Investigation (equal) and methodology (equal). A.O.O.: Investigation (equal) and methodology (equal). J.W.: Data curation (lead), formal analysis (lead), and writing—review and editing (equal). C.K.: Data curation (lead), formal analysis (lead), methodology (equal), and writing—review and editing (equal). J.M.M.: Methodology (equal), supervision (lead), validation (equal), and writing—review and editing (lead). A.K.N.: Methodology (equal), supervision (lead), validation (equal), and writing—review and editing (lead). A.N.: Project administration (lead), supervision (lead), resources (lead), methodology (equal), validation (equal), and writing—review and editing (lead).
Footnotes
Acknowledgments
The authors acknowledge the German Development Bank through the East African Community for funding this study. They are also thankful to the National Microbiology Reference Laboratory, Nairobi, Kenya, for the provision of host bacterial clinical isolates. The authors thank the Kenya Institute of Primate Research (KIPRE) for providing a conducive laboratory infrastructure for wet lab skills and Dr. Lilian Musila of the Microbiology Hub, WRAIR-A Kericho, Kenya, for the sequencing support. They also acknowledge Vanessa Natasha, Martin Georges, and Meshack Tweya of the Microbiology Hub, WRAIR-A, for their contribution to the phage genome sequencing procedure.
Author Disclosure Statement
No conflicts of interest were disclosed by the authors.
Funding Information
This study was sponsored by the Inter-University Council for East Africa (IUCEA) scholarship opportunity offered to I.K.L. The IUCEA received funds from the German Development Bank (KfW) through the East African Community.
Data Availability
All pertinent data for this study are included in the Supplementary Materials or the main text. The whole-genome sequence of the phage is found in the GenBank of the NCBI database with accession number PV469479.
Supplemental Material
References
Supplementary Material
Please find the following supplemental material available below.
For Open Access articles published under a Creative Commons License, all supplemental material carries the same license as the article it is associated with.
For non-Open Access articles published, all supplemental material carries a non-exclusive license, and permission requests for re-use of supplemental material or any part of supplemental material shall be sent directly to the copyright owner as specified in the copyright notice associated with the article.