
Abstract
No mechanistic actions for piracetam have been documented to support its nootropic effects. Voltage-gated calcium channels have been proposed as a promising pharmacological target of nootropic drugs. In this study, we investigated the effect of piracetam on CaV2.2 channels in peripheral neurons, using patch-clamp recordings from cultured superior cervical ganglion neurons. In addition, we tested if CaV2.2 channel inhibition could be related with the effects of piracetam on central neurons. We found that piracetam inhibited native CaV2.2 channels in superior cervical ganglion neurons in a dose-dependent manner, with an IC50 of 3.4 μmol/L and a Hill coefficient of 1.1. GDPβS dialysis did not prevent piracetam-induced inhibition of CaV2.2 channels and G-protein-coupled receptor activation by noradrenaline did not occlude the piracetam effect. Piracetam altered the biophysical characteristics of CaV2.2 channel such as facilitation ratio. In hippocampal slices, piracetam and ω-conotoxin GVIA diminished the frequency of excitatory postsynaptic potentials and action potentials. Our results provide evidence of piracetam's actions on CaV2.2 channels in peripheral neurons, which might explain some of its nootropic effects in central neurons.
Introduction
Nootropic compounds, like piracetam, are modulators of brain functions. Since the discovery of piracetam, related compounds have been developed for the treatment of cognitive disorders. These compounds have been classified as cognitive enhancers, antiepileptic drugs or unknown clinical efficacy drugs, based on their structure and clinical effect. 1 Piracetam belongs to the group of cognitive enhancers. However, the mechanism by which piracetam modulates brain function remains a major question in the field.
Since piracetam is a derivative of γ-aminobutyric acid (GABA) and their structures are closely related, one possible mechanism of piracetam is to mimic GABA's actions. 2 However, early studies showed that piracetam did not affect GABA receptors. 3 Other possible mechanisms of piracetam's action are to enhance membrane fluidity, 4 improving glucose uptake and ATP production and altering mitochondrial function. 5–7 However, these long-term effects could be explained by mechanisms acting on receptors, ion channels and transporters. For example, piracetam enhances the efficacy of α-amino-3-hydroxy-5-methyl-4-isoxazolepropionic acid (AMPA) receptors, 8 exerts inhibitory effects on adrenergic, cholinergic and opioidergic systems that facilitate antinociception, 9 and inhibits neuronal high-voltage-activated (HVA) calcium channels in invertebrates. 10 Levetiracetam, another nootropic compound, which is structurally related to piracetam but is used as an antiepileptic drug, inhibits CaV2.2 channels in CA1 hippocampal neurons and reduces intracellular calcium elevation in cortical neurons. 11,12 However, the effect of piracetam on neuronal CaV2.2 channels in vertebrates remains elusive.
The purpose of our study was to assess if piracetam could inhibit neuronal CaV2.2 channels in vertebrates and also whether or not this inhibition was mediated by a G-protein mechanism. We studied the piracetam effects in superior cervical ganglion (SCG) neurons, where CaV2.2 channels are a predominant population, and in hippocampal neurons, where the effect of other nootropic drugs has been already reported. 11,12 In SCG neurons, CaV2.2 currents were recorded in whole-cell voltage-clamp configuration before and during piracetam application. Additional experiments were designed to test the involvement of G-proteins in the mechanism of piracetam. Our results showed a clear-cut inhibition of CaV2.2 channels by piracetam in sympathetic neurons and provided evidence of a relation of CaV2.2 channel inhibition with a reduction in excitability in central neurons. Also, our experiments showed that piracetam inhibits CaV2.2 with characteristics similar to the classical voltage-dependent inhibition induced by G-proteins. However, we showed that the mechanism of inhibition induced by piracetam does not involve the G-protein-coupled receptor (GPCR) signaling pathway. This inhibition might represent a novel mechanism of regulation for CaV2.2 channels.
In a previous report, we showed that changes in intracellular calcium equilibrium during chronic epilepsy coexist with alterations in excitability. 13 Since piracetam has been proposed to be effective in some epilepsies, we hypothesized that targeting synaptic calcium channels might be a mechanism to restore calcium homeostasis and to reduce hyperexcitability. Here, we tested piracetam in a model of hyperexcitability induced by low magnesium and high potassium.
Materials and methods
SCG sympathetic neuron culture and electrophysiology
SCG neurons were isolated from five-week-old male Wistar rats as described previously.
14,15
Rats were obtained from the animal breeding facility of the School of Medicine at the Universidad Nacional Autonoma de Mexico (UNAM) and were handled according to the Mexican Official Norm for Use, Care and Reproduction of Laboratory Animals (NOM-062-ZOO-1999). Animals were anesthetized with CO2 and decapitated with a guillotine. Following dissection, ganglia were desheated and sliced into small pieces. Tissue was dissociated with 20 U/mL papain, 1 mg/mL collagenase and 10 mg/mL dispase for 60 min and neurons were dissociated by gently pipetting. Cell suspension was centrifuged at 180 g for three minutes and washed twice in Leibovitz's medium (L15) and once in Dulbecco's modified Eagle's medium (DMEM). Neurons were plated on glass coverslips coated with poly-
In our experiments, CaV2.2 current was defined as the component of calcium current that is sensitive to 100 μmol/L Cd2+ in the presence of 2 μmol/L nifedipine, since CaV2.2 channels carried above 90% of the calcium current in SCG neurons, with no detectable CaV2.1 (P/Q) component. 16 SCG neurons were constantly bathed during recording (1–2 mL/min) with a solution containing (in mmol/L) 165 tetraethylammonium chloride (TEA-CL), 2 BaCl2, 1 MgCl2, 10 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid (HEPES), 8 glucose, 0.002 nifedipine and 0.0001 tetradotoxin, pH adjusted to 7.4 with tetraethylammonium hydroxide, to isolate barium currents. The pipette solution (internal solution) contained (in mmol/L) 140 CsCl, 20 TEA-Cl, 5 MgCl2, 10 HEPES, 0.1 Cs4BAPTA (BAPTA = 1, 2-bis(o-aminophenoxy)ethane-N,N,N′,N′-tetraacetic acid), 4 Na2ATP, 0.3 GTP and 0.1 leupeptin, and 2 GDPβS (where indicated), pH adjusted to 7.2 with CsOH. Nifedipine-containing external solution was prepared daily from 25 mmol/L absolute ethanol stock solution and protected from light. Piracetam was added into the bath solution and was applied by using a focal perfusion system that exchanges the solution locally within 5–10 s. All experiments were performed at room temperature (23–25°C). Reagents were obtained as follows: BAPTA (Molecular Probes, Eugene, OR, USA), L15 and DMEM (Invitrogen Corp., Carlsbad, CA, USA); all other chemicals and reagents were purchased from Sigma Chemical Co. (St Louis, MO, USA).
Currents were recorded in whole-cell voltage-clamp configuration 17 with an HEKA EPC-9 patch-clamp amplifier (HEKA Elektronik, Lambrecht/Pfalz, Germany). Pipettes made of borosilicate glass and with a resistance of 1.8–2.2 MΩ were coated with Sylgard 184 (Dow Corning, midland, MI, USA). Capacity transients were cancelled and series resistance (2–7 MΩ) was compensated to >60%. Voltage pulse command protocols were generated, and data were digitized, recorded and analyzed using Pulse + PulseFit (HEKA Instruments, Inc., Bellmore, NY, USA). Steady-state currents were sampled at 10 kHz and were low-pass filtered at 3 kHz (3-pole Bessel filter).
Rat brain slice preparation and electrophysiology
To study CA1 pyramidal neurons from the hippocampus, we used male Wistar rats (250–300 g body weight), individually housed and maintained under controlled conditions (12 h normal light/dark cycles, 22–25°C, and 38% relative humidity) with food and water ad libitum. Five-week-old male Wistar rats were killed by decapitation under ether anesthesia. Brains were extracted and sliced (400 μm) with a vibratome. Slices were maintained at room temperature (24–26°C) for one hour in artificial cerebrospinal fluid (ACSF) containing (in mmol/L) 126 NaCl, 3 KCl, 1.5 CaCl2, 1.5 MgCl2, 1.25 Na2HPO4 and 10 glucose. ACSF was continuously bubbled with 95% O2/5% CO2 mixture and equilibrated to pH 7.4 with NaOH. Rate perfusion was 1–2 mL/minute. Subsequently, brain slices were transferred to a recording chamber and continuously superfused with ACSF or ACFS with 10 μmol/L piracetam warmed at 34°C. Cells were visualized by differential interference contrast microscopy using a ×40/1.0 objective in an upright microscope (FN1; Nikon, Melville, NY, USA). Patch pipettes were pulled from borosilicate glass and filled with an internal solution containing (in mmol/L) 145 KCl, 5 HEPES, 5 ethylene glycol tetraacetic acid, adjusted to pH 7.3 with KOH.
Membrane potential and current recordings were made in voltage and current clamp whole-cell configuration using a List EPC7 amplifier (Heka Elektronik). Capacity transients were cancelled using EPC7 circuitry and series resistances (4–5 MΩ) was compensated to >70%. Voltage pulse command protocols were generated, and data were digitized, recorded and analyzed using Pclamp6 (Axon Instruments, Molecular Devices, Sunnyvale, CA, USA). Membrane voltage and currents were digitized using a 12-bit A/D Tecmar Lab Master Board (Scientific Solutions, Inc., Mentor, OH, USA) with a maximum digitization rate of 125 kHz and sampled at 10 kHz. Recordings were filtered at 3 kHz (3-pole Bessel filter). Total currents were elicited with a voltage step from −60 to +30 mV during 20 ms. Current traces were not corrected for linear leak current. Voltage responses were generated with 800 pA square pulses during 20 ms. Also, spontaneous activity was recorded for 45 min and intervals of 20 s every 10 min were stored to count the number of action potentials (APs) or excitatory postsynaptic potentials (EPSPs) in the control conditions, under 10 μmol/L piracetam and under 1 μmol/L ω-conotoxin. ω-Conotoxin was used to determine indirectly if the piracetam effects could be explained by CaV2.2 channel inhibition.
Hyperexcitability model
To study the effect of piracetam on epileptiform discharges from hippocampal CA1, we used the hyperexcitability model based on the application of low magnesium and high potassium. 18 Brain slices were superfused with free Mg2+ ACSF containing (in mmol/L) 126 NaCl, 7 KCl, 1.5 CaCl2, 0 MgCl2, 1.25 Na2HPO4, 10 glucose and adjusted to pH 7.4 with NaOH. Sustained spontaneous neuronal discharges were evident within approximately 30 min of superfusion of free Mg2+ ACSF. Nine of 12 recorded neurons presented epileptic activity characterized by high-frequency discharges of APs. We recorded spontaneous activity for 45 min and quantified the number of APs during 20-s recordings in control, low-magnesium–high-potassium solution and low-magnesium–high-potassium plus piracetam (10 μmol/L) solution.
Data analysis
Current amplitudes were taken as the mean value of recorded points between 4 and 5 ms after onset of the test pulse prior to reaching the peak current. Due to the magnitude of the cell current being cell-size dependent, aggregate current data are presented as current densities normalized to cell capacitance.
Dose–response data for the piracetam effect on native CaV2.2 channels were fitted to the function: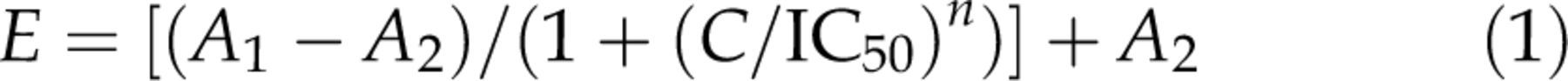
The conductance G for barium, G
Ba, was calculated utilizing the following equation: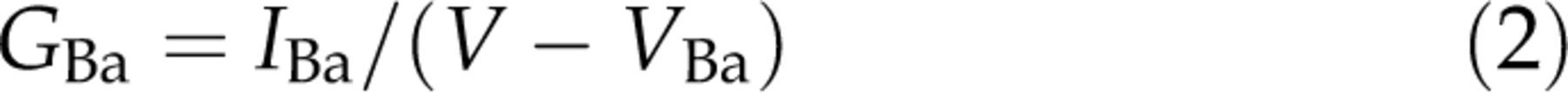
The Boltzmann equation was used as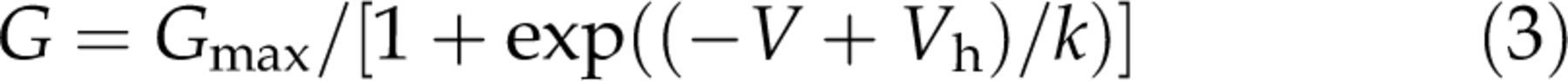
Origin 8.0 software was utilized to perform fit of data, and for statistical analysis (OriginLab Corporation, Northampton, MA, USA). All data are expressed as mean ± standard error of the mean (SEM) and comparisons between groups were analyzed using Student unpaired t-tests. P < 0.05 was considered to be statistically significant.
Results
Piracetam inhibits CaV2.2 calcium channels in sympathetic neurons
Cyclic derivatives of GABA affect calcium signaling in neurons. Levetiracetam inhibits CaV2.2 channels in hippocampal neurons
11
and piracetam inhibits neuronal HVA calcium channels in invertebrates.
10
However, it is not well established whether piracetam can act on CaV2.2 channels in vertebrates. The calcium current in SCG neurons is mainly mediated by omega conotoxin-sensitive channels (CaV2.2) in recordings on which L-type calcium channels have been blocked with 2 μmol/L nifedipine.
14,16
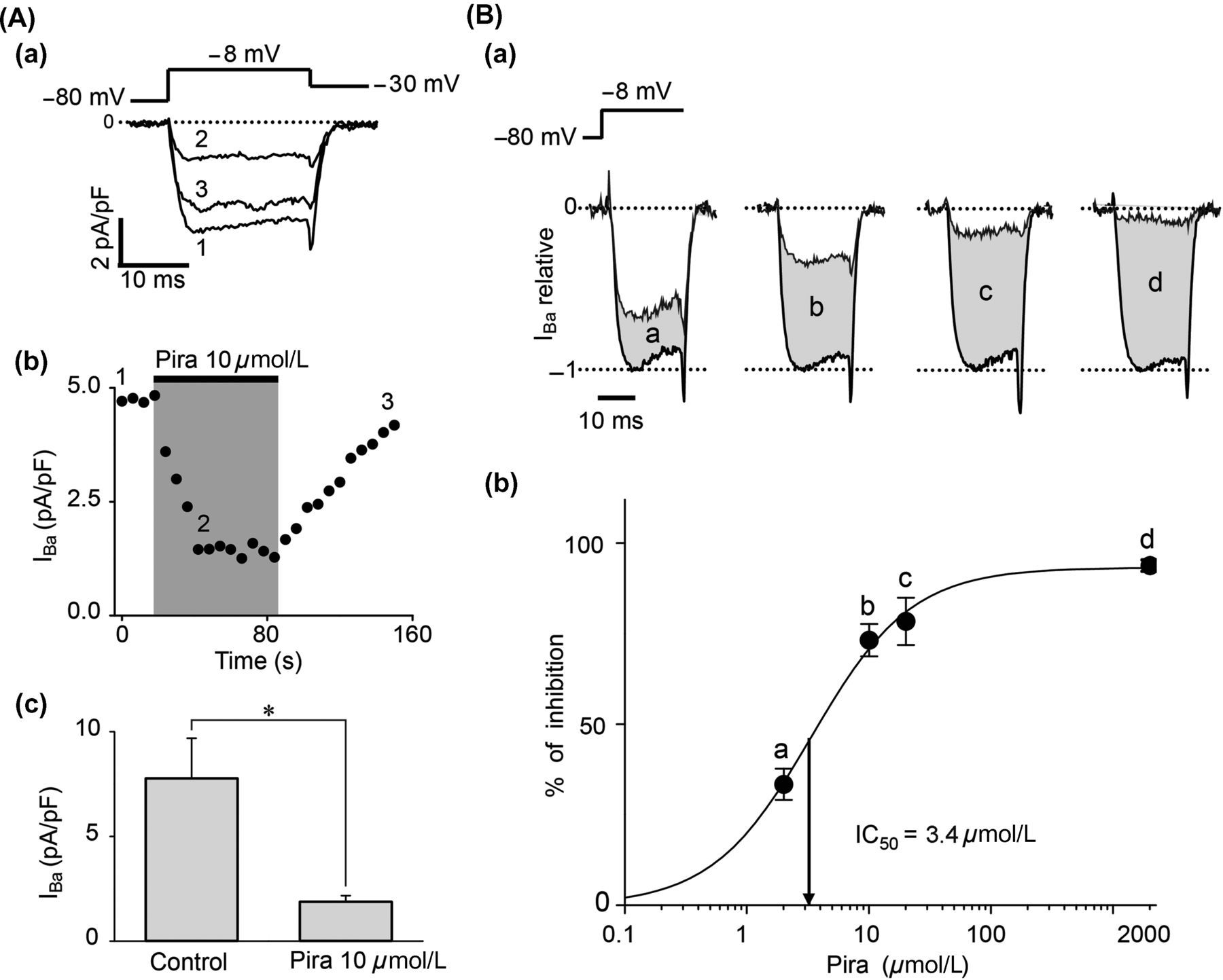
Figure 1 shows the percent of nifedipine- and ω-conotoxin-sensitive channels in SCG neurons (n = 8). In our hands, the 18 ± 3% of calcium current was blocked by 2 μmol/L nifedipine and the 78 ± 4% was blocked by 1 μmol/L ω-conotoxin GVIA. According to previous reports, these data show that SCG neurons express predominantly the CaV2.2 channels. Along this work, we recorded calcium currents in the presence of 2 μmol/L nifedipine, and in such conditions, the CaV2.2 current represents 90% of the calcium current. Here we characterized the effect of piracetam on CaV2.2 channels in rat SCG neurons. Voltage pulses were elicited every six seconds from −80 to −8 mV for 20 ms in voltage-clamp whole-cell configuration, using barium as the charge carrier. A rapidly activated inward barium current was inhibited by near 77% after bath application of 10 μmol/L piracetam (n = 7, Figure 2Aa). The effect of piracetam reached its maximum after 24 s, remained stable for one minute and reversed rapidly by washing-out (Figure 2Ab). Current amplitude was measured isochronally at 4–5 ms from depolarizing pulse onset. Figure 2Ac shows a summary of average currents amplitude density in control (7.79 ± 1.90 pA/pF) and under 10 μmol/L piracetam (1.90 ± 0.27 pA/pF). Therefore, piracetam induced strongly, rapidly and reversibly inhibition on CaV2.2 channels in SCG neurons. Also, inhibition of CaV2.2 current depended on piracetam concentration. We assayed piracetam concentrations of 2, 10, 20 and 2000 μmol/L (n = 4 for each concentration) and the dose–response curve was fitted to a sigmoid function by the least-squares method (Figure 2B). The half-maximal concentration of piracetam (IC50) was 3.4 ± 0.6 μmol/L with a maximal inhibition of 94 ± 2% at 2000 μmol/L (Figure 2Bb). The calculated Hill coefficient for piracetam effect was 1.1 ± 0.2 (Equation 1), suggesting a bimolecular interaction without cooperativity. Inhibition of CaV2.2 channels might be explained by direct interaction with piracetam or by a mechanism mediated by G-proteins; we explored both possibilities in this paper.
Percent of calcium current flowing through nifedipine- and ω-conotoxin-sensitive channels in superior cervical ganglion (SCG) neurons. (a) Time course of the effect of 2 μmol/L nifedipine and 1 μmol/L ω-conotoxin GVIA on calcium current. Inset shows representative superimposed traces under control conditions (1), nifedipine (2) and ω-conotoxin GVIA plus nifedipine (3). (b) Summary of the percent of calcium current sensitive to nifedipine, ω-conotoxin GVIA and the percent of calcium current resistant to both blockers (other). Data are shown as mean ± SEM. Piracetam inhibited CaV2.2 channels in superior cervical ganglion (SCG) neurons. (Aa) Representative voltage-clamp whole-cell recordings of calcium current before (1), during application of 10 μmol/L piracetam (2) and at wash-out (3) in isolated SCG neurons. (Ab) Time course of calcium current amplitude showing piracetam-induced inhibition. Piracetam was applied to the cell when indicated by the gray area. (Ac) Summary of the effect of piracetam on barium current in seven cells. (Ba) Representative superimposed current traces before and during application of 2 μmol/L (a), 10 μmol/L (b), 20 μmol/L (c) and 2000 μmol/L (d) piracetam. (Bb) Dose–response curve of the inhibition induced by piracetam (n = 4 for each dose). Percent of inhibition was determined as the difference between current amplitude before and during piracetam application divided by current before piracetam. Smooth curve represents sigmoid fit to data (Equation 1). The Hill coefficient obtained from the curve was 1.1, and IC50 = 3.4 μmol/L. Data are shown as mean ± SEM. *P < 0.05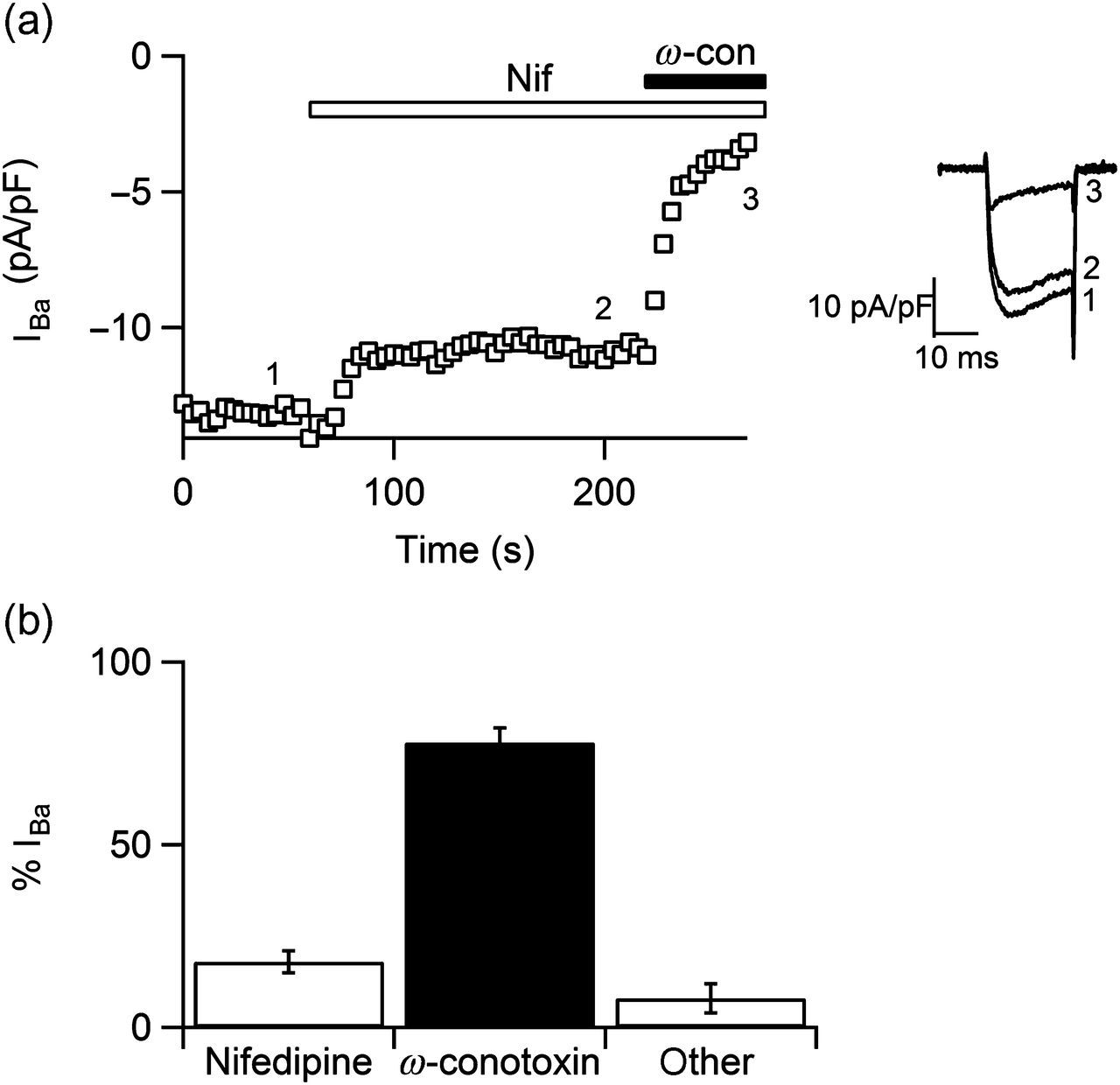
Piracetam enhances facilitation ratio of CaV2.2 channels
To explore the effect of piracetam on the biophysical characteristics of CaV2.2 channels, we examined possible alterations in the voltage-dependence profile. We elicited currents with voltage pulses at different potentials from −50 to +38 mV in steps of 8 mV. Figure 3A shows family currents before (a) and during 10 μmol/L piracetam application (b). The I–V curves show that CaV2.2 channels are activated between −40 and +40 mV, with the maximal current amplitude at −10 mV (n = 5, Figure 3Ac). Piracetam inhibited CaV2.2 current ranging from 55% to 75% depending on the activation voltage. The maximal inhibition by piracetam occurs at 6 mV, suggesting that this inhibition depends on voltage activation of the channel. We analyzed the conductance–voltage relationship and found that piracetam inhibited the conductance by near 70% (Figure 3Ad), induced a significant moderate shift of the half conductance (V
h) by −3 mV (Figure 3Ae, Table 1) and did not significantly affect the steepness factor k value (see Equations 2 and 3). As the effect of piracetam depended on voltage and shifted voltage dependence of channels, it resembles the effect of voltage-dependent inhibition mediated by G-proteins. To address if such mechanisms are related, we used the conditioning prepulse protocol to evaluate the extent of prepulse facilitation. This paradigm represents a widely used tool for identifying voltage-dependent inhibition mediated by G-proteins.
19,20
In these experiments, currents were evoked every 10 s with a pair of 20-ms depolarizing test pulses to −8 mV from a holding potential of −80 mV, one before (P1) and the other 50 ms after (P2) a 30-ms conditioning prepulse (PP) to +80 mV (Figure 3B upper panel). The current in control conditions, before piracetam application, was bigger in P2. This increase of calcium current after a conditioning prepulse is defined as tonic inhibition. Tonic inhibition is the basal inhibition of CaV2.2 channels without stimulation of any GPCR due to a free pool of Gβγ proteins bounded to the channels.
21
De-polarizing prepulses also relieved much of the piracetam-induced inhibition (Figure 3Ba). We analyzed the extent of voltage-dependent inhibition in piracetam-treated SCG neurons using a facilitation ratio by comparing currents in the second test pulse with those in the first pulse. In control conditions, this facilitation ratio was 1.48 ± 0.03 (n = 7) and during piracetam application it rose to 2.00 ± 0.12 (n = 7, Figure 3Bb). These results together suggest that piracetam induces a voltage-dependent inhibition of CaV2.2 channels and increases facilitation ratio like the membrane-delimited mechanism of G-protein inhibition.
CaV2.2 inhibition induced by piracetam was voltage-dependent. (A) Family of currents in the absence (Aa) and presence of 10 μmol/L piracetam (Ab) elicited with the protocol illustrated at the top. (Ac) Current–voltage relationship before (circles) and during piracetam application (squares). Symbols represent mean ± SEM values (n = 5). Relationship between conductance and voltage shows that piracetam reduces maximal conductance (Ad) and shifts significantly the voltage dependence of the channel by −3 mV (Ae). Conductance was calculated as stated in Materials and methods (Equation 2). Solid lines represent the best fit to a Boltzmann function (Equation 3) and are normalized in panel Ae. (B) Piracetam-induced inhibition is released by a conditioning pulse. (Ba) Representative superimposed current traces under control conditions and under piracetam. Barium currents elicited with a protocol (top) consisting of two identical test pulses (P1 and P2) to −8 mV from a holding potential of −80 mV. A conditioning pulse (PP) to +80 mV preceded P2. Note that current elicited by P2 is bigger than that elicited by P1 under both conditions. (Bb) Summary of facilitation ratio (n = 7). Facilitation ratio was calculated as current in P2 divided by current in P1. Facilitation ratio was 1.48 ± 0.03 in control conditions and 2.00 ± 0.12 under 10 μmol/L piracetam. Data are plotted as mean ± SEM. *P < 0.05 Parameters derived from Boltzmann fit to the G
Ba-voltage relationship, under control conditions and piracetam application (data from Figure 3Ad) Data are mean ± SEM from five cells in each condition. *P < 0.05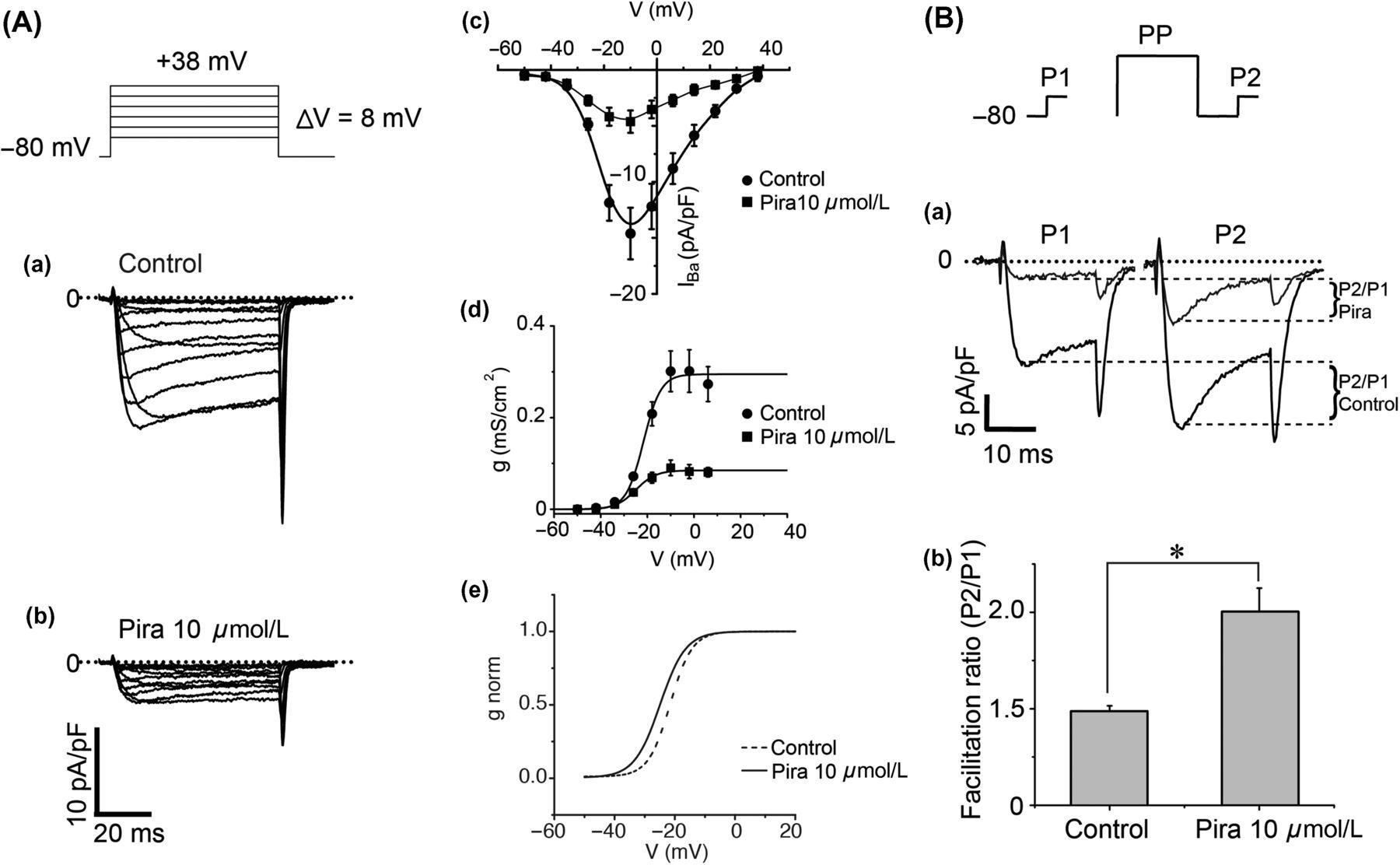
Piracetam-induced inhibition of CaV2.2 channels is independent of GPCR signaling pathway
GPCR activation induces a voltage-dependent inhibition of CaV2.2 channels which is mediated by the Gβγ subunits.
22,23
Since our previous experiments showed that piracetam induces a voltage-dependent inhibition like the G-protein-mediated inhibition of CaV2.2 channels, we hypothesized that piracetam acts through the G-protein signaling pathway. To address whether or not G-proteins are involved, we applied 2 mmol/L GDPβS into the recording pipette. This non-hydrolyzable analog of GDP prevents dissociation of G-protein subunits
24
and then CaV2.2 channels could not be inhibited by piracetam if Gβγ dimer is the underlying mechanism of this inhibition. In the absence of piracetam, dialysis of 2 mmol/L GDPβS decreased the facilitation ratio from 1.48 ± 0.03 to 1.21 ± 0.05, according to the block of tonic inhibition.
21
Surprisingly, in the presence of piracetam and 2 mmol/L GDPβS, the facilitation ratio was 1.87 ± 0.08 (Figure 4A), similar to that observed in neurons under piracetam and without GDPβS (2.00 ± 0.12). This result suggests that piracetam enhances the tonic inhibition of CaV2.2 channels by a mechanism independent of G-proteins or GPCR activation.
CaV2.2 inhibition by piracetam did not involve G proteins. (A) Piracetam effect is not hampered by dialysis of GDPβS. (Aa) Representative superimposed current traces in control conditions and under piracetam in the presence of 2 mmol/L GDPβS into the pipette solution. Currents were elicited with the protocol of double pulse, at the top. Note that intracellular dialysis of GDPβS avoided the current increase in P2 under control conditions. Despite the presence of GDPβS into the cell, the current in P2 under piracetam was bigger than in P1. (Ab) Summary of facilitation ratio with or without GDPβS for control conditions (1.21 ± 0.05) and under piracetam (1.87 ± 0.08, n = 7). Data are plotted as mean ± SEM. *P < 0.05. (B) Piracetam inhibition is not occluded by 100 μmol/L noradrenaline (NA). (Ba) Representative current traces in control conditions (1), under 100 μmol/L NA (2) and under 100 μmol/L NA plus piracetam (3). (Bb) Time course of current amplitude showing effect of NA and NA plus piracetam. Drugs were applied to the bath solution when indicated by the gray area. Currents were elicited with the protocol at the top. (Bc) Summary of percent of inhibition by 100 μmol/L NA (78 ± 0.04%) and 100 μmol/L NA plus 10 μmol/L piracetam (96 ± 0.01%, n = 5). Data plotted as mean ± SEM. *P < 0.05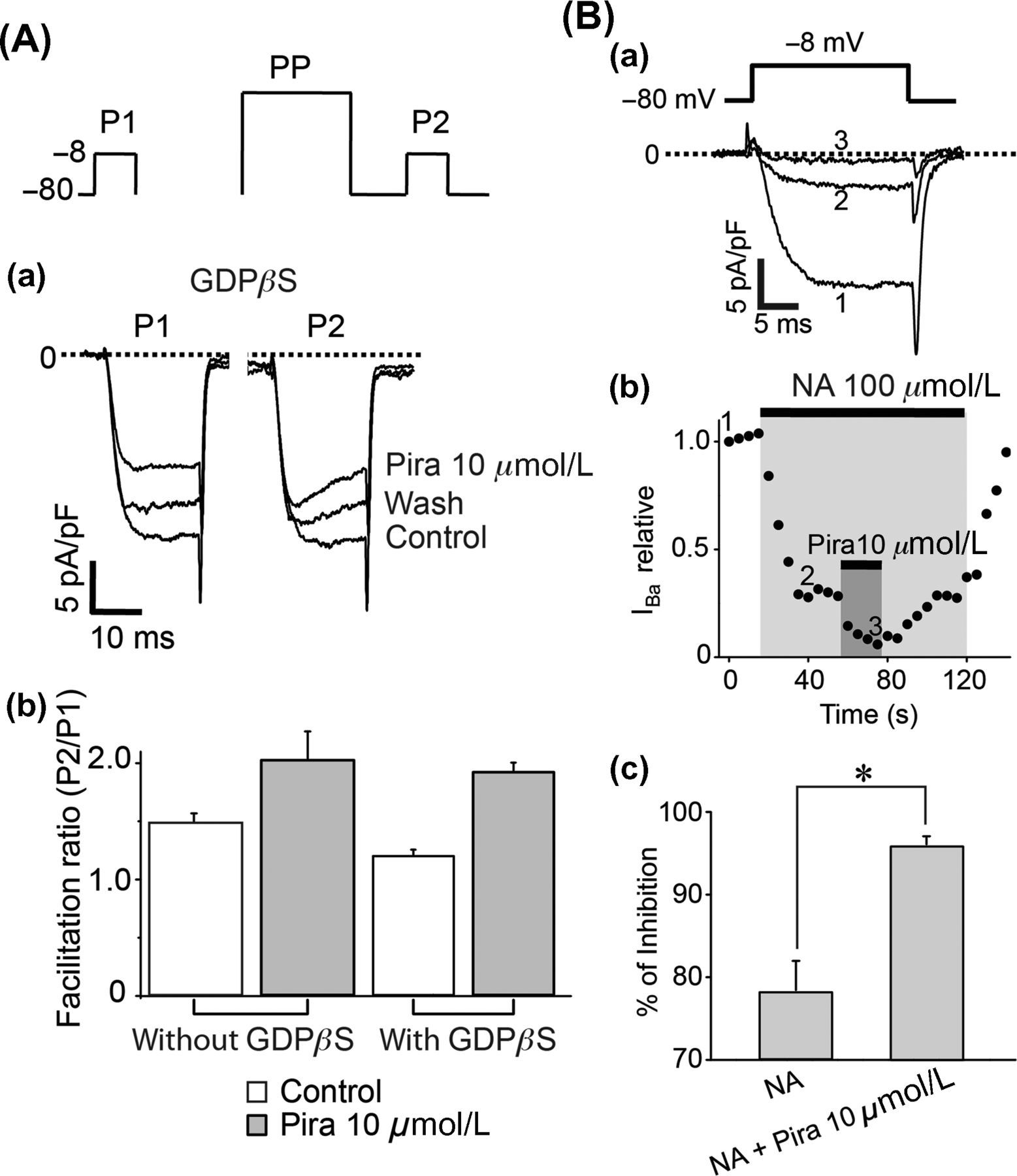
We further investigated if the mechanism of piracetam differs from GPCR activation by testing additive effects of piracetam and noradrenaline on the inhibition of CaV2.2 channels. Figure 4Ba shows representative superimposed current traces from control conditions, under 100 μmol/L noradrenaline, and after addition of 10 μmol/L piracetam in the presence of noradrenaline. We used saturating concentration of noradrenaline to rule out possible Gβγ proteins that were not activated at lower concentrations. Piracetam application on neurons bathed with noradrenaline inhibits even more CaV2.2 current. While the inhibitory effect of noradrenaline was 78 ± 4%, inhibition by piracetam plus noradrenaline was 96 ± 1% and therefore the effect of piracetam was not occluded by noradrenaline (Figure 4B). This result supports that piracetam-induced CaV2.2 inhibition is independent of the G-protein signaling pathway and raises the possibility that piracetam alters basic biophysical properties of CaV2.2 channels by a novel mechanism.
Piracetam reduces the frequency of APs and EPSPs in CA1 pyramidal cells
Having established the effect of piracetam on CaV2.2 channels in isolated sympathetic neurons, we tested the effect of piracetam on naturally connected central neurons in hippocampal slices. One of the clinical uses of piracetam is for treatment of some epilepsies. Previously, we showed that in chronic epilepsy, changes in intracellular calcium equilibrium coexist with alterations in excitability.
13
Since piracetam inhibited CaV2.2 channels in SCG neurons and some antiepileptic drugs target this channel in central neurons, we did a first approach to relate this piracetam-induced inhibition of CaV2.2 channels with its effects on hippocampal neurons. We first tested the effects of piracetam on the excitability of pyramidal neurons in slices of the CA1 region of hippocampus, comparing the shape of APs during a current stimulus. In Figure 5A, APs were evoked by a current stimulus of 800 pA during 20 ms. We found that piracetam decreased the duration of APs (black dotted line) when compared with control cells (black line), a result that is evident during the third AP. Piracetam also induced changes in resting membrane potential and in after hyperpolarization potential. Changes in the shape of APs are consistent with alterations in underlying ionic currents. Thus, total currents were elicited with voltage stimulus for 20 ms from −60 to +30 mV and were recorded from control- and piracetam-treated cells, revealing a slow ionic current component, consistent with calcium channels (Figure 5B). Total currents are presented without leak and capacitive subtraction. If piracetam acts on the calcium current in hippocampal neurons, subtraction of current in control and in piracetam should eliminate ionic currents carried by sodium and potassium. Accordingly, the total current subtraction (control minus piracetam-treated cells) revealed a typical current phenotype for HVA calcium channels (Figure 5Bb).
Piracetam reduced the frequency of action potentials (APs) and excitatory postsynaptic potentials (EPSPs) in hippocampal slices. (A) Piracetam effect on APs from hippocampal neurons before (solid line) and during piracetam application (dashed line). APs were induced by a current stimulus of 800 pA during 20 ms. (B) Subtraction of total current recorded under piracetam application from total current in control conditions reveals a current with a high-voltage-activated calcium current shape. (Ba) Representative total currents recorded from somas of hippocampal pyramidal neurons in control conditions, under piracetam and after wash-out. Currents were elicited with a voltage stimulus from −60 to 30 mV for 20 ms. (Bb) Subtraction of total currents. (C) Piracetam effect on spontaneous activity of hippocampal neurons. (Ca) Representative recordings of spontaneous activity from a single pyramidal neuron before and during piracetam application or in the presence of 1 μmol/L ω-conotoxin GVIA. Arrows point to EPSPs and APs. Summary of the number of APs (Cb) and EPSPs (Cc) counted during 20-s recordings in seven cells under control conditions, 10 μmol/L piracetam and 1 μmol/L ω-conotoxin. Data are presented as mean ± SEM. *P < 0.001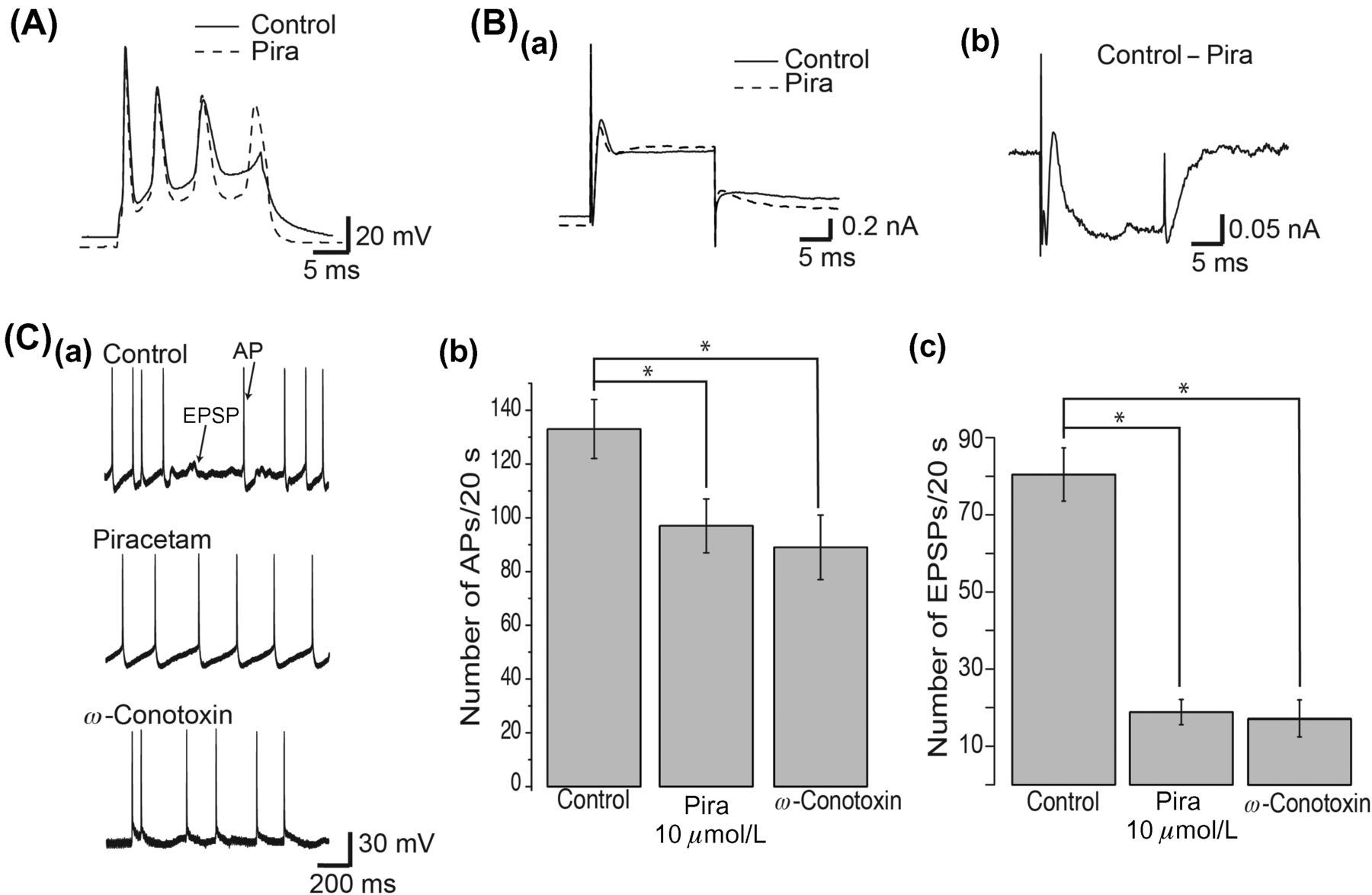
CaV2.2 currents are involved in neurotransmitter release, and therefore inhibition of CaV2.2 current by piracetam should alter the frequency of EPSPs recorded in CA1 pyramidal neurons. Accordingly, 10 μmol/L piracetam application revealed a remarkable blockade of synaptically mediated activity. Figure 5C shows representative one-second recordings of spontaneous activity, without eliciting a current stimulus, of CA1 pyramidal neurons in control conditions (n = 7), under piracetam (n = 7) and under 1 μmol/L ω-conotoxin (n = 4). Piracetam significantly decreased the frequency of APs (P < 0.05). While 133 ± 11 APs were counted during 20-s recordings under control conditions, 97 ± 10 and 89 ± 12 APs were counted under piracetam and ω-conotoxin, respectively (Figure 5Cb). Also, piracetam significantly decreased the frequency of EPSPs (P < 0.001). While 80 ± 7 EPSPs were counted during 20-s recordings under control conditions, 18 ± 3 and 18 ± 5 EPSPs were counted under piracetam and ω-conotoxin, respectively (Figure 5Cc). These results suggest that piracetam alters the firing pattern and synaptic transmission on hyppocampal pyramidal neurons. In addition, the fact that ω-conotoxin GVIA, a specific blocker of CaV2.2 channels, induced a similar effect of piracetam, reducing the frequency of APs and EPSPs in hippocampal neurons, suggests a relation between the piracetam-induced inhibition of CaV2.2 current, observed in SCG neurons, and the piracetam-induced reduction of excitability in hippocampal neurons.
Piracetam diminishes hyperexcitability
Since piracetam reduces the frequency of APs of CA1 pyramidal neurons under control conditions, likely through inhibition of calcium channels, and calcium homeostasis is relevant in hyperexcitability,
25
we investigated if piracetam is able to attenuate discharges in hyperexcited hippocampal neurons. We used the well-documented model of hyperexcitability based on low magnesium and high potassium. Low-magnesium-induced hyperexcitability in rat hippocampal slices has been extensively used to achieve pharmacological characterization of antiepileptic drugs and experimental analgesics.
18
In our experimental conditions, sustained spontaneous neuronal discharges were evident within approximately 30 min of low-magnesium and high-potassium application (n = 9). The number of APs during 20-s recordings in low magnesium–high potassium significantly increased (77.5 ± 0.9 APs/20 s), and decreased after adding 10 μmol/L piracetam to low-magnesium–high-potassium solution (37.3 ± 1.8 APs/20 s, Figure 6). Then, under the same hyperexcitability condition, piracetam significantly reduced the frequency of APs to near levels of that observed before hyperexcitability onset (33.0 ± 1.0 APs/20 s).
Piracetam diminished the frequency of action potentials (APs) in a model of hyperexcitability in hippocampal slices. (A) Representative 20-s recording of spontaneous activity from a single pyramidal neuron in control solution (a), low-magnesium, high-potassium solution (b), and low-magnesium, high-potassium solution plus 10 μmol/L piracetam (c). Control solution was artificial cerebrospinal fluid. (B) Summary of the number of APs counted during 20 s for each experimental condition in nine cells. Data are presented as mean ± SEM. *P < 0.01
Taken together, piracetam inhibits CaV2.2 current in sympathetic neurons and reduces the frequency of APs in normal and hyperexcited hippocampal slices. These effects might be causally related and could be underlying the antiepileptic effect of piracetam.
Discussion
Our experimental results demonstrate that piracetam inhibits CaV2.2 channels in peripheral neurons and shows that CaV2.2 inhibition could be related with the effects of piracetam on hippocampal neurons. We found that in SCG neurons, piracetam enhances the facilitation ratio of CaV2.2 channels. Piracetam-induced inhibition is not hampered by means of blocking G-protein signaling or occluded by activating previously a GPCR. In addition, piracetam reduced the frequency of EPSPs and APs in normal and hyperexcited CA1 pyramidal neurons.
CaV2.2 channel inhibition by piracetam in peripheral neurons
To properly study the actions of piracetam on CaV2.2 channels, we used a preparation of primary SCG neurons, in which the major component of the whole-cell calcium current is carried by CaV2.2 channels. 16,26,27 In here, the contribution of L-type Ca2+ (CaV1) channels was excluded with nifedipine application, and the contribution of P/Q-type (CaV2.1) and R-type (CaV2.3) channels, which represents a negligible portion of the whole Ca2+ current in cultured rat SCG neurons, was not considered. This preparation, therefore, allowed us to examine the effects of piracetam on a homogeneous neuronal population of CaV2.2 channels in vitro. We found that piracetam suppresses dihidropyridine-insensitive Ca2+ current by about 90%. We previously described a similar suppression response with 10 μmol/L ω-conotoxin GVIA, 14 indicating that the whole population of CaV2.2 channels in SCGs is sensitive to piracetam. HVA calcium channel inhibition by piracetam has been previously reported in neurons from snail Helix pomatia. 10 However, there are some subtle differences. Near 19% of HVA current was inhibited by 2 mmol/L piracetam after more than one minute in snail neurons while 10 μmol/L piracetam inhibited CaV2.2 current by 77% after 30-s application in SCG neurons. We cannot rule out if these discrepancies depend on differences in taxonomical groups or in technical approach.
In addition, piracetam inhibited CaV2.2 channels in primary SCG neurons in a dose-dependent manner, with an IC50 of 3.4 μmol/L, and a unitary value for the Hill coefficient, ruling out cooperative actions, which is consistent with a high selectivity and specificity for piracetam on CaV2.2 channels. In cultured hippocampal neurons, levetiracetam inhibits N-type calcium current with an IC50 of 14.7 μmol/L, 11 suggesting that both compounds act on the same concentration range and that piracetam could be even more potent than levetiracetam for inhibiting CaV2.2 channels. In fact, levetiracetam inhibits only half or less CaV2.2 channels at maximal doses, 11 while piracetam inhibited the whole population of CaV2.2 channels in our experiments. Even though levetiracetam and piracetam are compounds related in structure, they have some chemical differences that can explain variations in their effects. Piracetam has the simplest structure of all of the nootropic compounds. Levetiracetam, on the other hand, has an additional ethyl group. Accordingly, levetiracetam has been grouped into antiepileptic drugs while piracetam has been grouped into cognitive enhancers. 1 Subtle differences in chemical structures might explain their pharmacological differences and, in the same sense, might explain disparities between CaV2.2 channel inhibition induced by levetiracetam or by piracetam.
Possible mechanism of action of piracetam on CaV2.2 channels
We then sought to determine if the observed decrease in CaV2.2 current amplitude was caused by a direct action of piracetam on the channel, or if it was instead the result of indirect action involving a G-protein-mediated mechanism. CaV2.2 current traces in the presence of piracetam are kinetically different from traces in the absence of piracetam (see Figure 2Ba under letter b and Figure 3Ba under P1). Besides, piracetam induces a moderate shift of voltage-dependence and increases the facilitation ratio of CaV2.2 channels. Gβγ subunits coupled to GPCRs have similar effects. 22,23 However, when we tested G-proteins on piracetam action, we found no relation. Piracetam action is not hampered by dialysis of GDPβS or occluded by application of saturating concentration of a GPCR agonist, suggesting that piracetam acts directly on CaV2.2 channels. Previous studies have demonstrated that piracetam interferes with the pore region of some ion channels, as in the case of GABAA receptors. 28 In addition, piracetam has great affinity for polar heads of phospholipids, 29 inducing a new organization of lipids, and in such a way, piracetam increases neuronal membrane fluidity 4,30 and facilitates mobile phospholipid-complex formation. 31 Thereby, we propose that these changes in membrane properties can cause interference with the intrinsic channel gating or the binding of regulatory subunits (i.e. Ca2+ channel β-subunit and G-protein βγ-subunit). In conclusion, piracetam revealed a novel CaV2.2 inhibition, mimicking some characteristics of voltage-dependent inhibition by G-proteins likely acting directly on the channel molecule.
Piracetam effect on hippocampal CA1 pyramidal neurons
Piracetam also has effects on central neurons. In hippocampal CA1 pyramidal neurons, we observed that piracetam altered the firing patter in response to a specific current stimulus. Taking into account inhibition of CaV2.2 channels in SCG neurons, we assessed, by indirect measurements, if the piracetam effects in CA1 neurons were a consequence of inhibition of CaV2.2 channels. We recorded total currents in the somatic region and found that most likely calcium currents are altered by piracetam, since subtraction of currents recorded in the absence or presence of piracetam gave a current with a HVA phenotype. However, CaV2.2 channels are mainly located in synaptic ends, being necessary for neurotransmitter release. 32 Accordingly, we found that the frequency of EPSPs diminishes in the presence of piracetam. We compared the effect of piracetam with the effect of a specific blocker of CaV2.2 channels, the ω-conotoxin GVIA. ω-Conotoxin GVIA also diminished the frequency of APs and EPSPs as piracetam did. Together, these results suggest that the effect of piracetam on the excitability of hippocampal CA1 neurons is related with the inhibition of CaV2.2 channels. Previous results have shown that nootropic drugs, such as levetiracetam, inhibit CaV2.2 channels, 11,12 which can explain the reduction of vesicle release in synapses after a levetiracetam bath. 33 Piracetam and levetiracetam might share this mode of action to diminish hyperexcitability, yet they are commonly used for different pathologies treatment. On the other hand, the fact that piracetam reduced excitability can be explained by additional alterations on other conductances such as sodium or potassium channels. For instance, it has been previously reported that piracetam inhibits calcium-activated potassium channels in invertebrate neurons. 10 We also observed that during a current stimulus, piracetam also induced changes in resting membrane potential, after hyperpolarization potential and AP duration, suggesting that piracetam could be targeting potassium channels such as delayed rectifiers, calcium-activated potassium channels or M-current potassium channels. Further studies should address the effect of piracetam on other ion channels which will lighten the whole mechanism of piracetam as a nootropic compound.
We found that piracetam also diminishes the frequency of APs in hyperexcited CA1 pyramidal neurons. Hyperexcitation is caused when excitatory neurotransmitter release is augmented or inhibitory neurotransmitter release is diminished. 34,35 Since neurotransmitter release requires activation of voltage-gated calcium channels and CaV2.2 channels have been found in the hippocampus, 36 inhibition of CaV2.2 channels could explain the effect of piracetam on hyperexcitability. Other antiepileptic drugs also target calcium channels 37 or related subunits, such as alfa2delta auxiliary subunit. 38–40 These results together support that piracetam diminishes the number of APs in control and hyperexcited states, and suggest a likely mechanism through the inhibition of CaV2.2 channels.
Our results provide evidence of a novel inhibition of CaV2.2 channels showing an increased facilitation ratio that is not mediated by G-proteins. This finding advances our understanding in calcium channel regulation, bridging the gap with clinical applications.
Footnotes
ACKNOWLEDGEMENTS
This work was supported by UNAM-DGAPA-PAPIIT IN200710 grant, and a grant from The Alexander von Humboldt Stiftung, Germany. We thank Manuel Hernandez and Guillermo Luna for technical help, Ing. Gustavo Díaz for software support and Dr Enrique Pinzon for the excellent care of rats.