
Abstract
The transcription factor PRDM16 regulates differentiation of brown adipocyte tissue in mice. Recently, however, it has been demonstrated that genetic knockout of Prdm16 in mice leads to a complete cleft of the secondary palate in offspring. To identify genes whose promoters bind PRDM16 in mouse embryonic palate/maxillary mesenchymal cells, we have conducted a chromatin immunoprecipitation-promoter microarray analysis (ChIP-Chip). One hundred and twenty-two gene promoters were identified as capable of binding PRDM16. These could be functionally grouped to include those on genes linked to muscle development
Introduction
PRDM16 (MEL1, PFM13) is a 1247 amino-acid protein (140 kDa) with six distinct domains that include the namesake PR domain (PRDI-BF1 and RIZ1 homologous), two DNA-binding domains, a proline-rich, repressor and acidic domains. The PR domain is similar to the SET domain found in many chromatin-modifying proteins. The Prdm gene family contains 17 members in humans (16 in mice), and the Prdm16 gene is similar to the Mds1/Evi1 gene and the oncogene Evi1. 1 Prdm16 is also expressed as a shorter splice variant that is missing most of the PR domain. 2 The function of the shorter form is unknown.
Early studies in (1;3)(p36;q21)-positive myeloid leukemias revealed that ectopic expression of Prdm16 led to hyperproliferation, suggesting a role in cell cycle progression. 1,2 Recently, Prdm16 was shown to be a master gene controlling differentiation of Myf5-expressing mesenchymal cell precursors into brown adipose tissue via a transcriptional complex with C/EBP-β. 3,4
Prdm16 is expressed in multiple embryonic 5 and adult 6 mouse tissues. We have previously demonstrated expression of Prdm16 in the murine embryonic secondary palate, 7 suggesting a role for this transcription factor in the development of this structure. Indeed, Prdm16 knockout mouse embryos display a completely penetrant cleft palate. 8 The mechanism by which Prdm16 contributes to this phenotype is unknown. We have previously demonstrated that PRDM16 binds to multiple Smads and may modulate signaling through the transforming growth factor β (TGF-β) signaling pathway. 7 Signaling via TGF-β is essential for normal palate development. 9 In an effort to dissect the molecular pathways that are influenced by PRDM16 and essential for development of the secondary palate in mice, genes whose promoters bind PRDM16 in mouse embryonic palate/maxillary mesenchymal cells were identified by high-density promoter arrays following chromatin immunoprecipitation.
Materials and methods
Animals
Hsd:ICR (CD-1®) mice were obtained from Harlan Laboratories, Inc (Indianapolis, IN, USA) and Prdm16+/– mice (strain B6;129S5-Prdm16 Gt683Lex/Mmcd) were purchased from the Mutant Mouse Regional Resource Center (UC-Davis, Davis, CA, USA). Both strains were housed at an ambient temperature of 22°C with a 12-h on/12-h off light cycle and access to food and water ad libitum. Timed matings were achieved by caging one mature Prdm16+/– male and two nulliparous Prdm16+/– female mice overnight. The presence of a vaginal plug was taken as evidence of mating and that morning was designated embryonic day 0.5 (E0.5). Pregnant mice were euthanized by carbon dioxide asphyxiation/cervical dislocation on E13.5 for preparation of primary palate mesenchymal cell cultures. All procedures for the humane use and handling of mice were approved by the University of Louisville Institutional Animal Care and Use Committee and encompass guidelines as set out in the EC Directive 86/609/EEC for animal experimentation.
Establishment and culture of primary embryonic palate mesenchymal cells
Primary cultures of mouse embryonic palate mesenchymal (MEMM) cells were established from secondary palatal tissue microdissected from E13.5 mouse embryos. The dissected tissue was dissociated by a 10-min digestion with 0.05% (w/v) trypsin and suspended in OptiMEM (Invitrogen Corp, Gaithersburg, MD, USA) supplemented with 5% fetal bovine serum (FBS; Sigma Chemical Co, St Louis, MO, USA). Cells were passed through a 70-μm filter, and 9 × 105 cells were plated in a 10-cm-diameter tissue culture dish. Cell cultures were incubated at 37°C in a humidified atmosphere of 5% CO2.
Reverse transcription and realtime polymerase chain reaction
RNA from palate tissue was isolated using the RNeasy system (Qiagen, Valencia, CA, USA). Complimentary DNAs (cDNAs) were synthesized from 400 ng of total RNA with the SuperScript First Strand cDNA Synthesis System (Invitrogen Corp). Realtime polymerase chain reaction (PCR) assays were performed with probe:primer sets obtained from Applied Biosystems (Foster City, CA, USA) on the ABI Prism 7000 Sequence Detection System platform (Applied Biosystems). The signal from 18S RNA was used as the internal control. Fold-change values were determined according to Livak and Schmittgen. 10
Transfection
Ten culture dishes (10 cm, 60–70% confluent) were transfected (10 μg/dish pcDEF3-PRDM16-6X-myc or empty vector) using Effectene transfection reagent (Qiagen) and incubated for 48 h before crosslinking.
Crosslinking
Cells were trypsinized from each dish and like samples pooled. Protein–DNA crosslinking was effected in OptiMEM growth medium + 5% FBS by first incubating cells with 1.5 mmol/L ethylene glycol bis-succinimidyl succinate (EGS; Pierce Chemical Co, Rockford, IL, USA) for 20 min at room temperature
Chromatin immunoprecipitation
Cells were lysed and isolated nuclei digested in nuclease digestion buffer (Active Motif, Carlsbad, CA, USA) containing protease inhibitors and 200 units/mL micrococcal nuclease for five minutes at 37°C. Digestion was terminated with ethylenediaminetetraacetic acid (EDTA). This procedure led to fragments of DNA ranging in size from 200 to 1000 bp. The sheared chromatin was immunoprecipitated with 6.7 μg/mL anti-c-Myc (mouse monoclonal clone 9E10; BD Pharmingen, Franklin Lakes, NJ, USA), 100 μL protein G magnetic beads, and protease inhibitor cocktail (Complete, EDTA-free; Roche Diagnostics, Indianapolis, IN, USA) for four hours at room temperature with constant mixing. Protein G beads were washed once with ChIP buffer 1 (Active Motif) and two times with ChIP buffer 2 (Active Motif). Bound chromatin was eluted and de-crosslinked overnight at 65°C. Samples were then digested with proteinase K (Roche Diagnostics) for two hours at 37°C and purified using Affymetrix cDNA cleanup columns (Affymetrix, Santa Clara, CA, USA). Purified chromatin was then taken through two rounds of linear amplification using the WGA2 and WGA3 genomic DNA amplification systems, respectively (Sigma, St Louis, MO, USA). During the final round of amplification, a mixture of 8 mmol/L deoxythymidine triphosphate and 2 mmol/L deoxyuridine triphosphate was used in order to prepare the samples for further fragmentation with uracil-DNA glycosylase and purinic/apyrimidinic (AP) endonuclease (Affymetrix), which yields an average fragment size of ∼60 nucleotides. The integrity of the DNA at each point was determined by analysis on the Agilent 2100 Bioanalyzer (Agilent Technologies, Santa Clara, CA, USA) and nucleic acid concentrations determined on the NanoDrop ND1000 spectrophotometer (Thermo Fisher Scientific, Waltham, MA, USA).
Chip hybridization
Fragmented chromatin was hybridized to Affymetrix GeneChip® mouse promoter 1.0R arrays (p/n 900890; Affymetrix) according to the manufacturer's instructions. Following hybridization, washing and staining, chips were scanned with the GeneChip® scanner 3000 (Affymetrix) and processed using GCOS1.2 software. The resultant CEL files containing the raw data were imported and analyzed using Partek Genomics Suite (Partek, St Louis, MO, USA). A cut-off of P < 0.05 was used to generate the list of enriched gene promoters.
PCR confirmation
To verify putative gene targets identified from the promoter array, PCR primers were designed using Primer-Blast (NCBI, NIH, Bethesda, MD, USA) and tested on control- and PRDM16-transfected MEMM cells that were immunoprecipitated with anti-c-Myc. Each reaction consisted of 2 ng DNA, 1 μmol/L each of forward and reverse primers, 10 units/mL JumpStart REDTaq (Sigma) and 100 μmol/L dNTP mix. Amplification conditions were: 95°C, five minutes; 30–35 cycles of 95, 57 and 72°C/30 s; and a final extension step of 72°C for seven minutes. Amplified products were analyzed on 2% agarose gels. The sequence of primers used for these analyses are reported in Supplementary Table 1 (please see
Whole-mount in situ hybridization
A cDNA clone for mouse osteopontin (in the vector pCMV6-Kan/Neo) was purchased from OriGene Technologies, Inc (Rockville, MD, USA). In order to synthesize both sense and antisense riboprobes suitable for in situ hybridization, a 610-bp fragment of the cDNA was subcloned into pBluescript II SK (+) (Agilent Technologies). Digoxigenin-labeled riboprobes were synthesized with the DIG RNA kit (Roche Diagnostics) and purified by repeated LiCl precipitation. Wild-type and Prdm16−/− mouse fetuses were removed on E14.5 and E15.5 and the mid-facial region isolated by removing tissue anterior and posterior to the maxilla/palate. Tissue was fixed at 4°C overnight in 4% paraformaldehyde, and then prepared for in situ hybridization as described previously. 7
Results
Genes identified by ChIP-Chip analysis that may be regulated by PRDM16 in mouse embryonic palate/maxillary embryonic mesenchymal cells*
*Mouse embryonic palate/maxillary mesenchymal cells were transfected with PRDM16-myc-expressing plasmid and a ChIP-Chip analysis performed as described in the Materials and methods section. One hundred and twenty-two genes were identified that had a P value < 0.05
†Chr, chromosome number
‡Region start and ¶region end are the chromosomal positions for the promoter region bound by PRDM16
#Model-based Analysis of Tiling-arrays (MAT) score calculated using the algorithm described by Johnson et al. 12
To confirm the validity of these results, 20 candidate genes were selected and PCR primers designed using Primer-Blast (NCBI). In addition, PCR primers were synthesized for PGC1-α, Angiotensinogen and Resistin because these three genes are directly regulated by PRDM16.
13
Each primer set was used to test chromatin immunoprecipitated with anti-Myc antibodies from control- or PRDM16-transfected MEMM cells. The results from PCR analysis of 16 gene promoters selected from Table 1 are presented in Figure 1. In each reaction, there was enrichment in chromatin from PRDM16-transfected cells demonstrating that PRDM16 bound to each promoter. Four genes either gave no enrichment or no amplification of the input material. Additionally, there was enrichment of PGC1-α and Resistin, but not Angiotensinogen.
Polymerase chain reaction (PCR) confirmation of putative PRDM16-regulated genes in mouse embryonic palate mesenchymal (MEMM) cells. Input (1%) or immunoprecipitated, purified chromatin (2 ng) from control- or PRDM16-expressing MEMM cells was subjected to standard PCR reactions as described in the Materials and methods section. For each primer set, lane 1 represents the amplification from 1% of the amount of chromatin subjected to immunoprecipitation, lane 2 is that from chromatin from control-transfected MEMM cells, lane 3 from chromatin from PRDM16-Myc-transfected MEMM cells and lane 4, water control. In each case, there was increased amplification from PRDM16-Myc-transfected versus control-transfected MEMM cells. In some cases, the amplification from ‘input’ was weak, but detectable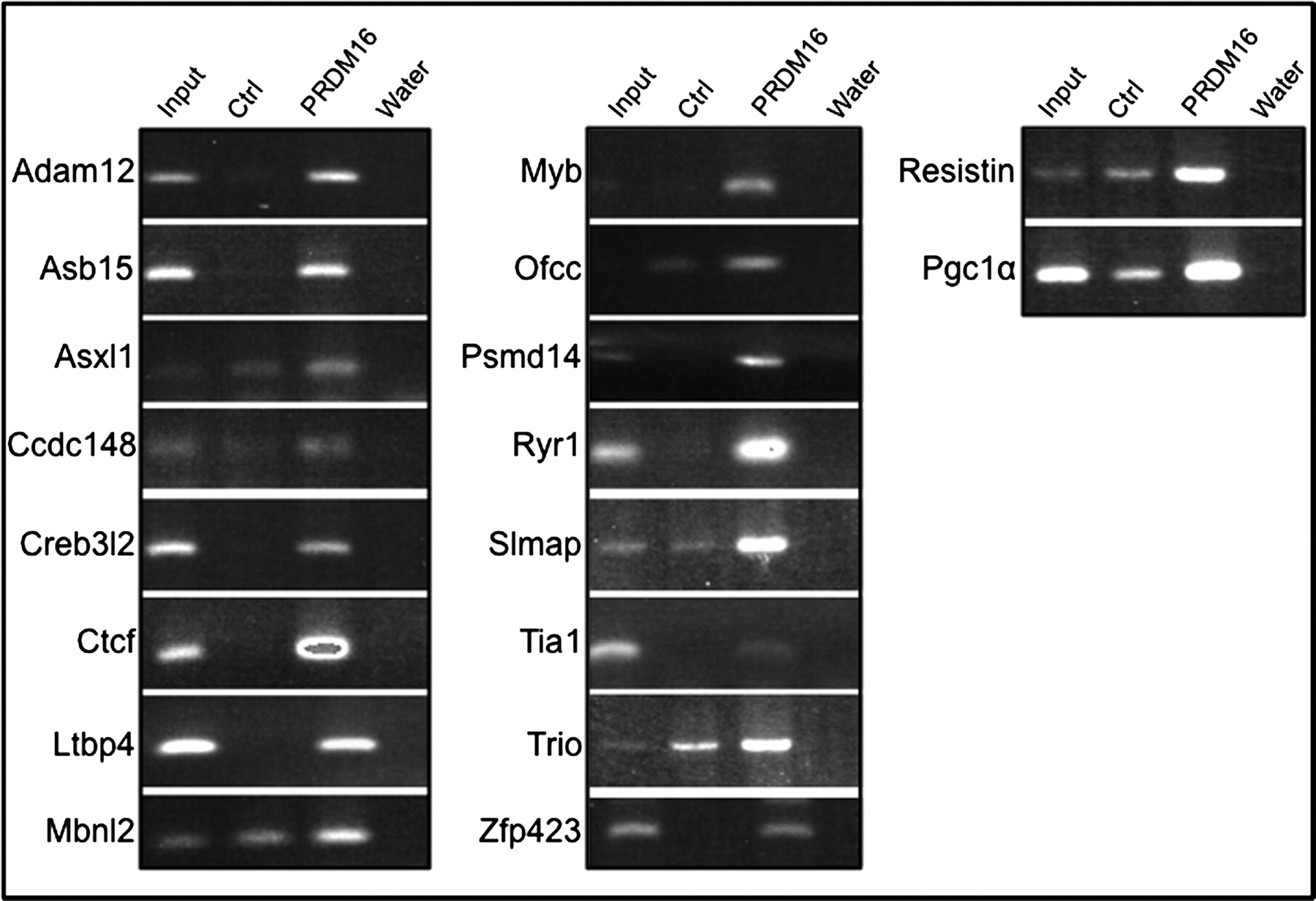
Functional classification of genes regulated by PRDM16 in MEMM cells*
*The function(s) of the genes reported in Table 1 were obtained from searching the existing literature using PubMed Bioinformatics Resource (PubMed identification numbers and GenRIF summaries). In some cases, genes have overlapping function (e.g. Asb15). Genes were classified according to an established function
Because Prdm16 is expressed to a greater extent in the anterior (future bony) palate,
7,8
has been demonstrated to prevent differentiation of mesenchymal precursors into a muscle phenotype,
4
and predicted to regulate genes necessary for both bone and muscle development, we hypothesized that loss of Prdm16 expression would alter expression of genes important for these processes. Osteopontin (Opn; also known as secreted phosphoprotein-1) is a marker for bone formation,
15
and in a previous large-scale gene expression study of mouse palate development, was found to undergo dramatic upregulation from E12.5 to E14.5.
16
To test the hypothesis that Opn expression is regulated by Prdm16, secondary palates from wild-type and Prdm16−/−
fetuses were isolated on days 13.5 and 14.5 of gestation, RNA purified and cDNAs synthesized. Expressions of Opn and a muscle cell marker, Myf4, were analyzed by realtime PCR, and expression levels on both days were determined and data presented as change in expression on E14.5 relative to E13.5 (Figure 2). As expected, in wild-type palate tissue, the expression of Opn was dramatically greater on E14.5 compared with the expression level on E13.5 (Figure 2). In contrast, the expression of Opn in palate tissue isolated from Prdm16−/−
fetuses was significantly less compared with wild-type levels of each on E13.5 and E14.5 (P < 0.01). The muscle marker, Myf-4, was decreased on E14.5 in wild-type palates when compared with expression on E13.5, but was significantly increased in Prdm16−/−
palate tissue on E14.5 (Figure 2). These data suggest that both muscle and bone development in the secondary palates of Prdm16−/−
fetuses is affected.
Expression of Opn and Myf-4 in Prdm16−/−
secondary palate tissue. The secondary palates from wild-type and Prdm16−/−
E13.5 and E14.5 fetuses were isolated and total RNA purified, from which cDNAs were synthesized. The expression of Opn and Myf-4 was assayed by semiquantitative realtime polymerase chain reaction (PCR) and normalized to the expression of 18S RNA. The expression level on both days was determined and the data presented is the change in expression on E14.5 relative to E13.5. The expression of Opn was significantly upregulated on E14.5 in wild-type palate tissue; however, in the absence of Prdm16, Opn expression was significantly decreased (*P < 0.001, one-way analysis of variance [ANOVA]). Conversely, the expression of Myf-4 was decreased in wild-type tissue but increased in Prdm16−/−
palate tissue (**P < 0.05, one-way ANOVA)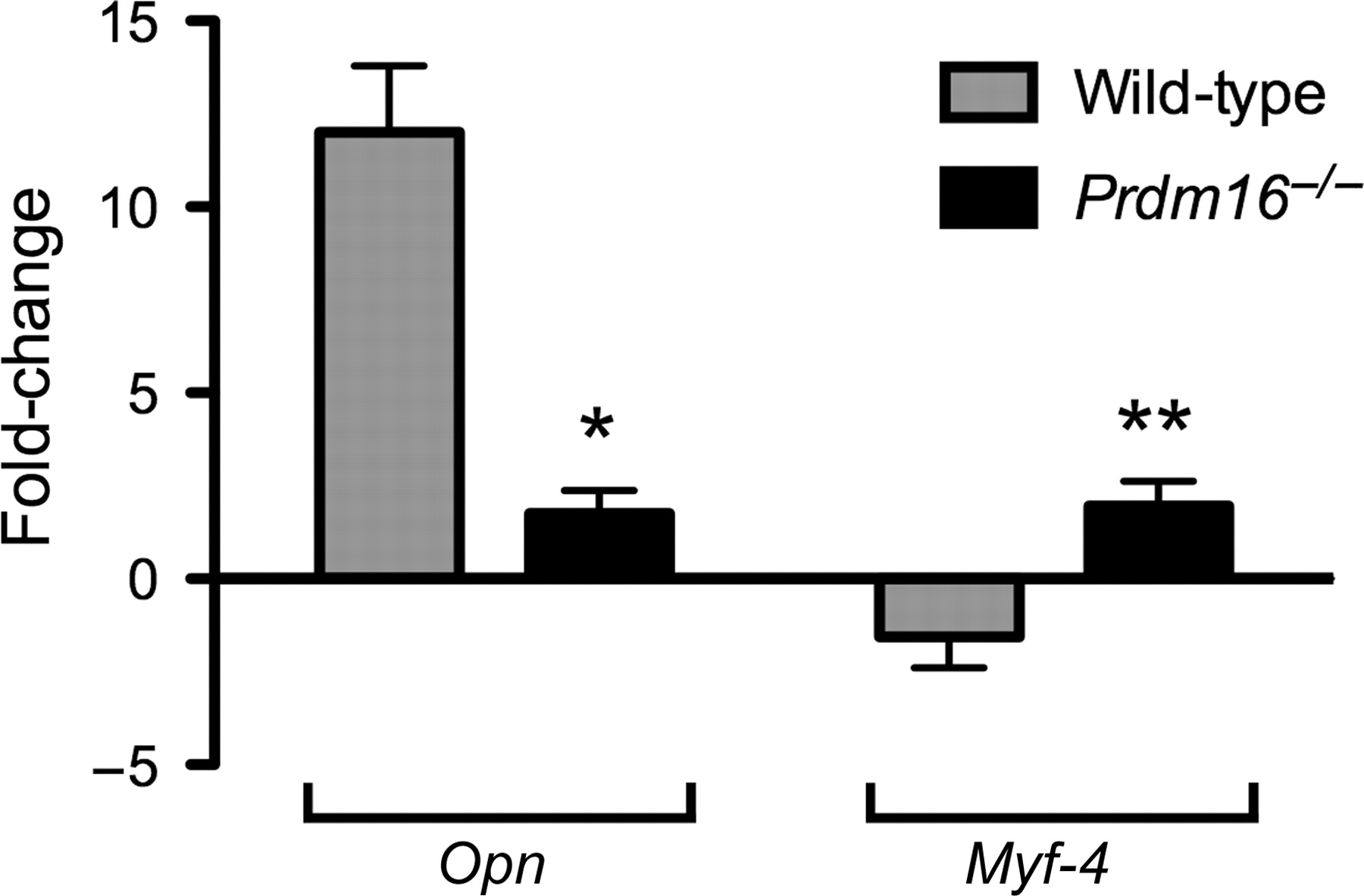
To confirm that the expression of Opn was altered in Prdm16−/−
palate tissue, in situ hybridization was conducted on E14.5 (Figures 3a–f) and E15.5 (Figures 3g–j) maxillary and palate tissue. Opn has a unique expression pattern in secondary palate tissue in that it is found in small, isolated clusters of cells. In wild-type palate tissue on E14.5, these clusters were located primarily along the medial edge seam on the oral side of the palate (Figure 3a, yellow arrows). On the nasal surface of the palate, scattered Opn-positive areas were observed, particularly at the posterior end of the secondary palate (yellow arrow in panels b and c). As shown in Figures 3d and e, Prdm16−/−
mutant fetuses have a wide cleft, and expression of Opn on both the oral and nasal aspects of the secondary palate was essentially absent. On E15.5, Opn expression was detected in the surfaces between the primary and secondary palates of wild-type fetuses (panel g, yellow arrow). On the nasal surface of wild-type fetuses, Opn was primarily expressed in the anterior two-thirds of the secondary palate (panel h, SP). On E15.5, Prdm16−/−
fetal tissue Opn was found to be misexpressed in palatal rugae (panel i, red arrow) while expression in the posterior palate persisted (yellow arrowheads). There was little detectable expression on the nasal surface of the secondary palate (panel j). These data demonstrate a unique expression pattern for Opn in the secondary palate and that the expression of Opn in Prdm16−/−
palate tissue is both reduced in level and aberrant in localization.
In situ hybridization analysis of Opn expression in wild-type and Prdm16−/−
E14.5 and E15.5 fetuses. Wild-type and Prdm16−/−
fetuses were isolated on E14.5 (a–f) and E15.5 (g–j), and maxillary and palatal tissues were dissected and processed for whole-mount in situ hybridization with a digoxigenin-labelled riboprobe specific for mouse Opn. Panels a and b represent E14.5 wild-type samples viewed from the oral and nasal aspect, respectively. Only a few clusters of Opn-positive cells were observed on the oral surface, primarily along the medial edge seam (MES, yellow arrows). On the nasal surface, scattered Opn-positive areas were observed, particularly at the posterior end (yellow arrow in b). Panel c is the boxed area in panel b at higher magnification. Yellow arrows indicate clusters of Opn-positive cells. Panels d and e are Prdm16−/−
maxillary and palatal tissue viewed from the oral and nasal aspects, respectively, demonstrating a wide cleft with no detectable expression of Opn in the palatal shelves. Panel f is wild-type tissue hybridized to an Opn sense riboprobe (negative control), revealing only background staining. Panels g and h represent maxillary/palatal tissue from E15.5 wild-type fetuses viewed from the oral and nasal aspects, respectively, demonstrating Opn expression between the primary and secondary palates (g, yellow arrow). On the nasal surface, Opn was primarily expressed in the anterior two-thirds of the fused secondary palate (SP). Panels i and j are oral and nasal views, respectively, of maxillary and palate tissue from E15.5 Prdm16−/−
fetal tissue. On the oral surface of the secondary palate, Opn was found to be misexpressed in palatal rugae (red arrow) while expression in the posterior palate persisted (yellow arrowheads). These data demonstrate that the expression of Opn in Prdm16−/−
palate tissue is both reduced and altered in distribution. PP, primary palate; PS, palatal shelf; MES, medial edge seam
Discussion
The role of the putative transcription factor PRDM16 is beginning to be elucidated in multiple tissues, most notably, in the development and differentiation of brown adipose tissue, where it drives Myf5-expressing (muscle)-precursor cells into the adipose lineage. 3 Interestingly, a significant number of PRDM16-regulated genes identified in the current screen have been linked to development of muscle tissue. For example, Asb15 is primarily expressed in skeletal muscle and is important for myoblast differentiation. 17,18 Trio (triple functional domain protein) is a member of the Dbl-homology family of guanine nucleotide exchange factors and is also necessary for skeletal muscle development. 19 Likewise, the sarcolemma-associated protein (Slmap) is essential for myotube formation 20 and Ryr1 is found predominantly in skeletal muscle where it plays a central role in excitation–contraction coupling. 21 Four genes identified have been demonstrated to contribute to chondro and/or osteogenesis (Creb3l2, Bag1, Kdr and Tsc22d1). Analysis of additional gene markers for early, mid and late bone development revealed a generalized impairment/delay (manuscript in preparation). Evidence for this hypothesis was obtained from an analysis of the expression of a marker for bone (Opn) and muscle (Myf-4) development. The expression of Opn was significantly reduced in palate tissue from Prdm16−/− fetuses, while that for Myf-4 was significantly increased. Opn has been previously linked to human cases of orofacial clefting. 22 Thus, Prdm16 may play an important contributory role to myo-, chondro- and/or osteogenesis in the developing orofacial region, in addition to regulating other processes important for normal development.
Development of the secondary palate in both mice and humans occurs through a series of morphogenetic events that include reorientation of the palatal processes from a position lateral to the tongue to a position superior to the tongue, fusion of the bilateral shelves to one another and the nasal septum, and finally terminal differentiation into the bony, anterior and soft, muscular posterior palate. Only one of the PRDM16-regulated genes identified in the current screen (Ofcc1) is similar to a gene in humans that has been linked to orofacial clefting in humans. 23 Although the function of this gene is unknown, it has been demonstrated to be expressed in migrating neural crest cells and craniofacial bones. 24 In addition to potentially regulating both muscle and bone development in the secondary palate, PRDM16 likely regulates other, more general aspects of cellular physiology. A number of other genes were identified as PRDM16-regulated, including those linked to epigenetic regulation, protein stability and intracellular trafficking. Therefore, the role of Prdm16 during development of the secondary palate appears to be complex, potentially regulating the expression of numerous genes that control several key processes essential for normal palate development (e.g. cell proliferation/differentiation).
The results presented in the current report suggest that Prdm16 functions to control cell differentiation during development of the secondary palate in mice by either promoting differentiation or preventing premature differentiation. It is possible that defects in palate development seen with the Prdm16 knockout mouse are caused by abnormal muscle and/or bone development that leads to altered morphogenesis of the nascent palatal processes leading to failure of reorientation and subsequent separation of the oral and nasal cavities.
Footnotes
ACKNOWLEDGEMENTS
The authors thank Dr Takeshi Imamura at the JFCR Cancer Institute, Tokyo, Japan, for the pcDEF3-PRDM6-6X-myc plasmid and Dr Guy Brock for help with bioinformatics. This study was supported by NIH grants DE018215, HD053509 and P20 RR017702 (to RMG) from the COBRE program of the National Center for Research Resources.