
Abstract
There is a wide pathological spectrum of kidney sarcomas that show characteristic histology, ontogeny, and clinical-biological behavior. While leiomyosarcomas commonly arise from the capsule, solitary fibrous tumors and clear cell sarcomas typically show renal sinus and medullary epicenter, respectively. Although distribution and imaging findings of some sarcomas may be characteristic, definitive diagnosis warrants histopathological examination following surgery. Renal sarcomas manifest advanced disease at presentation and portend poor prognosis.
Keywords
Renal cell carcinoma (with its histological subtypes) is the most common malignant neoplasm of the kidney that constitutes more than 90% of kidney tumors. Renal sarcomas are rare malignancies that constitute less than 1% of all malignant renal tumors (1). The kidney is the second most common site for genitourinary tract sarcomas that account for 5% of all soft tissue sarcomas. Renal sarcomas may arise from variegated progenitor cell lines present in the renal parenchyma, vasculature, sinus fat, capsule, or embryonic mesenchyme (Table 1). The diagnosis of renal sarcomas is one of exclusion after excluding sarcomatoid transformation of renal cell carcinoma (RCC), metastatic renal involvement from sarcoma elsewhere in the body and renal involvement by a primary retroperitoneal sarcoma, the so-called
Primary renal sarcomas
Synopsis of primary renal sarcomas
Taxonomy of primary renal sarcomas
According to the 2004 World Health Organization (WHO) classification scheme, the primary renal sarcomas are classified based on histopathology into three broad categories: mesenchymal neoplasms; mixed mesenchymal and epithelial tumors; and neuroendocrine tumors (Table 3).
Classification of primary renal sarcoma (based on WHO histological classification)
Malignant mesenchymal neoplasms can be further categorized into pediatric and adult types. Clear cell sarcoma, rhabdomyosarcoma, and extra-skeletal Ewing's sarcoma constitute pediatric sarcomas. Synovial sarcoma and PNET/Ewing's sarcoma represent mixed mesenchymal and epithelial and neuroendocrine tumors, respectively. Adult sarcomas include leiomyosarcoma, liposarcoma, angiosarcoma, solitary fibrous tumor (hemangiopericytoma), malignant fibrous histiocytoma, and osteosarcoma. Leiomyosarcoma is the most common renal sarcoma (40–60%) followed by liposarcoma (10–15%) (4). Most primary renal sarcomas demonstrate unique immunohistochemical and/or molecular abnormalities that permit accurate diagnosis after resection.
Pediatric primary renal sarcomas
Clear cell sarcoma of kidneys
Clear cell sarcoma of the kidney (CCSK), also known as ‘bone metastasizing renal tumor of childhood’, was first reported by Morgan and Kidd in 1978 (7). CCSK is a rare aggressive renal tumor closely resembling Wilm's tumor in clinical presentation. Clear cell sarcoma is the second most common renal malignancy in children with annual incidence of approximately 20 cases in the US. Gene expression profiling has suggested that mesenchymal cell with neural markers to be the probable cell of origin for clear cell sarcoma. CCSK is rarely seen before 6 months or after 14 years of age; the mean age of presentation is 36 months (8). There is a male predilection (male:female ratio 1.6:1). However, CCSK has also been reported in adults up to 30 years; no significant clinical and pathological differences have been reported between adult and pediatric patients (9).
Common clinical presentations include fever, hypertension, flank mass, pain, and hematuria. CCSKs are usually large (mean size 11 cm), often unilateral and monocentric tumors of medullary pyramids; cystic changes are common. Microscopically, numerous histologic variants of CCSK have been reported, with the myxoid variant being the most common. The classical pattern consists of cords of cells separated by regularly spaced, arborizing fibrovascular septa; uniform proliferation of small round cells is diagnostic. The diffuse accumulation of mucopolysaccharide matrix material between neoplastic cells gives rise to the cystic appearance.
On imaging, CCSK appears as a large, well-circumscribed, solid mass with cystic changes; venous invasion is infrequent (5%) but extracapsular spread commonly occurs (70%) (Fig. 1) (10). In addition to frequent bone metastasis, CCSK also metastasizes to lymph nodes, liver, and lung. Prompt surgery coupled with radiation and chemotherapy has significantly brought the mortality down to 30–40% with a 2-year survival rate of 39–49%. Addition of doxorubicin (adriamycin) to vincristine and actinomycin chemotherapy regimens has improved survival rates. The prognosis is poor with an overall survival of 57%. Recurrence may be seen up to 10 years after surgery; 20% of recurrences occur within 3 years after diagnosis and bone metastasis is a common mode of recurrence.

A 5-year old girl with clear cell sarcoma. Axial contrast-enhanced CT scan demonstrates a predominantly hypodense lobulated mass arising from right renal hilum (white arrow); there are ill-defined internal areas of heterogenous enhancement (*); inferomedially the mass is causing scalloping over normally enhancing renal perenchyma (black arrowhead) without evidence of any obvious parenchymal invasion; the medulla have been obliterated medially with only a thin rim of cortex preserved.The inferior vana cava shows presence of tumor thrombosis (black arrow)
Rhabdomyosarcoma
Rhabdomyosarcoma (RMS) arises from skeletal muscle progenitor cells. The majority (60%) of pediatric RMSs originate in the head and neck region or the urogenital tract. Primary renal RMSs are extremely rare. The diagnosis of primary pediatric renal RMS is controversial; while some authorities believe that it might represent ‘rhabdoid’ differentiation in Wilm's tumor or a part of spectrum of a monomorphic nephroblastoma, others believe that pure RMS of the kidney in childhood is a pathologically distinct entity (11, 12). The histological subtypes of RMS include embryonal (botryoid as a subtype of embryonal subtype), alveolar, and pleomorphic variants (8). Due to its origin, immunohistochemical stains are positive for desmin, myoglobin, and myogenin (13). Less than 10 adult cases of primary renal RMSs have also been reported in the literature (14). In adults, renal RMSs mostly show pleomorphic histology (15). Fifty percent of adult patients of kidney RMSs are in the 7th or 8th decade without any sex predilection (15). Clinical presentation is usually with flank pain and palpable mass.
RMSs present as large non-specific soft tissue masses on CT and show poor contrast enhancement. RMS originating from renal pelvis may cause hydronephrosis and delayed contrast excretion and are indistinguishable from transitional cell carcinoma. Diagnosis is made on histopathology following surgical excision. Radical nephrectomy is the treatment of choice. RMSs are very aggressive neoplasms with dismal prognosis resulting in death within 2 years (14).
Primary neuroendocrine renal sarcoma
Extraosseous Ewing sarcoma/primitive neuroectodermal tumor
Primitive neuroectodermal tumors (PNETs) are derived from the neural crest and belong to a family of ‘small round blue cell tumors’. Primary PNET of kidney is a rare tumor with 50 cases reported in the literature (16). PNETs are commonly seen in childhood or adolescence; two-thirds of cases occur before the age of 35 years (median age 20 years) (17). It is very difficult to differentiate PNET and extraosseous Ewing Sarcoma as separate entities. Both share common stem-cell precursor and unique chromosomal abnormality t(11; 22) (q24; q12) (16, 18). However, the stage of differentiation in which the stem-cell precursor are blocked is different in both the tumors, explaining their different biological behavior and prognosis (16). Tumor cells demonstrate neurosecretory granules on electron microscopy; diffuse CD99 positivity in the cytoplasm of the tumor cells are characteristic (16). Tumors also exhibit diffuse strong membrane positivity for MIC2, which allows confident differentiation from other primary renal neoplasms (19). At histology, Homer-Wright type rosettes are a hallmark of PNETs and may differentiate them from extra-skeletal Ewing's sarcoma.
On CT, renal PNETs are large heterogeneous tumors that may replace the kidneys. Diffuse calcification, areas of internal hemorrhage or necrosis, and peripheral hypervascularity are common (Fig. 2) (17, 20). On MR, PNETs demonstrate heterogeneous intermediate to high T2 signal intensity (21). Venous extension into the renal vein, inferior vena cava and right heart has been described (20). PNETs exhibit aggressive course characterized by early metastatic disease (25–50% at the time of presentation) and death. Metastases commonly occur in the lungs, bones, and the liver (22). PNETs demonstrate high response (94% complete response) to a combination of surgery, irradiation, and chemotherapy (23).
A 41-year-old man with extraosseous renal Ewing's sarcoma. Axial contrast-enhanced CT scan demonstrates a large well-circumscribed heterogeneously enhancing lobulated solid soft tissue density mass (white arrow) arising from the posteromedial aspect of the left renal lower pole region. The heterogeneity is due to internal areas of necrosis or hemorrhage (*) and the periphery of the mass (black arrowhead) shows enhancement pattern corresponding to the retained renal parenchyma (back arrow), splayed along the anterior aspect of mass
Adult primary renal sarcomas
Leiomyosarcoma
Leiomyosarcoma is the most common histological subtype of renal sarcoma comprising 58% of all kidney sarcomas (6). Leiomyosarcoma (LMS) arise either from the renal capsule or from the smooth muscle of the renal pelvis or the renal vasculature (24). LMSs commonly affect patients during the 6th decade with a female preponderance (25, 26). Common presenting symptoms include flank pain, abdominal mass and hematuria; spontaneous rupture has also been reported (24, 27). Histologically, LMS shows interlacing spindle-shaped cells with cellular atypia, closely interspersed with variable amount of connective tissue (8, 28). LMSs are positive for vimentin and muscle specific antigens, while sarcomatoid variants of RCC are not (26, 29).
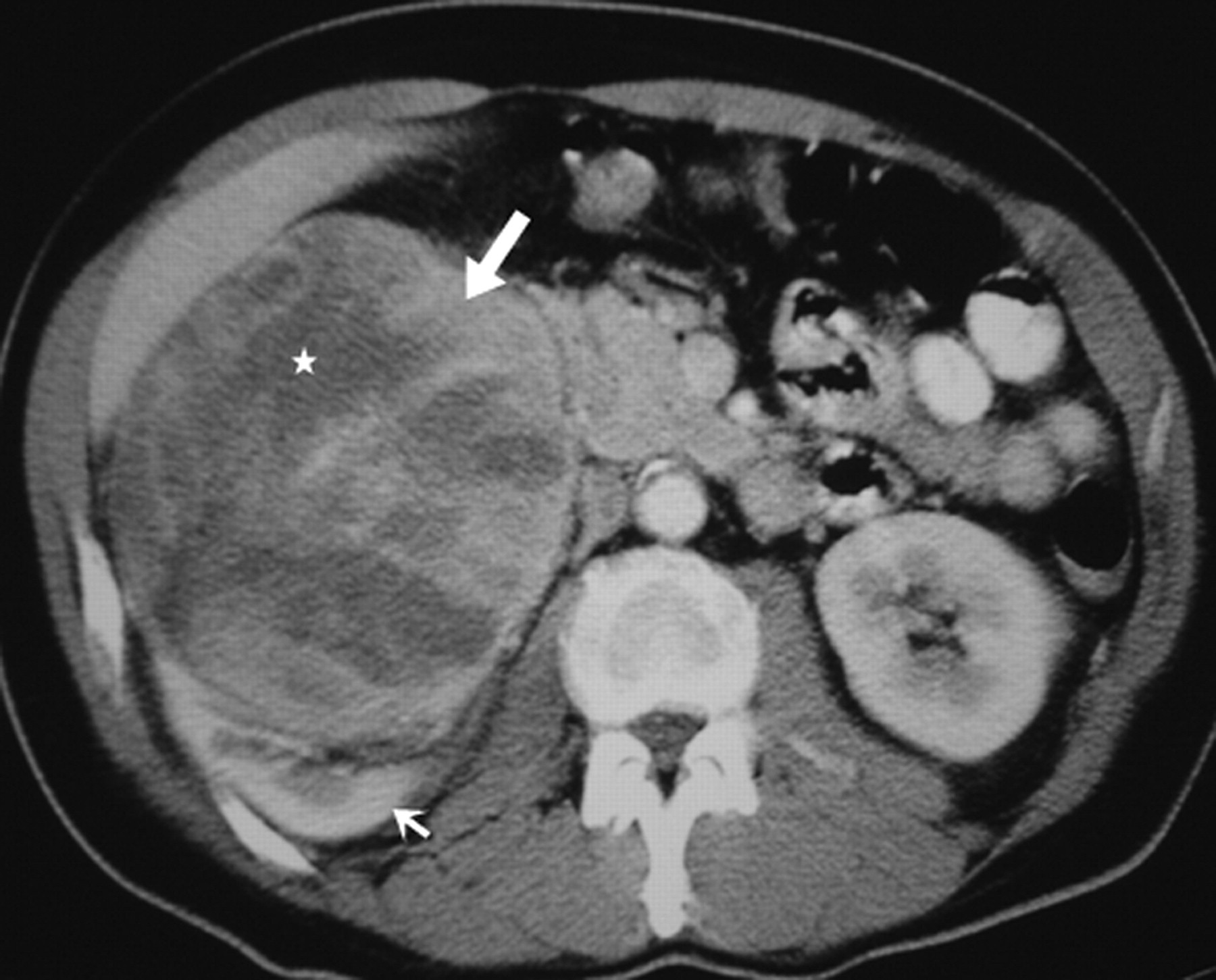
On cross-sectional imaging studies, renal LMS manifests as a large, well-circumscribed, encapsulated, heterogeneous soft tissue mass that usually project exophytically from the kidney cortex (Fig. 3). Ten percent of tumors show calcification. On non-contrast CT, LMS appears isoattenuating to the paraspinal muscles and hyperdense to the renal parenchyma. On contrast-enhanced CT, LMS shows variable enhancement of the fibrous stroma (Fig. 4) (30). LMS complicated by rupture appears as a heterogeneous mass with foci of hyperattenuation; hematoma in the perinephric space may be an associated finding. Presentation of a multi-loculated cystic mass with peripheral enhancement is distinctly rare. While LMS arising in the renal pelvis may prolapse into the collecting system, tumors originating in the renal vein may grow into the inferior vena cava. LMSs appear iso- to hypointense on T1-weighted (T1W) images, iso- to hyperintense on T2-weighted (T2W) images and show delayed enhancement following administration of contrast due to abundant fibrous connective tissue (Fig. 5) (30).
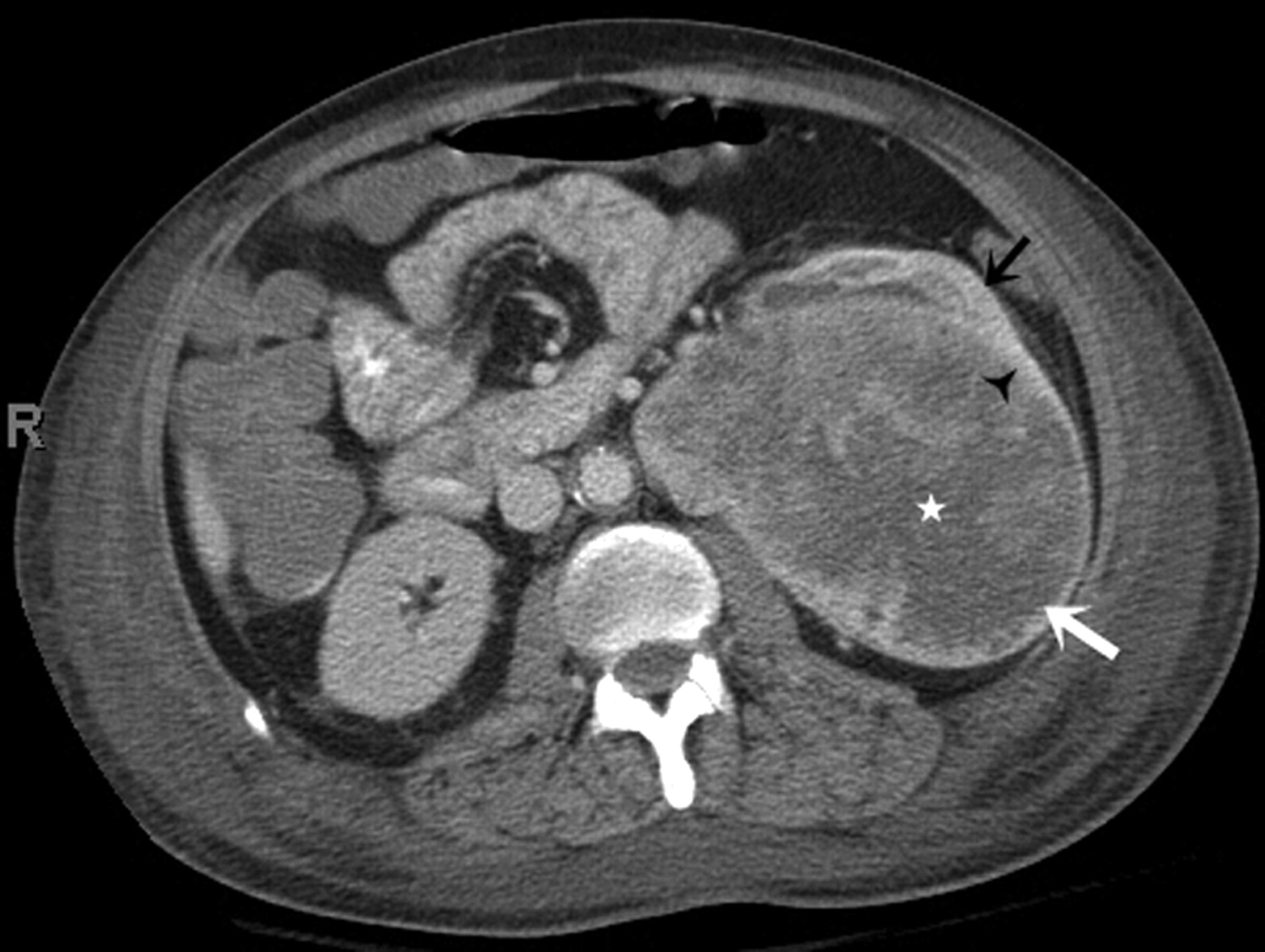
A 49-year-old man with capsular leiomyosarcoma. Axial contrast-enhanced CT scan (arterial phase) demonstrates a well-circumscribed hypodense non-enhancing mass (large white arrow) arising from the anterior aspect of left renal upper pole with peripheral capsular rim enhancement (white arrowhead). There is scalloping of underlying renal parenchyma without evidence of any invasion (small black arrow). The growing mass is invading anterior pararenal space and the pancreatic tail (small white arrow); the pancreas is pushed anteromedially. There is evidence of fat stranding along the posterolateral aspect of mass (*). Incidentally, an enhancing right adrenal nodule is seen (black arrowhead)
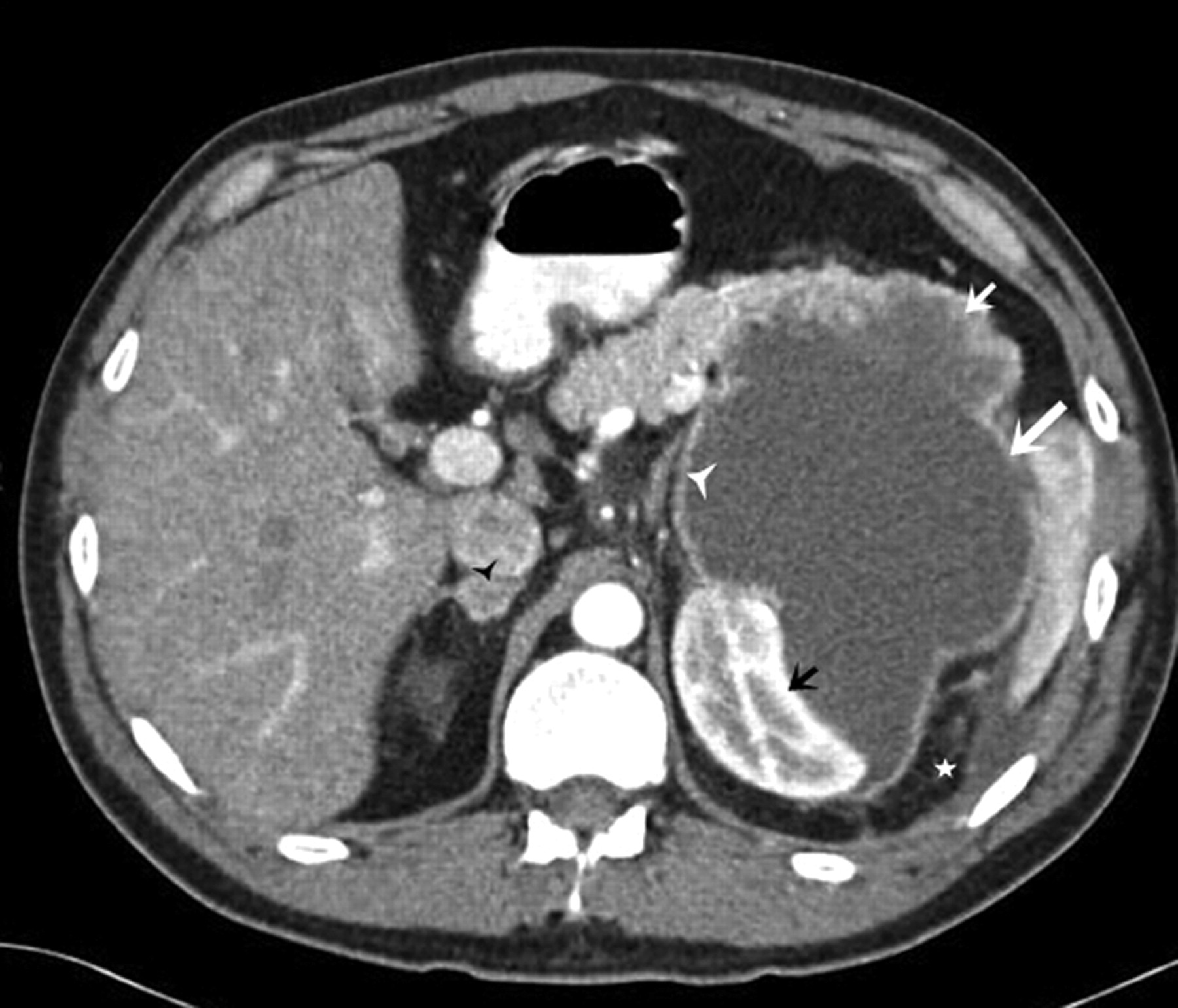
A 60-year-old man with renal leiomyosarcoma. Axial contrast-enhanced CT scan demonstrates a large, heterogeneously enhancing solid soft tissue density mass arising from the anterior aspect of the right renal midpolar region and splaying the renal parenchyma (solid white arrow). The heterogeneity is due to internal areas of necrosis (*) as the mass outgrows its vascular supply. The adjacent normally enhancing renal parenchyma, seen posteriorly (small white arrow)
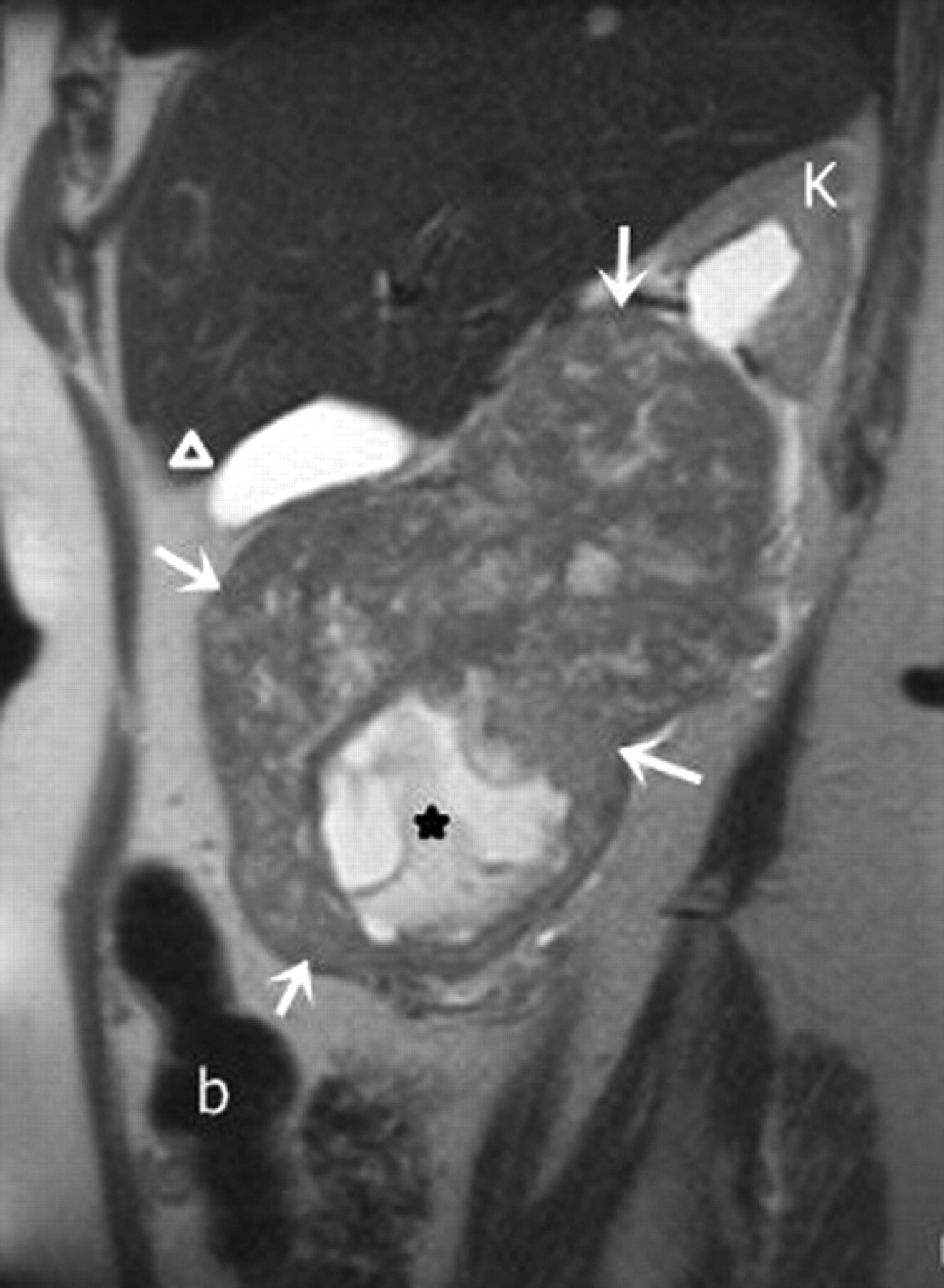
A 74-year-old woman with leiomyosarcoma. Sagittal T2-weighted MR image shows a large, iso- to hyperintense heterogeneous solid mass (arrows) arising from inferior aspect of right kidney; growing anteromedially and displacing the otherwise normal-appearing splayed upper pole renal parenchyma towards left side (K). Inferiorly, a well-defined focal hyperintense area (*) is consistent with necrosis. Bowel (b); gallbladder (arrowhead)
Treatment of choice is radical resection; irradiation and chemotherapy are not effective. Favorable prognostic factors include size <5 cm, low histological grade, absence of lymph node metastasis and complete surgical excision (31). Overall, LMS demonstrates an aggressive and rapidly progressive natural history; local and distant recurrences are common. The prognosis is uniformly poor with a 5-year survival rate of 29–36% (32).
Liposarcoma
Primary retroperitoneal liposarcomas (LPSs), the most common retroperitoneal sarcomas, may arise in peri- or pararenal space and encase, surround or displace the kidney. On the other hand, primary LPSs of kidneys are rare sarcomas that originate from the lipoblasts of renal sinus fat and renal capsule and distort the architecture of the kidney (33, 34). Less than 50 cases of primary renal LPSs have been documented in English literature to date. Primary renal LPSs are commonly seen in 4th and 6th decades (34). Like other renal sarcomas, LPSs remain clinically asymptomatic till they are large. Common presenting symptoms include pain, hematuria, abdominal mass, or loss of weight (35). Histologically, LPSs have been classified as myxoid (60%), well-differentiated (25%), and pleomorphic (10%) types; the majority of published cases of primary LPSs are well-differentiated tumors (35).
On CT and MRI, LPSs often appear as well-circumscribed, heterogeneous, soft tissue tumors that demonstrate variable proportion of macroscopic fat. However, undifferentiated LPSs with minimal or no fat content are difficult to distinguish from other sarcomas. LPSs may extend into the renal vein (36). Primary fat-containing LPSs cannot reliably be distinguished from the more common angiomyolipomas (AMLs) of the kidney. However, recently described ‘angular interface sign’ is a useful sign suggesting benign nature of the mass. This sign describes that benign lesions shows a definable apex within the renal parenchyma and an exophytic component beyond the renal capsule (37). In addition, angiomyolipoma commonly show enlarged vessels on contrast-enhanced CT; this feature may differentiate it from well-differentiated liposarcomas, especially when the AML originates from the renal sinus (38). Epithelioid cell (progenitor cells of AMLs) and its cellular derivatives show immunoreactivity for melanocyte markers such as HMB-45 that helps to distinguish AMLs from primary renal LPSs (39).
Treatment of choice is radical nephrectomy, with or without radiotherapy. The prognosis varies with differentiation, grade, size and stage of the tumor. Late recurrences have been seen with LPSs up to 13 years following resection (40).
Angiosarcoma
Angiosarcoma (AGS) originates from the endothelium of the blood and lymphatic vessels. Most renal AGSs are metastatic in origin. Primary renal AGSs are extremely rare with less than 30 cases reported to date in English literature (41).The mean age of presentation is 58 years (range 30–77 years) (8). An androgenic factor has been postulated due to its strong male preponderance. Extra-renal AGSs are known to be associated with exposure to vinyl chloride, Thorotrast and radiation; these risk factors have not been described with primary renal AGSs. Common symptoms include flank pain and hematuria; AGSs may also manifest as large kidney masses that may rupture spontaneously (42). Histologically, the tumor cells form closely packed sinusoidal vascular channels lined by polygonal or spindle-shaped tumor cells. Immuno-histochemistry exhibits positivity for endothelial cell markers such as factor VIII, antibodies anti-CD34 and CD31 (8).
On imaging, AGSs appear as large, heterogeneously enhancing soft tissue masses with hypervascular and non-enhancing hemorrhagic components. Metastases are common at the time of diagnosis. Treatment of AGSs consists of radical nephrectomy followed by chemotherapy with isofamide or Taxanes (paclitaxel or docetaxel). The size of the tumor and presence or absence of metastases at the diagnosis play crucial role in determining prognosis. Size exceeding 5 cm signifies approximately 13% 5-year survival (41). AGSs metastasize to bone and liver; the mean survival rate after diagnosis of metastatic disease remains as low as 13 weeks (41).
Solitary fibrous tumor/hemangiopericytoma
Solitary fibrous tumor (SFT) and hemangiopericytoma represent a histological gamut of fibroblastic-type of mesenchymal neoplasms with overlapping clinical, imaging, and cytopathological characteristics. Most SFTs were erroneously characterized as hemangiopericytomas and were thought to originate from and/or display pericytic differentiation (43, 44). The new WHO classification of soft tissue tumors now classifies most hemangiopericytomas as solitary fibrous tumors (45).
Primary renal SFTs are rare and less than 40 cases have been reported in English literature (46). Most SFTs show a benign clinical course and originate in renal capsule, interstitial or peripelvic connective tissue. However, the malignant nature and sarcomatous transformation of SFTs have been described (46, 47). The age of presentation ranges between 28–72 years; 50% of reported cases occur between 3rd and 4th decades and 70% of patients were women (15). SFTs manifest with a painless lump, flank pain, or hematuria (15, 48). Microscopically, SFTs show spindle cells with variable density and admixture of dense collagen bundles (15).
On CT scans, SFTs manifest as centrally located soft tissue masses with both early and persistent contrast enhancement (Fig. 6) (49, 50). On MRI, SFTs are isointense to the kidney on T1W and show hypointensity on T2WI. SFTs typically demonstrate hypervascularity and persistent enhancement following contrast administration. A ‘spoke-wheel pattern of enhancement’ has also been described, akin to that seen with renal oncocytomas (51). A characteristic hypervascular pattern in the early arterial phase characterized by large vessels encircling the tumor with displacement of the main arteries around mass has been described on catheter angiography with hemangiopericytoma (52). Due to lack of definitive pre-operative diagnosis and overlapping imaging features with renal cell carcinomas, most SFTs are surgically resected. Although SFTs usually show indolent course, metastasis has been described even in histopathologically characterized ‘benign’ SFTs, necessitating a long-term follow-up (53).
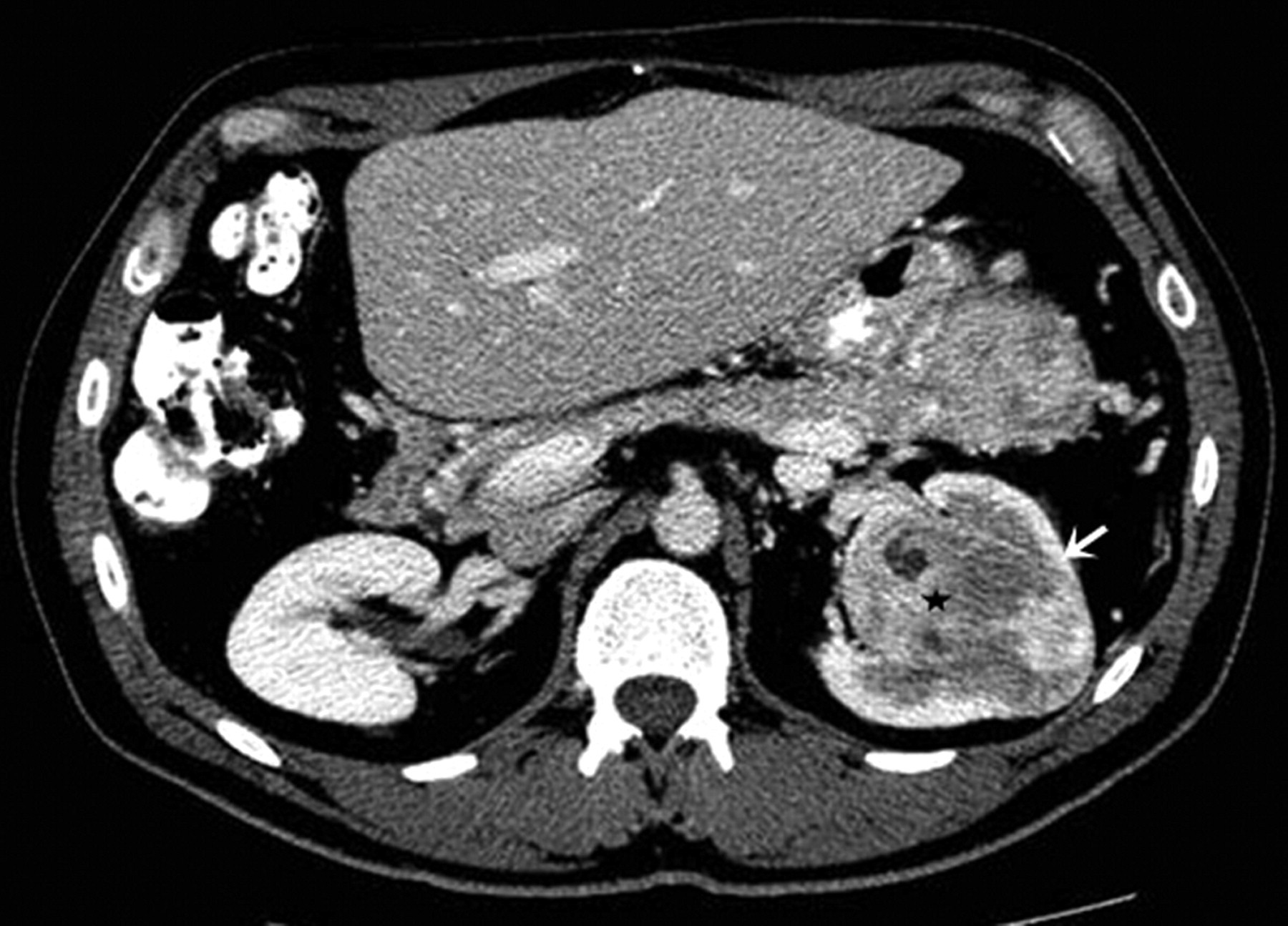
A 45-year-old woman with solitary fibrous tumor. Axial contrast-enhanced CT scan demonstrates a heterogeneously enhancing centrally located left renal mass which invades the adjacent parenchyma (*); the partially retained renal parenchyma shows normal enhancement pattern (white arrow)
Osteosarcoma
Primary osteosarcoma (OS) of kidney is extremely rare with only 24 cases reported to date in English literature (15). Renal OSs originate from osteoblasts of embryonic mesenchyme (54). Primary OS of the kidneys usually manifest between 43–82 years of age; mean age of occurrence is 61.5 years. There is no sex predilection (55). The clinical presentation is non-specific (8). Histologically, OS is characterized by a pleomorphic pattern with spindle cells and multinucleated giant tumor cells producing neoplastic osteoid and immature bone (8, 54).
Renal OS manifests as a heterogeneous soft tissue mass with calcification or ossification (Fig. 7). Extensive ossification in a ‘sunburst’ pattern may suggest the diagnosis (54, 55). It is noteworthy that sarcomatoid variant of RCC and metastatic osteosarcoma (Fig. 8) to the kidney (seen in 12% cases of primary osseous osteosarcoma on autopsy) may also contain ossification (6). The mainstay of treatment is radical nephrectomy with radiation and chemotherapy, but the benefit of chemotherapy is questionable. Distant metastases to lung and liver are common and prognosis is very poor. Mean survival after the diagnosis is 8–22 months and the longest documented survival is 3 years after nephrectomy (54, 56).
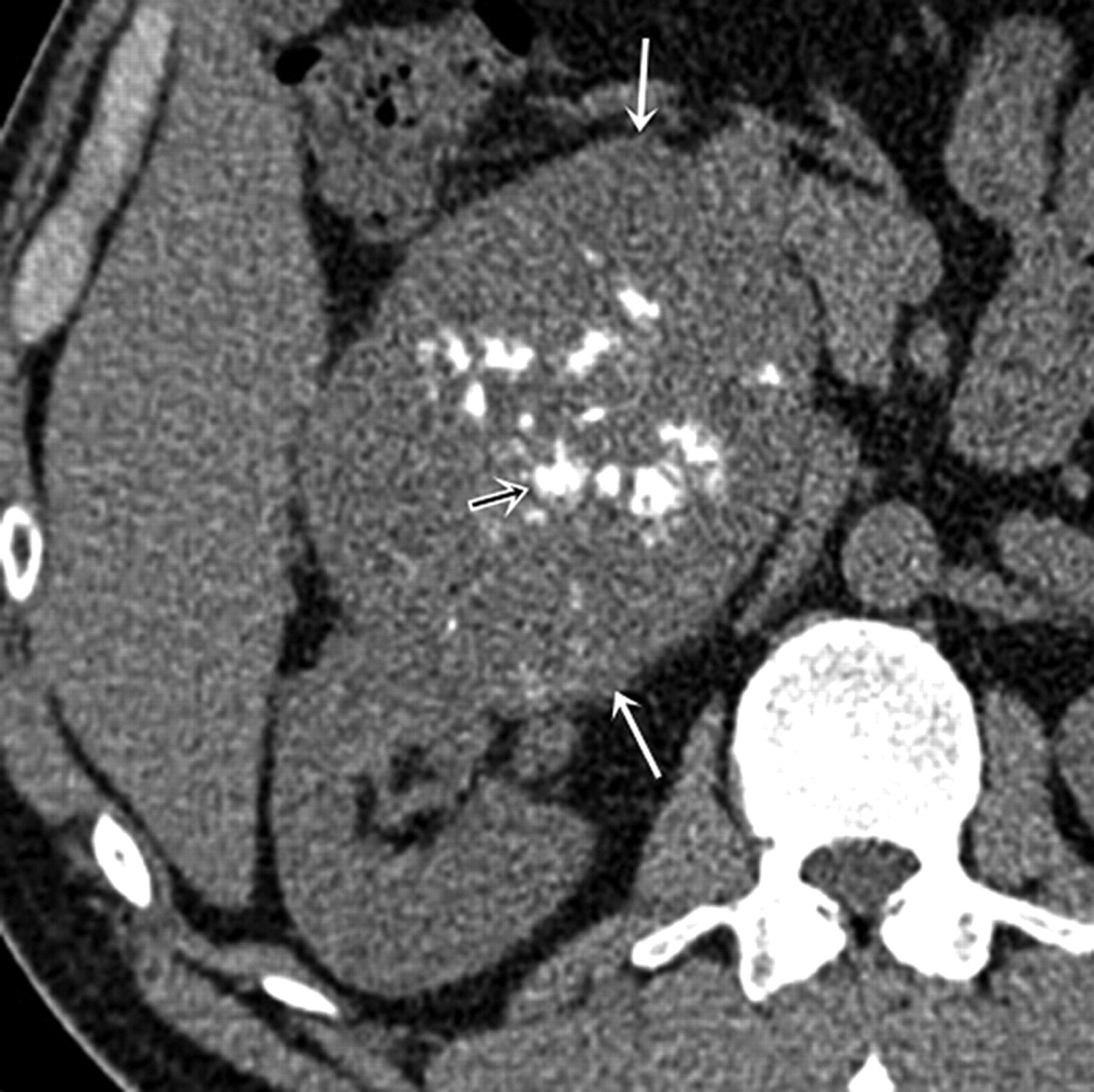
A 58-year-old man with primary renal osteosarcoma. Axial non-contrast CT scan demonstrates a large exophytic soft tissue sold mass arising from anterior lip of right renal interpolar region (white arrows); punctate hyperdense foci (small black arrow) in center are consistent with ossification
A 60-year-old woman with primary osteosarcoma in thigh and metastatic osteosarcoma in right kidney. Axial contrast-enhanced CT scan demonstrates heterogeneously enhancing and expansile mass in right kidney (arrow). There is complete replacement of the normal kidney by tumor as well as an associated tumor thrombus in the right renal vein (arrowhead)
Malignant fibrous histiocytoma
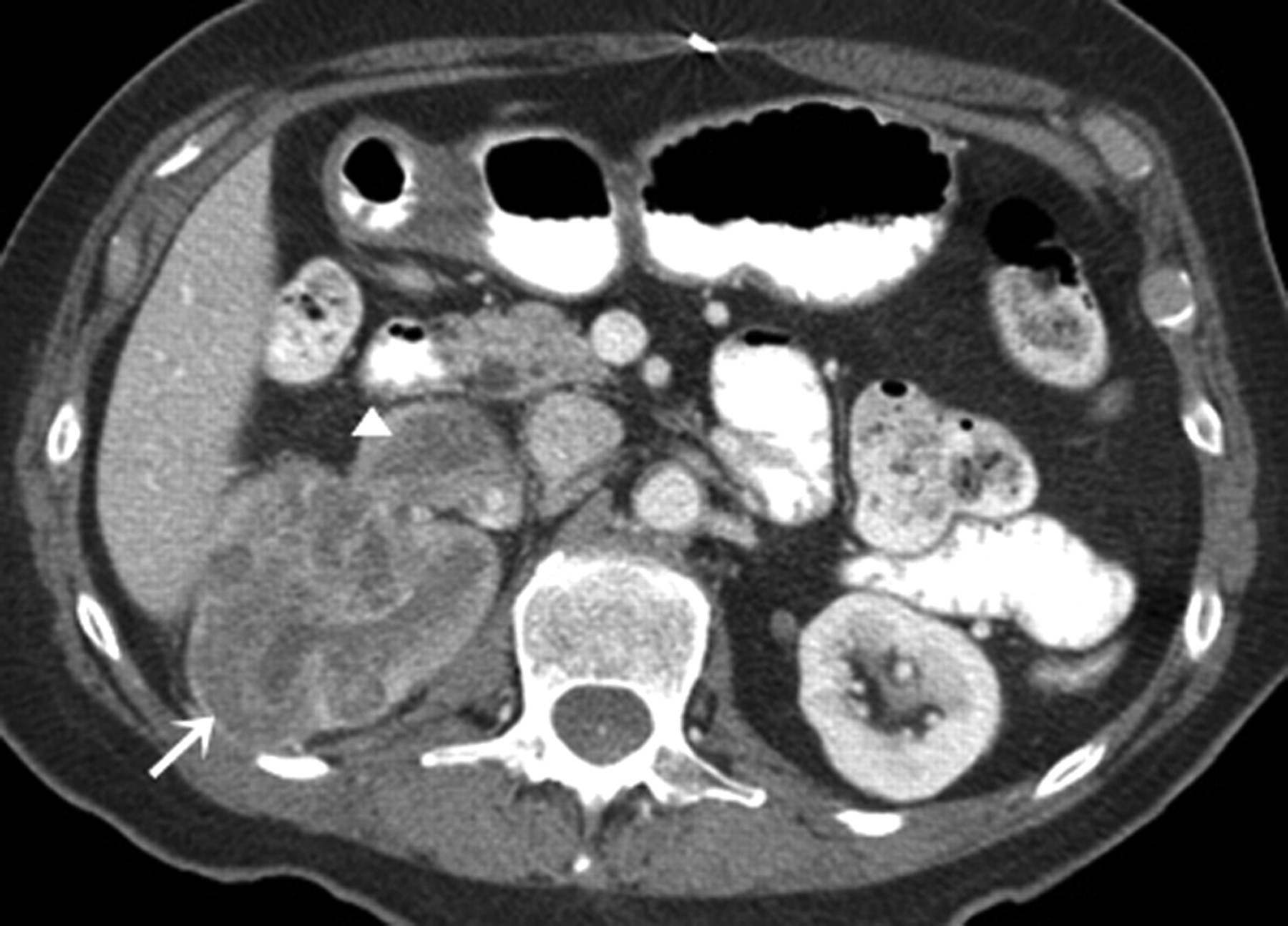
Malignant fibrous histiocytoma (MFH) is a primordial mesenchymal tumor with both histiocytic and fibroblastic differentiation. MFH, the most common soft tissue sarcoma seen in adults, commonly occur in the extremities. Abdominal MFHs commonly arise in the retroperitoneum. Primary renal MFHs are rare; less than 50 cases have been reported to date in English literature (57). Renal MFHs commonly originate from the fibroblasts of the renal capsule. Ninety percent of the patients are aged 40–79 years with a male predilection (60%) (15, 58).
Most frequent clinical presentations are weight loss, flank pain, and abdominal mass. Microscopically, MFH shows storiform arrangement of spindle cells with intermingled histocyte like cells, multinucleated giant cells and xanthoma cells. Imunohistochemical studies are positive for CD68 and anti-alpha-1 antichymotrypsine (15).
Cross-sectional imaging does not show any specific characteristics (Fig. 9). Fibrous component in the tumors is depicted as hypointense areas on T2-weighted images (59). Renal vein and inferior vena cava invasion has been documented (57); 50% of MFHs present with necrosis and calcification. Radical nephrectomy is the treatment of choice; role of adjuvant chemotherapy and radiotherapy is doubtful. MFHs show local recurrence in 50% cases; distant metastases to the lungs and bone are common (57). Prognosis is poor with 5-year survival rate of only 14% (15, 60).
A 55-year-old man with malignant fibrous histiocytoma. Axial contrast-enhanced CT scan demonstrates a large well-circumscribed heterogeneously enhancing solid left renal mass (arrow) with internal non-enhancing areas(*) consistent with necrosis.; otherwise normally enhancing splayed kidney is seen medially (K)
Fibrosarcoma
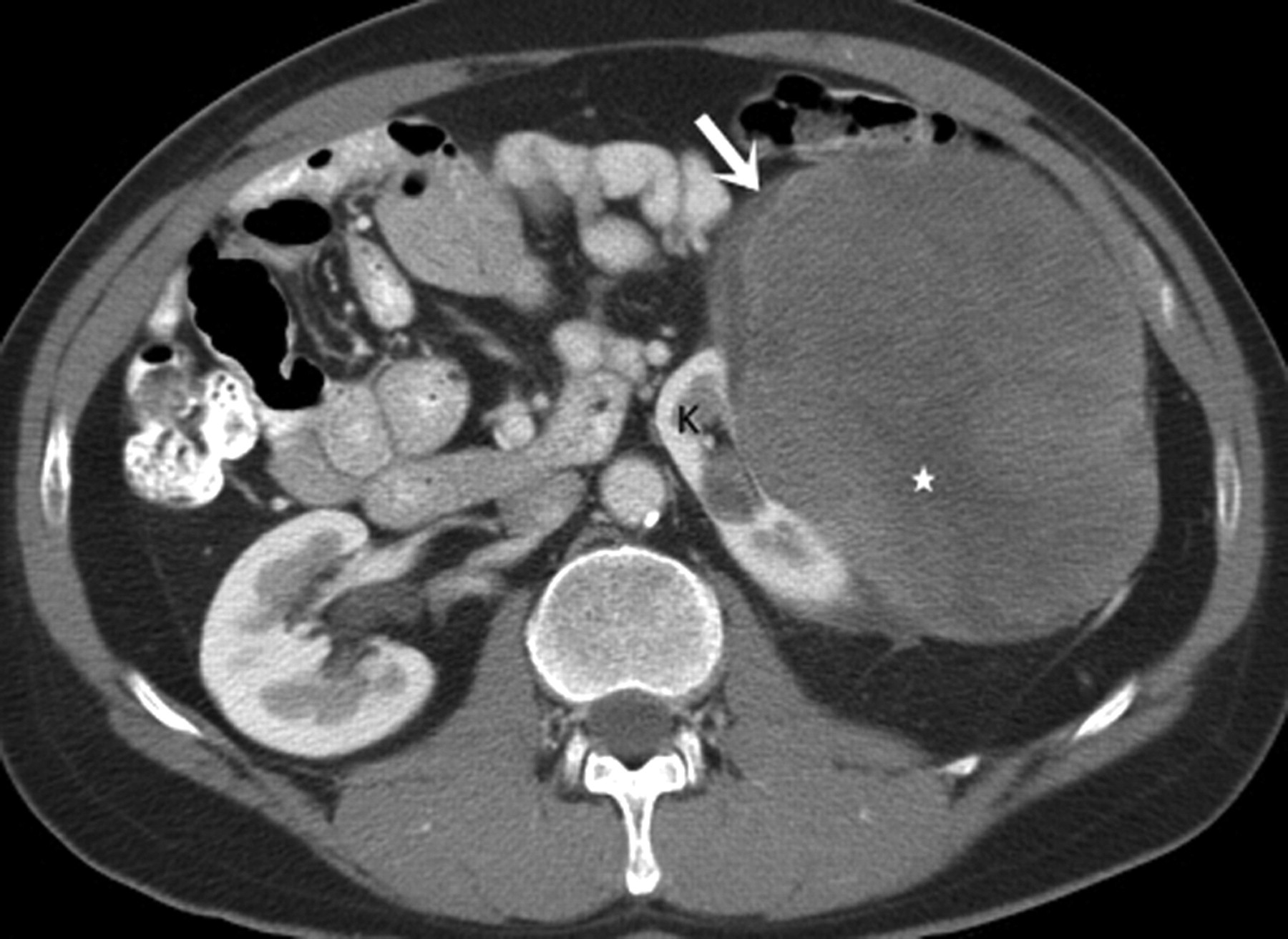
Primary renal fibrosarcoma (FS), mostly arises from fibroblasts in renal capsule (5). FSs are mainly seen in adults (40–60 years age) without any sex predilection. Less than 20 cases have been described in literature (15). Clinically, FSs present with non-specific symptoms (61). Microscopically, FSs show elongated spindle cells with tapered nuclei and delicate cytoplasm. Elongated spindle cells in broad fascicles focally produce a typical ‘herring bone’ pattern.
Imaging characteristics of renal FSs are non-specific. FSs of the kidneys manifest as large, heterogeneously enhancing soft tissue masses (Fig. 10). FSs may invade the renal vein in 40% of cases (62). Radical nephrectomy is the treatment of choice; FSs are resistant to chemotherapy and radiotherapy. The prognosis is poor with a 5-year survival of 10% (8).
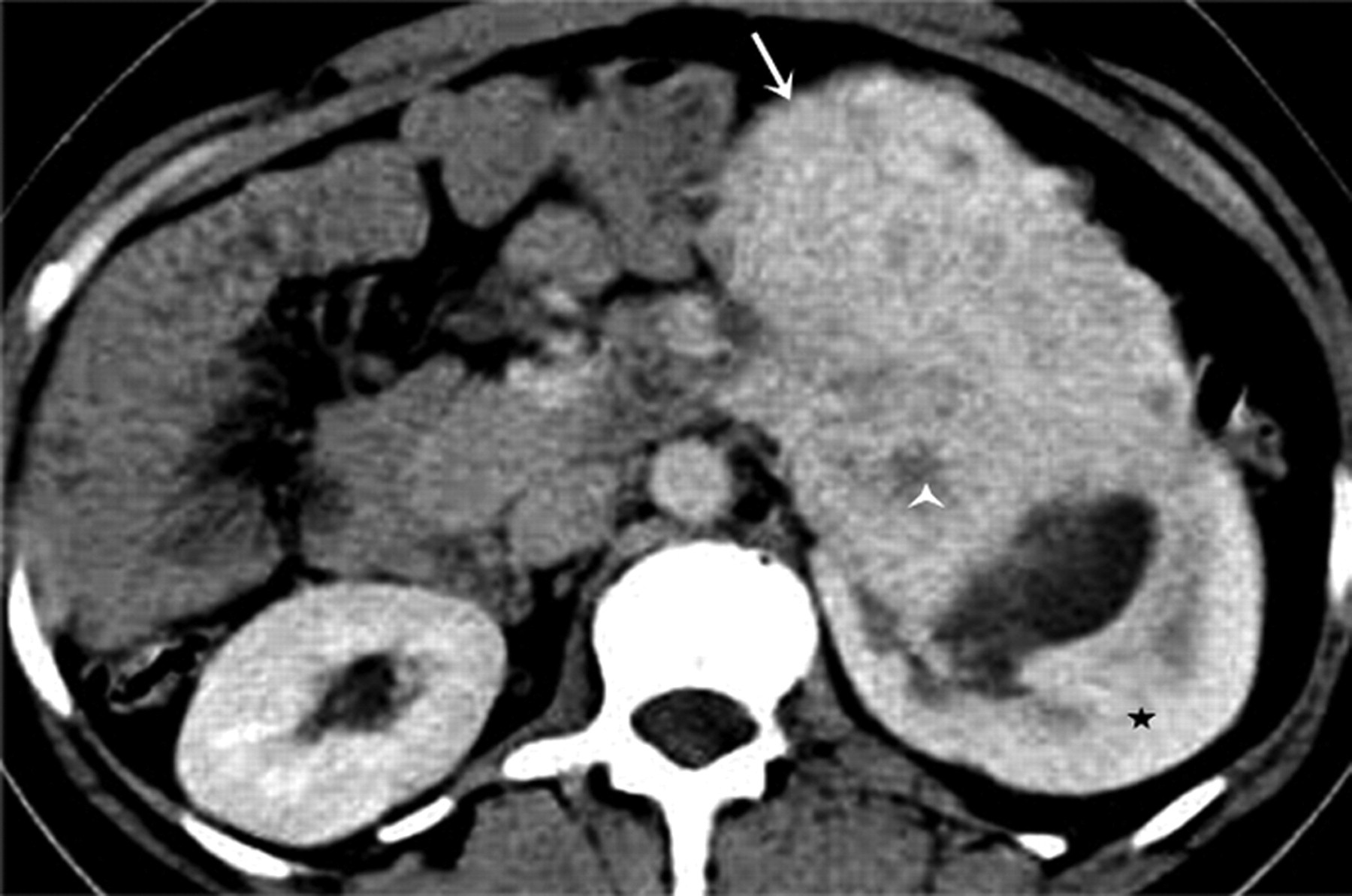
A 45-year old man with fibrosarcoma. Axial contrast-enhanced CT scan demonstrates a large lobulated heterogeneously enhancing exophytic soft tissue mass (arrow) arising from left renal lower pole (*). There are multiple non-enhancing internal areas of focal hypodensities (arrowhead) consistent with necrosis
Chondrosarcoma
Extra-skeletal chondrosarcomas (CSs) have a propensity to arise in the head and neck region and the lower extremities. The kidney is a rare target site of occurrence of CSs. Primary renal chondrosarcoma was first described in 1981 (63); only seven cases of renal CSs are reported in literature (15). It is hypothesized that mesenchymal cells from the developing renal blastema that preserve their multipotential capabilities give rise to mesenchymal tumors such as CSs of the adult kidney (64). Most primary renal CSs range in size from 7–23 cm (65). The clinical presentation is non-specific and include abdominal or flank pain and gross hematuria. At microscopy, CSs show undifferentiated mesenchymal cells and well-differentiated nodules of cartilage (15).
On imaging, CSs present as large soft tissue tumors with central ‘chondroid’ calcifications. However, large non-specific renal mass with both rim and amorphous calcification has also been described (63). Renal CSs are considered to be relatively slow-growing tumors but may have unpredictable outcome. Recurrence or distant metastasis even after 18 years of nephrectomy has been described (66).
Mixed (mesenchymal and epithelial) primary renal sarcoma
Synovial sarcoma
Synovial sarcomas (SSs) are mixed epithelial and stromal tumors that are usually seen in para-articular regions of the extremities. Primary renal synovial sarcoma was first described in 1999 by Faria et al.; less than 35 cases have been reported to date (67). SSs are seen to affect patients between 17 to 61 years (mean age 35 years) with a male:female ratio of 1.7:1 (68). SSs measure 5–19 cm at the time of diagnosis with majority (67%) demonstrating grossly identifiable cysts (68). SSs have an unique chromosomal translocation resulting in the fusion of SYT gene on chromosome 18 with an SSX family gene on chromosome X (69). This unique translocation t(X;18) (p11.2;q11.2) is detected with polymerase chain reaction and is diagnostic for SS.
Histologically, two different forms of SSs are seen: biphasic and monophasic. The monophasic form is composed of only spindle cells whereas biphasic form consist both glandular elements and spindle epithelial cells. Spindle cells are immunoreactive for vimentin, CD99, and bcl2. The microscopy shows cysts in 82% cases, which are lined by cytokeratin positive ‘hobnail epithelium’ (mitotically inactive polygonal eosinophilic cells with apically located nuclei) (8, 70). None of the immunohistochemical markers is diagnostic of SS.
On CT scans, SSs appear as a large, well-defined, soft tissue mass that may extend into renal pelvis or perinephric region. SSs show heterogeneous enhancement; hemorrhage, calcification, fluid levels, and septations are also seen (Fig. 11). On MRI, SSs are T1 isointense to paraspinal muscles and show heterogeneous T2 hyperintensity. Intermixed areas of low, intermediate, and high signal intensity impart the so-called ‘triple sign’– representing areas of hemorrhage, calcification, and fluid levels (71, 72). Radical nephrectomy is the treatment of choice in most cases. SSs are chemosensitive and complete remission of metastatic lung lesions have been reported within a 4-weeks course of Ifosamide and doxorubicin (73). SSs have better prognosis than other renal primary sarcomas with a 5-year survival rate of 42–89%. Size exceeding 5 cm, male gender, age more than 20 years, extensive tumor necrosis, high grade, more than 10 mitotic figures per 10 high powered fields, neurovascular invasion, and SYT-SSX1 variant are predictors of poor prognosis (74).

A 26-year-old man with synovial sarcoma. Axial contrast-enhanced CT scan demonstrates a well-defined hyposense soft-tissue mass (white arrow) with ill-defined internal areas of heterogenous enhancement (*) arising from right renal mid pole region. Renal pelvis (arrowhead)
Conclusion
Primary renal sarcomas are rare malignancies that encompass a broad histological spectrum. The imaging characteristics of most sarcomas of the kidney are indistinguishable from renal cell carcinoma, the most common malignant kidney neoplasm. Accurate diagnosis warrants histopathological evaluation after surgical resection. Most primary renal sarcomas demonstrate unique immunohistochemical and molecular features that facilitate diagnosis. Renal sarcomas show aggressive biological behavior and poor prognosis. Prompt diagnosis and characterization permits optimal patient management.