
Abstract
Although intermittent hypoxia is often associated with hypertension, experimental and clinical studies have demonstrated definite antihypertensive effects of some intermittent hypoxia conditioning (IHC) regimens. Mechanisms of this antihypertensive response are unknown. Endothelial dysfunction related to disturbed synthesis and/or reduced availability of nitric oxide (NO) has been linked to hypertension. Thus, experiments were conducted to determine if IHC can improve endothelium-dependent relaxation and formation of releasable vascular NO stores of young (4–8-week-old) spontaneously hypertensive rats (SHR). Rats were subjected to either IHC (9.5–10% O2, 5–10 min, 5–8 times per day, 20 d) or to sham conditioning. Endothelium-dependent relaxation to acetylcholine was measured in norepinephrine-precontracted, isolated aortic rings, and the size of NO stores was evaluated by percent relaxation to N-acetylcysteine (NAC), which releases stored NO. The capacity of aortic rings for NO storage was evaluated by the relaxation to NAC after prior incubation with an NO donor. IHC significantly suppressed the development of hypertension in young SHR. Endothelial function decreased from 54.7 ± 4.6% to 28.1 ± 6.4% relaxation to acetylcholine after 20 d of sham IHC, whereas endothelial function was sustained (60.3 ± 6.0% relaxation) in IHC rats. IHC also induced formation of available NO stores and enhanced the capacity of aortic rings to store NO. Therefore, the antihypertensive effect of IHC in young SHR is associated with prevention of endothelial dysfunction and with increased accumulation of NO stores in vascular walls.
Keywords
Introduction
Development and progression of hypertension are associated with endothelial dysfunction and eventual injury to target organs, such as the heart, kidneys, eyes and brain. Attenuated nitric oxide (NO) bioavailability, the main characteristic of endothelial dysfunction, is caused by increased scavenging of NO by reactive oxygen species. 1
In addition to reduced availability of NO, impaired vascular storage of NO may also contribute to endothelial dysfunction. Preformed pools of S-nitrosothiols (RS-NO) or dinitrosyl iron complexes (DNIC) exist in many mammalian tissues, including the vasculature, where they act as stores for otherwise short-lived NO. 2–4 Thus, NO availability for vascular regulation is determined not only by the balance between NO synthesis and degradation, but also by this NO storage. The formation of NO stores is an important mechanism of cardiovascular adaptation. On the one hand, storage of excessive NO protects tissues from NO toxicity and, on the other hand, NO stores serve as a reservoir of NO when NO production is depressed. 5,6 RS-NO and DNIC are the main forms of vascular NO storage. 4,7,8
NO bioavailability can be improved pharmacologically, 1 but, in addition, intermittent hypoxia conditioning (IHC) is an effective non-pharmacological way to improve NO bioavailability and storage. 9 Numerous studies have demonstrated that hypobaric IHC stimulates NO synthesis and slows the development of hypertension in young spontaneously hypertensive rats (SHR). 10 Also, a number of clinical studies conducted in Russia and Ukraine have demonstrated a definite, long-lasting antihypertensive effect of IHC. 10–15 However, experimental studies on mechanisms of the antihypertensive effect of normobaric IHC are scarce, and the role of NO-dependent mechanisms in the antihypertensive effect of normobaric IHC is unknown. Thus, this study was conducted to determine if IHC improves endothelial function and formation of releasable, vascular NO stores in young SHR.
Methods
Experimental animals
Experiments were carried out on 4–5-week-old male SHR. Rats were purchased from Charles Rivers Lab Inc (Wilmington, MA, USA). Three groups were included in the study: (1) 4–5-week-old SHR before IHC (n = 5); (2) 7–8-week-old SHR after IHC (n = 12); and (3) 7–8-week-old SHR after sham IHC (n = 9). The study was approved by the Animal Care and Use committees of the University of North Texas Health Science Center, and was conducted in accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals.
Intermittent hypoxia conditioning
Rats were subjected to normobaric IHC protocol, as described earlier 16 (5–8 cycles/d for 20 days, fraction of inspired oxygen (FiO2) 9.5–10% for 5–10 min/cycle, with intervening 4 min normoxia; Table 1), a regimen that may have clinical applications. 10,17 Control rats were sham-conditioned with 21% O2. Blood pressure (BP) was measured in conscious rats using the tail-cuff method (IITC Life Sciences, Woodland Hills, CA, USA).
Intermittent hypoxia conditioning protocol
*FiO2 (%) = fraction of inspired oxygen; †Replications = number of cycles of hypoxia/normoxia per daily session; ‡Σ Hypoxia = total minutes of hypoxia per session
Preparation of aortic rings
On the day following completion of the IHC conditioning program or the sham conditioning program, rats were sacrificed by decapitation. Aorta was isolated, and aortic rings (3.5–4.0 mm length) were mounted on two stainless steel wires passed through the vessel lumen and suspended in a temperature-controlled, isolated organ bath, containing Krebs bicarbonate buffer at 37 ± 0.5°C and aerated with 95% O2–5% CO2. The aortic rings were incubated for equilibration at a resting tension of 1.2 g for 60 min.
Endothelium-dependent and -independent relaxation
After equilibration, the aortic rings were precontracted with norepinephrine (NE; 0.5 μmol/L; Abbott Laboratories, Abbott Park, IL, USA). Endothelium-dependent relaxation was induced by acetylcholine (10 μmol/L; Sigma, St Louis, MO, USA). To determine if NO stores contributed to endothelium-dependent relaxation by acetylcholine, the aortic rings from each experimental group were treated with N
ω-nitro-
Endothelium-independent relaxation of aortic rings was induced by the NO donor, DNIC 10−6 mol/L, a dose which induces submaximum relaxation of rat aorta. DNIC with glutathione ligand was synthesized by treating ferrous sulfate and glutathione solutions with gaseous NO in 10 mmol/L (4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid) buffer (pH 7.4) with a molar ratio of Fe2+ to glutathione 1:2 with an NO pressure of 100–200 mmHg, as described earlier. 18,19
The percent relaxation to acetylcholine or DNIC was defined as the percentage decline in developed tension of the NE-mediated precontraction.
NO stores
NO stores in blood vessel walls can be revealed using low-molecular thiols, such as N-acetylcysteine (NAC), which penetrate cells and extract vasoactive NO from protein-bound DNIC and RS-NO. This reaction releases NO and induces vasodilation in proportion to the size of the NO stores (Figure 1a). 5,6
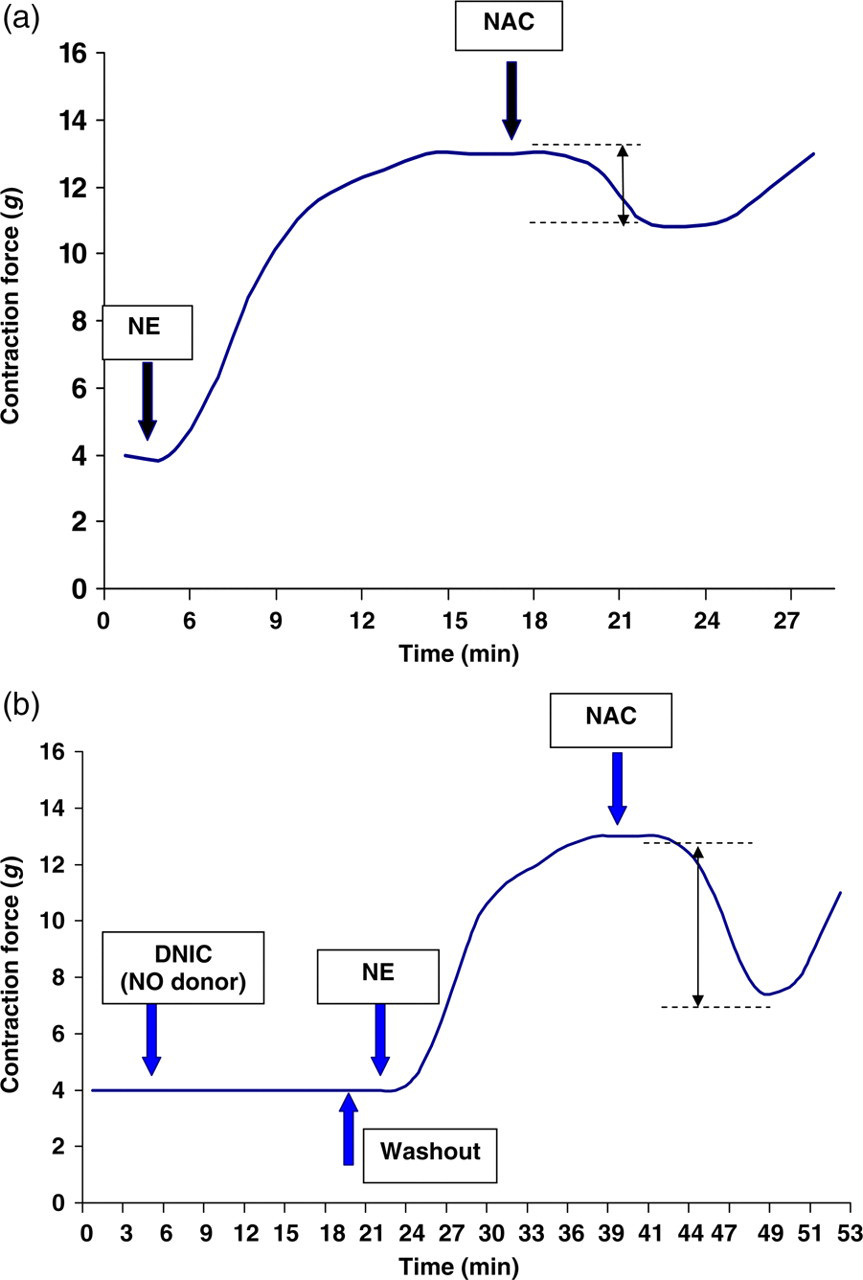
Schematic diagram illustrating detection of available NO stores (a) and evaluation of potential capacity of NO stores (b) in rat aorta. Abscissa: time (min); ordinate: contraction force of aortic segments (g). Arrows show estimated size of NO stores. DNIC, dinitrosyl iron complex, an NO donor; NAC, N-acetylcysteine; NE, norepinephrine; NO, nitric oxide
For measuring available NO stores, the aortic rings after one-hour stabilization were precontracted with NE (0.5 μmol/L). When the contraction reached a stable plateau, NAC (10−3 mol/L; Sigma) was added to the organ bath. NO stores were quantified by the relaxation response to NAC. This relaxation was expressed as a percent of the NE-induced precontraction.
The capacity of NO storage was assessed from the maximum amount of NO, which can be potentially stored in the wall of isolated vessels after incubation with an excess of NO donor (Figure 1b). The aortic rings were incubated with the NO donor, DNIC (10−5 mol/L), 20–22 for 20 min after the NE-induced contractile response had plateaued. The DNIC concentration of 10−5 mol/L was selected since we found previously that relaxation of the isolated rat aorta was not increased at concentrations of DNIC greater than 10−5 mol/L. 21–23 At the same time, vessel tone could spontaneously recover after decomposition of DNIC 10−5 mol/L in the organ bath. After washout of DNIC, total NO stores were evaluated by the relaxation response induced by NAC. 20–22
Statistical analysis
Data are presented as means ± SE. An analysis of variance was used to compare findings from IHC and sham rats and to compare the effect of time on measurements made in 4–5-week-old unconditioned rats and 7–8-week-old sham conditioned rats. Differences were considered significant at P < 0.05.
Results
The pretreatment systolic BP of 5-week-old SHR was 131 ± 2 mmHg. Figure 2 shows that the systolic BP of IHC rats was significantly lower than of age-matched sham-IHC rats (162 ± 2 mmHg in IHC rats versus 202 ± 3 mmHg in sham rats, P < 0.05).
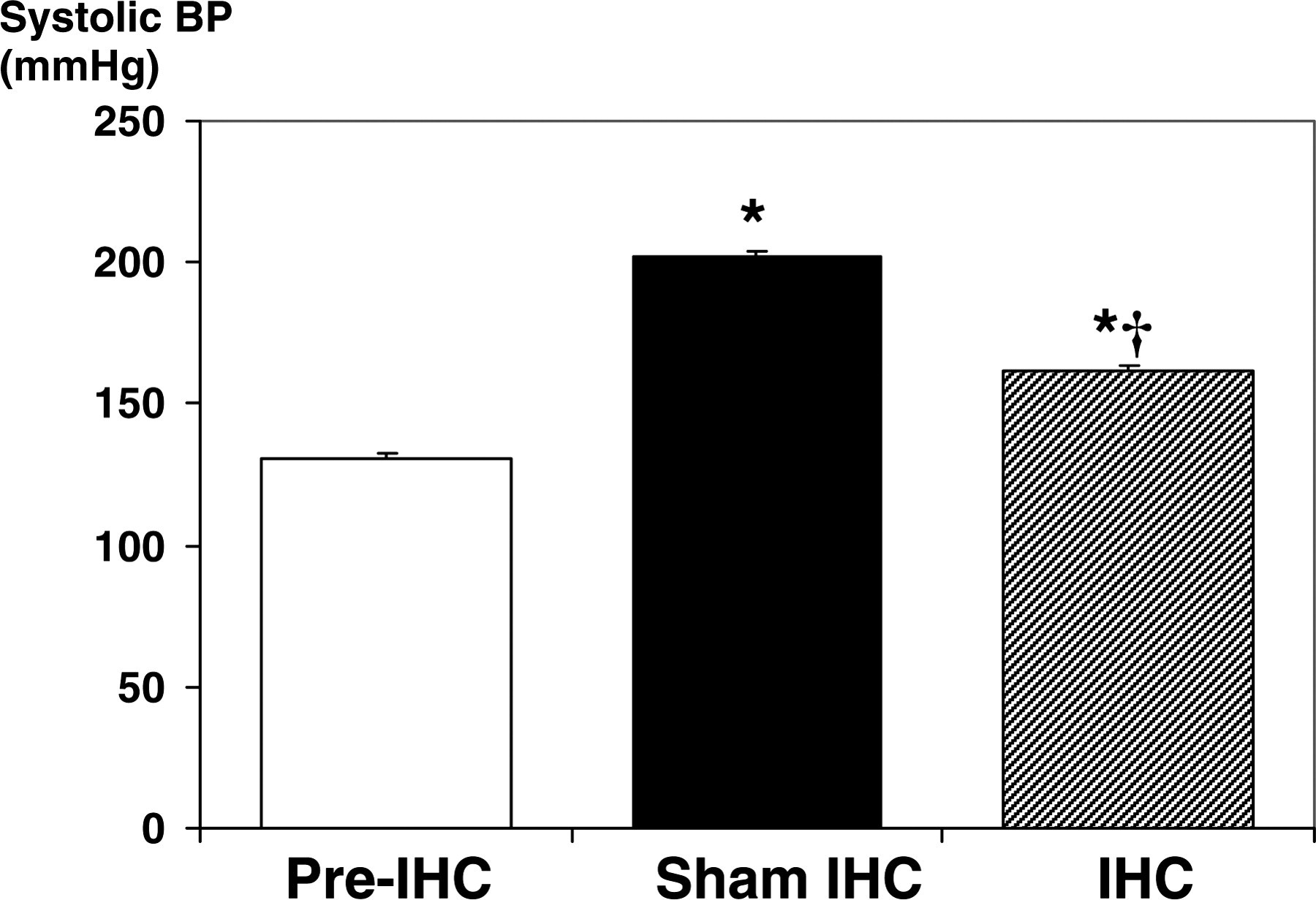
Effect of intermittent hypoxia conditioning (IHC) on systolic blood pressure (BP) in spontaneously hypertensive rats. *Significantly higher than pre-IHC, P < 0.05; †Significantly lower than sham-IHC, P < 0.05
As hypertension developed in sham rats, acetylcholine-induced, endothelial-dependent relaxation was impaired, from 54.7 ± 4.6% to 28.1 ± 6.4%, P < 0.002 (Figure 3). IHC improved this index of endothelial function (60.3 ± 6.0% relaxation in IHC rats versus 28.1 ± 6.4% in sham rats, P < 0.01). No acetylcholine-induced relaxation of the aorta was observed in vessels from any group of rats after inhibition of NOS with
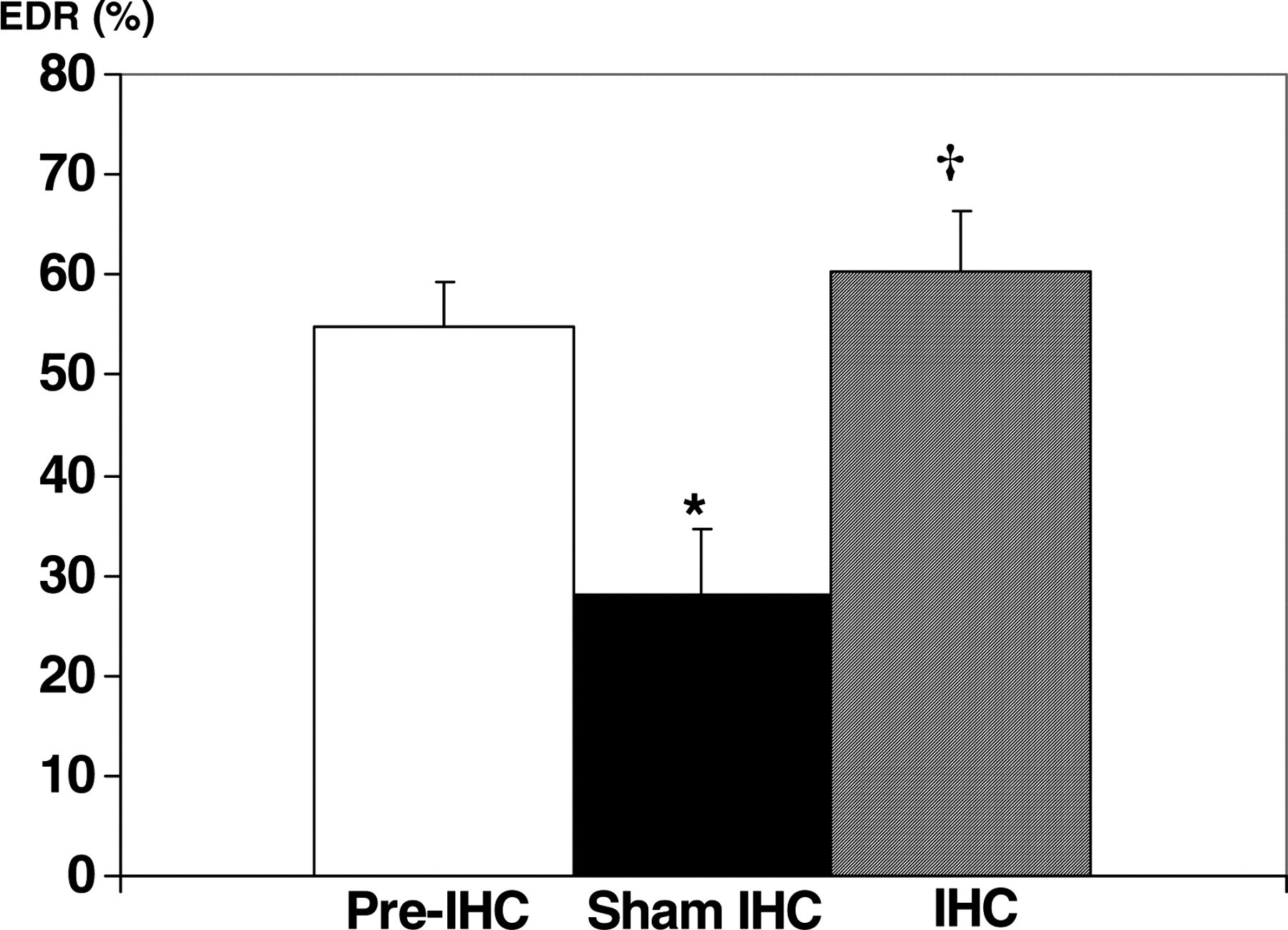
Effect of intermittent hypoxia conditioning (IHC) on endothelium-dependent relaxation (EDR) of isolated aortic rings from spontaneously hypertensive rats. EDR is expressed as percent relaxation of norepinephrine-induced precontraction. *Significantly less than pre-IHC, P < 0.05; †Significantly greater than sham-IHC
Treatment of the aortic rings with DNIC caused large endothelial-independent relaxation. This response to DNIC did not differ between aortic rings from SHR at baseline, after IHC and after sham IHC (Figure 4).
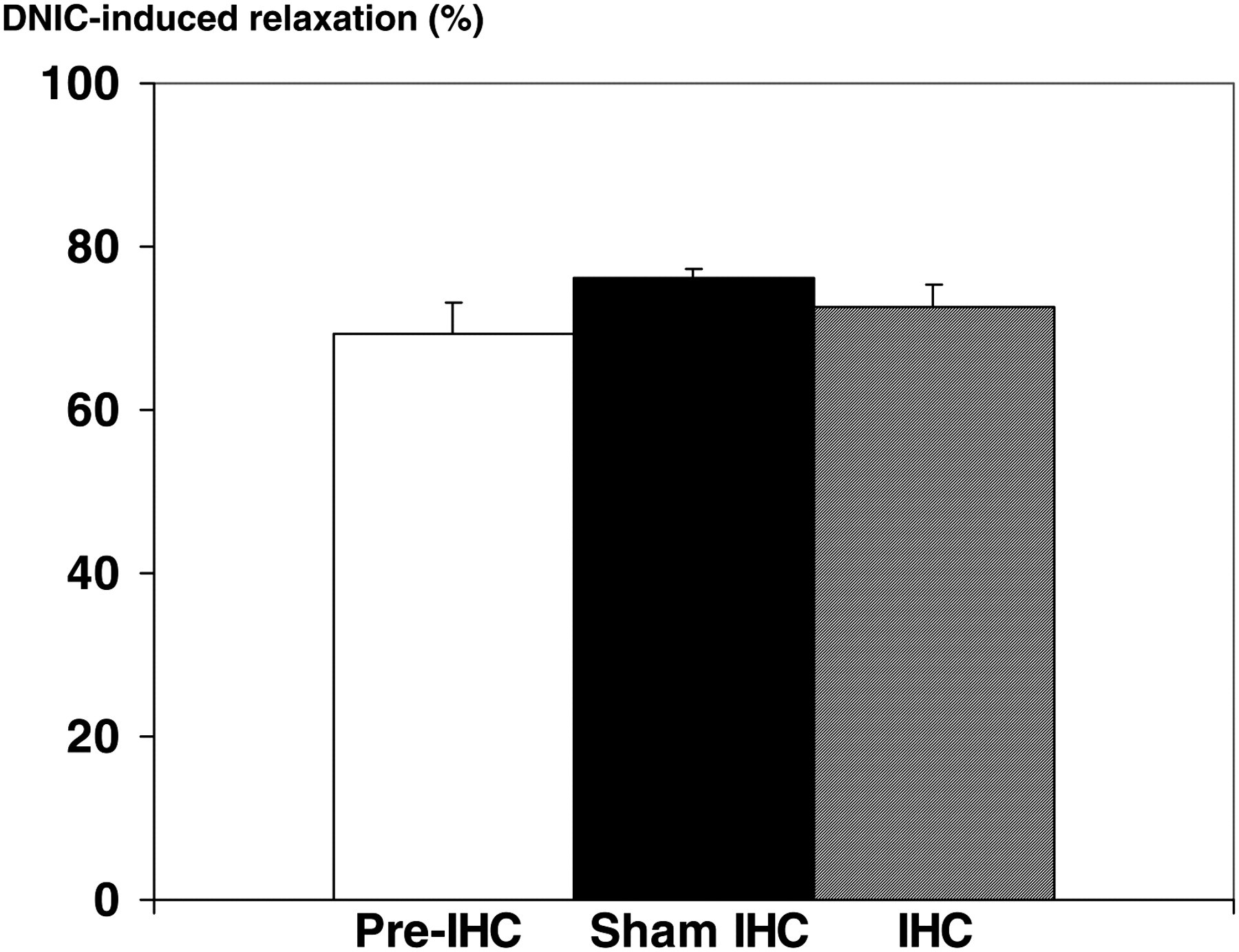
Effect of intermittent hypoxia conditioning (IHC) on endothelium-independent relaxation of isolated aortic rings from spontaneously hypertensive rats. Relaxation is expressed as percent relaxation of norepinephrine-induced precontraction. DNIC, dinitrosyl iron complex
In prior experiments, NO stores were evaluated in the absence and presence of the NOS inhibitor, N
ω-nitro-
The available NO stores in the aorta of 4–5-week-old SHR produced 16.8 ± 2.0% relaxation of precontracted aortic rings (Figure 5). In 7–8-week-old sham SHR, NO stores were similar (13.5 ± 3.2% relaxation, P > 0.05). IHC significantly increased formation of NO stores in the vascular wall (51.8 ± 5.2% relaxation in IHC rats versus 13.5 ± 3.2% in sham rats, P < 0.01).

Effect of intermittent hypoxia conditioning (IHC) on formation of nitric oxide (NO) stores in the aorta of spontaneously hypertensive rats. Size of NO stores is expressed as percent relaxation to N-acetylcysteine (NAC) of norepinephrine-induced precontraction (see Figure 1). *Significantly greater than pre-IHC and sham-IHC, P < 0.05
After incubation with the NO donor, DNIC, aortic walls accumulated NO stores (Figure 6). In 4–5-week-old unconditioned rats, addition of NAC after incubation with DNIC induced vasorelaxation of 43.7 ± 6.0%. In 7–8-week-old sham rats, this relaxation fell to 23.0 ± 3.6% (P < 0.05), whereas relaxation was sustained in IHC rats (59.6 ± 6.4%, P > 0.05 versus 4–5-week-old rats, P < 0.005 versus 7–8-week-old sham rats).
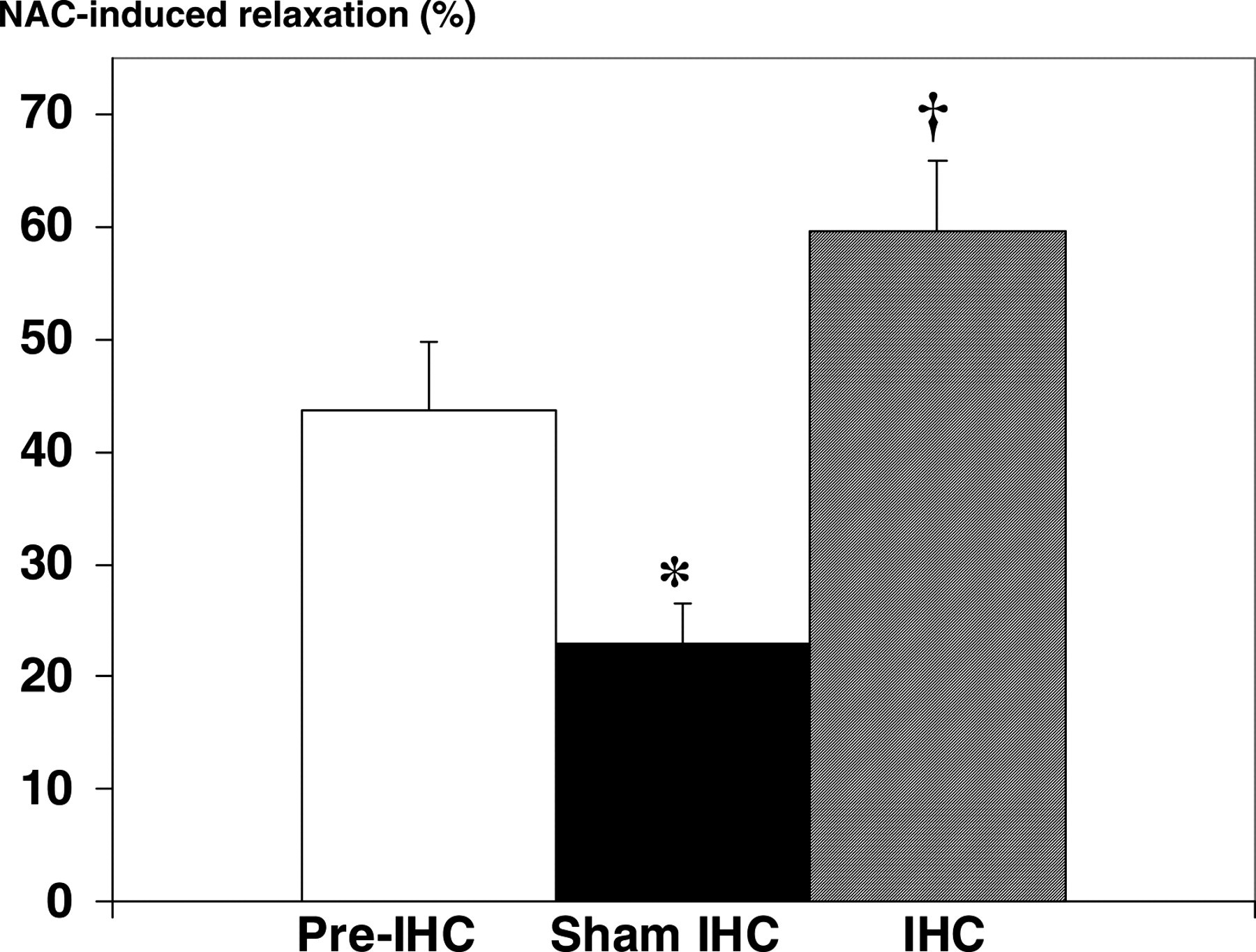
Effect of intermittent hypoxia conditioning (IHC) on ability of the aorta of spontaneously hypertensive rats to accumulate nitric oxide (NO) stores, i.e. the potential capacity of NO stores. Size of NO stores is expressed as percent relaxation to N-acetylcysteine (NAC) of norepinephrine-induced precontraction following incubation with dinitrosyl iron complex, an NO donor (see Figure 1). *Significantly less than pre-IHC, P < 0.05; †Significantly greater than sham-IHC
Discussion
The effects of intermittent hypoxia on systemic BP are controversial. On the one hand, chronic intermittent hypoxia induces hypertension in animals and in patients with sleep apnea.
10,24–26
On the other hand, multiple studies have demonstrated that adaptation to hypoxia prevents development of experimental hypertension (for a review see reference
10
). A major reason for this divergence is that the cardiovascular response to hypoxia strikingly depends on the hypoxic regimen, specifically, on duration and intensity of hypoxia exposure, the number of hypoxia/reoxygenation bouts per day and the total days of the protocol.
10,27
Protocols which induce systemic hypertension and impair endothelium-dependent vasorelaxation have generally employed frequent, often severe, hypoxia exposures for prolonged periods.
26–31
For example, Tahawi et al.
30
showed that the endothelium-dependent relaxation to acetylcholine of arterioles in rats exposed to a prohypertensive regimen of intermittent hypoxia (2–3% O2 for 3–6 s interspersed with 20.9% O2 for 15–18 s, 8 h/d for 35 d) was significantly attenuated compared with that of controls, and the degree of vasoconstriction in response to the NOS inhibitor,
There is much evidence that intermittent hypoxia modifies synthesis of vasodilatory factors, primarily NO (for a review see reference 9 ). Recent studies have demonstrated that adaptation to hypoxia is protective against both NO deficiency and NO overproduction. 5,6,9 Either of these NO disorders can contribute to hypertension, so the bidirectional moderating effects on the NO system observed in this study may be an antihypertensive mechanism of IHC.
Surprisingly, NO overproduction rather than NO deficiency is often observed in SHR.
35
The NO pathway is apparently up-regulated in blood vessels by a mechanism involving induction of endothelial NOS (eNOS)
36
and/or inducible NOS (iNOS).
37,38
However, Nava et al.
36
showed that levels of cyclic guanosine monophosphate (cGMP) in the heart and blood vessels of SHR are not increased compared with the normotensive control. Furthermore, no difference was observed between the pressor effects of the NOS inhibitor,
Chronic treatment with the iNOS inhibitor, aminoguanidine, suppressed development of hypertension and lessened vascular hyper-reactivity in SHR. 38 In early hypertension, iNOS induction may be sufficient to restrict BP elevation. Later, however, excessive NO inhibits eNOS and directly damages vascular cells by suppressing mitochondrial respiration and DNA synthesis. These effects eventually lead to endothelial dysfunction and increased BP, which, in turn, damages endothelium-dependent relaxation even further, creating a vicious cycle. 39,41
For revealing and quantifying NO stores, a low-molecular weight, cell-penetrating thiol, NAC, is used to extract NO from protein-bound DNIC and RS-NO.
6,21,42
The response to NAC is associated with activation of soluble guanylate cyclase
43
and the effect of NAC is partially
21
or completely
42
abolished by inhibition of soluble guanylate cyclase. This suggests that the NAC-induced relaxation is mediated by NO release. Previous experiments showed that the presence of the NOS inhibitor,
An
In the present study, NO stores revealed by NAC in the aorta of SHR exposed to IHC were significantly larger than in sham-IHC rats. IHC has been previously demonstrated to increase NO stores in rat vessels, 5,6,9,20,46,47 and the size of NO stores was positively correlated with plasma nitrite plus nitrate concentrations. 46 Therefore, the formation of NO stores may be regarded as an indirect sign of NO overproduction. Our data on increased NO storage in IHC rats are consistent with stimulating effects of intermittent hypoxia on NO synthesis, which has been observed by other authors and in our experiments (for a review see references 6,9 ).
The ability of blood vessels to form NO stores is impaired in some conditions associated with reduced NO bioavailability, such as hypertension in stroke prone-SHR 20 and metabolic syndrome. 48 Adaptation to hypoxia can expand the capacity of NO stores, 20,47 so this mechanism may contribute to the antihypertensive action of IHC. Accumulation of NO stores during adaptation to hypoxia may protect against potentially harmful effects of excess NO synthesized during repeated hypoxia exposures. On the other hand, as non-enzymatic NO sources, NO stores may compensate for decreased production of NO by endothelial cells, or provide NO for feedback inhibition of NO overproduction.
Recently, we have obtained new data supporting the important role of NO stores in the protective effects of IHC. A 20-d IHC program similar to that used in the current study afforded remarkable cardioprotection from ischemia–reperfusion injury in dogs. 49–51 This protection was associated with restriction of NO overproduction during reperfusion without compromising reactive hyperemia. 49–51 IHC induced formation of NO stores in coronary vessels of the same dogs and, moreover, significantly increased the ability of blood vessels to store NO, i.e. expanded the NO store capacity. When NAC was added to the diet each morning two hours before a hypoxia session, the cardioprotection was completely abolished, NO stores were reduced by 50% and the capacity for NO storage was significantly impaired. 52
Although the antihypertensive mechanisms of hypoxia adaptation are still not completely understood, they likely impact several major steps in the pathogenesis of sustained hypertension, including sympathetic nervous activity, Ca2+ loading of vascular smooth muscle, water and salt metabolism, oxidative stress, rarefaction of the microcirculation, endothelial dysfunction, and reduced synthesis and/or availability of NO. The roles of these putative mechanisms in the antihypertensive effect of IHC need further study.
Perspectives
This study has demonstrated that IHC, in contrast to the intermittent hypoxia experienced by persons with sleep apnea, is antihypertensive. Elucidating the antihypertensive mechanisms of IHC may lead to the use of IHC as a new or adjunctive treatment for essential hypertension that may reduce requirements for medication. IHC could be utilized by hypertensive patients who cannot or will not alter their lifestyle. Evidence that IHC lessens endothelial dysfunction and increases vascular NO stores in a model of essential hypertension is highly innovative, since presently there are no specific medications to treat endothelial dysfunction and increase NO availability.
Conclusion
IHC blunts the development of hypertension in SHR, and this antihypertensive effect of IHC is associated with prevention of endothelial dysfunction and with accumulation of vascular NO stores.
Footnotes
ACKNOWLEDGEMENTS
The expert technical assistance of Linda Howard and Arthur G Williams, Jr, is gratefully acknowledged. This research was supported by an intramural grant funded by the Cardiovascular Research Institute of the University of North Texas Health Science Center and by the Russian Foundation for Basic Research (Grant No. 09-04-00868a).