
Abstract
Ischemic myocardium exhibits inflammation, local angiotensin II (Ang II) generation and up-regulation of LOX-1, a lectin-like ox-LDL receptor. To define the inter-active roles of Ang II and inflammation in furthering tissue injury, cultured HL-1 cardiomyocytes were treated with Ang II. Ang II treatment up-regulated the expression of Ang II type 1 (AT1R) and type 2 (AT2R) receptors as well as LOX-1. Ang II also activated p44/42MAPK, p38MAPK, c-Jun and NF-kB, and increased the expression of inflammation-related genes (interleukins-6, interleukins-10, tumor necrosis factor-a, intercellular adhesion molecule-1). To study how inflammation per se might affect expression of Ang II receptors and LOX-1, cultured, cardiomyocytes were treated with lipopolysaccharide (LPS). Like Ang II, LPS increased the expression of AT1R, AT2R and LOX-1. LPS also activated mitogen-acticated protein kinase (MAPKs), c-Jun and NF-kB, and pro-inflammatory genes. The selective inhibitors of MAPKs, c-Jun and NF-kB each blocked the transcription of LOX-1 and pro-inflammatory genes in response to Ang II as well as LPS. These observations suggested a positive feedback between Ang II and inflammation. To delineate the role of AT1R and AT2R in LOX-1 expression, another set of cardiomyocytes were transfected with AT1R or AT2R cDNA. Forced over-expression of AT1R resulted in activation of MAPKs, c-Jun and NF-kB, up-regulation of inflammatory genes and LOX-1; on the other hand forced AT2R over-expression induced up-regulation of pro-apoptotic signals (pro-IL-1b and IL-1b), and decreased LOX-1 expression. These studies show that both Ang II and inflammation mediator LPS up-regulate AT1R, AT2R and LOX-1 expression. Up-regulation of AT1R promotes inflammation and LOX-1 expression, whereas up-regulation of AT2R promotes apoptosis signals and decreases LOX-1 expression.
Introduction
Angiotensin II (Ang II) is the major product of renin–angiotensin system (RAS) activation, which initiates the major pathophysiological actions of RAS and plays an important role in the pathogenesis of several cardiovascular disease states. Ang II exerts its effects by activating its receptors, primarily type 1 (AT1R) and type 2 (AT2R). Several studies suggest that Ang II is also an important pro-inflammatory mediator that induces inflammatory response in the blood vessel wall, kidney, heart and cultured vascular smooth muscle and endothelial cells.1–5
In general, AT1R activation by Ang II is thought to promote inflammatory response by enhancing STAT3 signal transduction and increasing tumor necrosis factor-α (TNF-α) production, whereas AT2R activation is believed to oppose the actions of AT1R. 6 However, there is evidence that both AT1R and AT2R are up-regulated in inflammatory disease states and may play an interactive role with the inflammatory process. For example, Okada et al. 7 observed that the expression of both AT1R and AT2R was increased in the cultured splenocytes exposed to lipopolysacchride (LPS). Ruiz-Ortega et al. 8 reported that blockade of both AT1R and AT2R was necessary to completely stop the inflammatory process in an animal model of kidney injury.
It is known that activation of nuclear factor-kappaB (NF-κB) plays a critical role in Ang-II mediated inflammation. Wolf et al. 2 reported that activation of both AT1R and AT2R stimulates NF-κB in COS7 cells, and this activation could be blocked by the AT2R blocker PD123319, but not by the AT1R blocker losartan. Esteban et al. 9 observed that Ang II activated NF-κB and enhanced inflammatory cell (monocyte/macrophage) infiltration in the kidneys of AT1R knockout mice, and this activation was blocked by PD123319. However, other in vitro and in vivo studies have suggested that AT2R activation does not affect NF-κB activation and the subsequent inflammatory response in the mice kidney tubular epithelial cells. 10 Some investigators, with the use of primary human and murine dermal fibroblasts, have even suggested that AT2R activation may inhibit NF-κB activation. 11 These observations imply that the activation of AT1R and AT2R may have variable effect depending on cell type, organ and animal species.
Work from several laboratories, including ours, has indicated the existence of a cross-talk between LOX-1, a lectin-like 52kD receptor for ox-LDL, and AT1R with downstream activation of mitogen-acticated protein kinase (MAPKs) and NF-κB as the signal bridge.12–14 It is of note that, LOX-1 is an important mediator of acute and chronic inflammatory responses. 15
However, the relationship of AT2R with LOX-1 is not known. Further, how inflammation per se affects Ang II receptors expression and activation is not known. This study was designed to study the complex interaction between inflammation and Ang II receptor expression, and their interactions with LOX-1, MAPKs and NF-κB in cultured cardiac myocytes.
Materials and methods
Cell culture and study protocol
Mouse HL-1 cardiomyocytes were seeded in T25 flasks or multiwell plates precoated with 0.02% gelatin and 5 μg/mL fibronectin, and cultured in Claycomb medium (SAFC Biosciences, Erie, PA, USA) supplemented with 10% fetal bovine serum, 2 mmol/L L-glutamine and 0.1 mmol/L nor-epinephrine at 37°C under 5% CO2. When cells reached 80% confluence, they were transfected with PCMV-SPORT6 plasmid with green fluorescent protein (GFP), AT1R or AT2R cDNAs (Invitrogen, Carlsbad, CA, USA). The ratio of cells expressing GFP was used to determine the transfection efficiency. When HL-1 cells reached 80% confluence, they were exposed to different concentrations of Ang II or LPS for 24 h. Based on preliminary data on dose-response, 100 nmol/L/L Ang II and 10 ng/mL LPS were used to study inflammation and AT1R, AT2R and LOX-1 expression and intracellular signaling (p44/42MAPK, p38MAPK, c-Jun and NF-κB). In parallel experiments, HL-1 cardiomyocytes were incubated with U0126 (10 μmol/L, p44/42MAPK activation blocker), SB203580 (5 μmol/L, p38MAPK activation blocker), SP600125 (0.1 μmol/L c-jun activation inhibitor) or JSH-23 (10 μmol/L, NF-κB activation blocker II) before treatment with Ang II or LPS.
Realtime-polymerase chain reaction
Total RNA was isolated from HL-1 cells using RNeasy Mini-Kits (Invitrogen), and cDNA was synthesized with SuperScript II 1st Strand DNA Synthesis Kit (Invitrogen) according to the manufacturer's instructions. Realtime poly-merase chain reaction (RT-PCR) was performed using a 20μL reaction volume containing 100 ng cDNA and 0.3μmol/L primers. The primer sequences are shown inTable 1.
Primers for realtime polymerase chain reaction

TNF-α, tumor necrosis factor-α VCAM-1, vascular cell adhesion molecule-1; ICAM-1, intercellular adhesion molecule-1; IL-1β, interleukins 1β;
IL-6, interleukins IL-6
Western blotting
Total protein was extracted from HL-1 cardiomyocytes. The samples were loaded and separated by sodium dodecyl sulfate polyacrylamide gel electrophoresis (SDS-PAGE) on 12% gels, and then transferred to the poly-vinylidene fluoride (PVDF) membranes (Bio-Rad, Hercules, CA, USA). The membranes were blocked with 5% non-fat milk in tris-buffered saline/0.1% tween (TBS-T), and then washed three times with TBS-T, and incubated with IL-1β, IL-6, AT1R, AT2R, phosho-p38MAPK, β-actin (ABcam, Cambridge, MA, USA), intercellular adhesion molecule-1 (ICAM-1) and vascular cell adhesion molecule-1 (VCAM-1) (Santa Cruz, Santa Cruz, CA, USA), p44/42MAPK, phospho-p44/42MAPK, phospho-c-Jun, phospho-NF-κB P65 (Cell signaling, Danvers, MA, USA) and LOX-1 (gift from Prof T Sawamura, Osaka, Japan) antibodies (1:1000) at 48C overnight. The blots were incubated horseradish peroxidase (HRP)-conjugated second antibody (1:10000) for one hour at room temperature. The immunoreactive bands were visualized by incubation with enhanced chemi-luminescence (ECL) Western-blotting substrate (Thermo Scientific, Rockford, IL, USA).
Cell adhesion assay
Monocytes (ATCC, Manassas, VA, USA) were preincubated with Cell Tracker (Invitrogen) for 30 min at 37°C. The predyed monocytes (5 × 104 cells/well) were seeded onto HL-1 cells cultured in 12-well plates. And then, they were incubated together for 30 min. Cells were washed three times gently with phosphate-buffered saline and randomly imaged with a fluorescence microscope.
Statistical analysis
Data are presented as mean ± standard deviation (SD) from at least three independent experiments, each in duplicate. Statistical analysis was performed with SPSS 11.5 software. Multiple comparisons were analyzed by one-way analysis of variance. P < 0.05 was considered statistically significant.
Results
Both Ang II and LPS induce inflammatory cytokine expression and promote monocyte adhesion
To study the direct effects of Ang II as inflammatory mediator, cultured cardiomyocytes were treated with Ang II (10-10,000 nmol/L) for 24 h. The Ang II treatment increased IL-6 and TNF-α expression in a concentration-dependent manner (Figures 1a and b). In parallel experiments, cells were treated with LPS, a potent proinflammatory stimulus (1-1000 ng/mL for 24 h). The LPS treatment had qualitatively similar effect as Ang II. It is of note that 100 nmol/L of Ang II and 10 ng/mL of LPS gave most consistent data, and these concentrations were chosen to study the effect of Ang II and LPS in subsequent studies.

Effect of Ang II and LPS on the expression of inflammatory cytokines and cell adhesion. (a) and (b) Incubation of HL-1 cardiomyocytes with Ang II (10 to 10,000 nmol/L/L for 24 h) and LPS (1 to 1000 ng/mL for 24 h) enhances the expression of IL-6 and tumor necrosis factor (TNF-α) mRNA in a concentration-dependent manner. (c) Expression of pro-inflammatory cytokines (IL-1β, IL-6 and TNF-α mRNA) in cells exposed to 100nmol/L Ang II and 10 ng/mL LPS. (d) Adhesion of monocytes to cardiomyocyte monolayer and, (e) VCAM-1 and ICAM-1 mRNA expression after exposure of cardiomyocytes to Ang II and LPS. Bar graphs represent mean ± SD (n = 3 per group).
Both Ang II and LPS increased IL-1β, IL-6 and TNF-α mRNA expression (P < 0.05) (Figure 1c). Both Ang II and LPS up-regulated the expression of ICAM-1 mRNA and enhanced monocyte adhesion to HL-1 cells (LPS >> Ang II) (Figures 1d and e). LPS treatment also increased VCAM-1 mRNA (P< 0.05) (Figure 1e).
Ang II and the expression of AT1R, AT2R and LOX-1
Next, we examined if Ang II itself would influence AT1R and AT2R expression in cultured cardiomyocytes. We observed that Ang II increased the expression of both AT1R and AT2R (both P < 0.01, AT1R.AT2R) (Figure 2). Ang II also increased LOX-1 protein expression (P < 0.01; Figure 2).
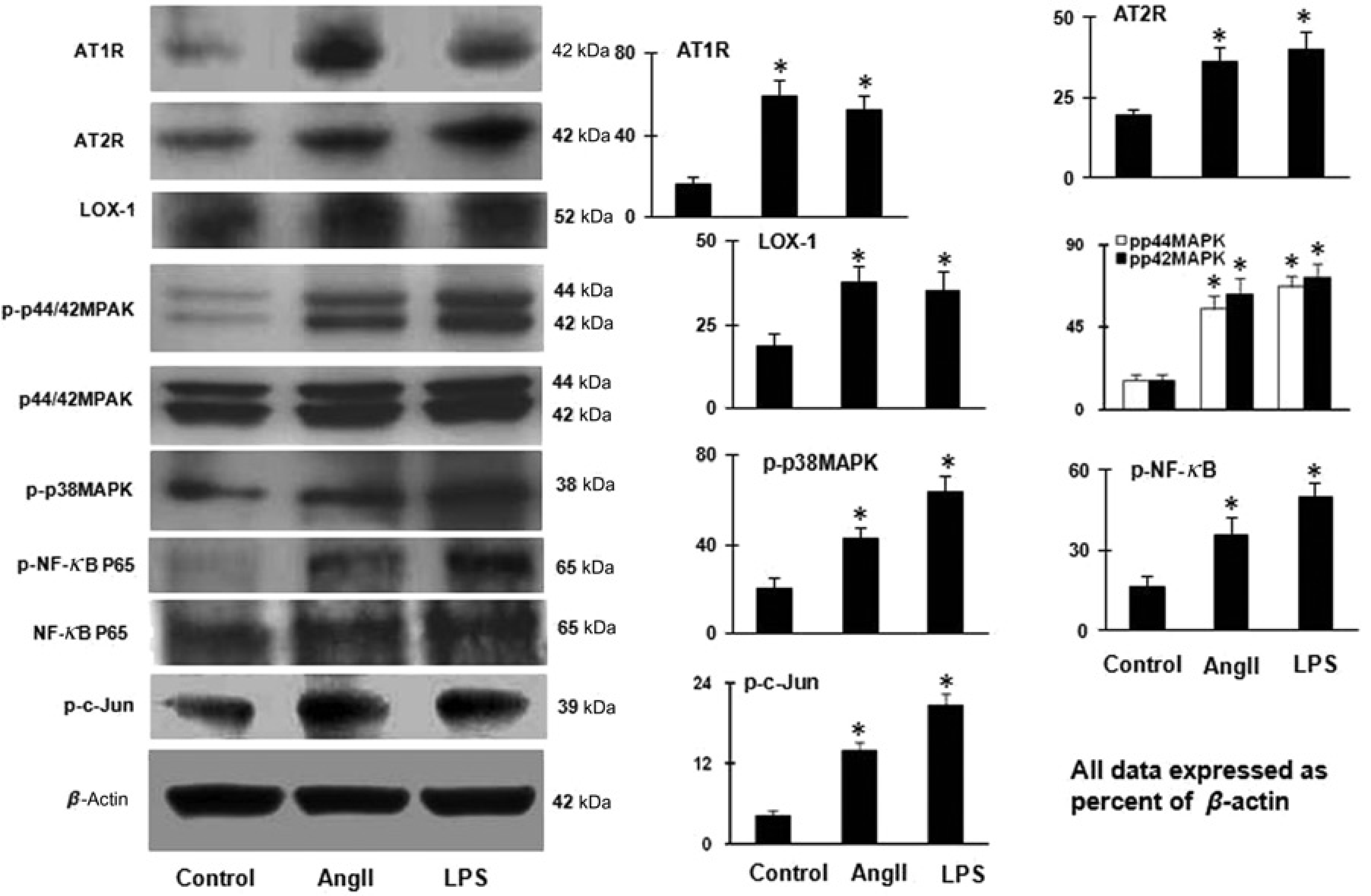
Ang II and LPS and the expression of AT1R, AT2R and LOX-1 and activation MAPKs, c-Jun and NF-κB. Ang II (100 nmol/L/L) and LPS (10 ng/mL) markedly increased AT1R, AT2R, LOX-1, phospho-p44/42MAPK, phospho-p38MAPK, phospho-NF-κB and phospho-c-Jun expressions. Bar graphs represent mean ± SD (n = 3 per group).
Further, Ang II enhanced the expression of phospho-p44/42MAPK, phospho-p38MAPK, phospho-NF-κB and phospho-c-Jun (P < 0.05; Figure 2).
LPS and the expression of AT1R, AT2R and LOX-1
Next, we examined if the inflammatory stimulus LPS would affect the expression of Ang II receptors and LOX-1. As shown in Figure 2, LPS treatment of cardiomyocytes significantly increased the expression of AT1R, AT2R and LOX-1 (P < 0.01). These effects of LPS were similar to the effects of Ang II. Further, LPS, like Ang II, enhanced phospho-p44/42MAPK, phospho-p38MAPK, phospho-NF-κB and phospho-c-Jun protein (P < 0.05; Figure 2). Further, Ang II enhanced the expression of phospho-p44/42MAPK, phospho-p38MAPK, phospho-NF-κB and phospho-c-Jun (P < 0.05; Figure 2).
AT1R and AT2R over-expression and expression of pro-inflammatory genes
In the basal state, AT1R or AT2R mRNA was undetectable in cardiomyocytes transfected with empty PCMV-SPORT6 plasmids. After transfection, however, AT1R (1078 bp) and AT2R transgenes (1092 bp) were expressed at a high level (Figure 3a).

The expression of inflammatory cytokines and adhesion molecules, and monocyte adhesion to HL-1 cardiomyocytes over-expressing AT1R and AT2R. (a) Expression of AT1R and AT2R after exogenous gene transfection. (b) Expression of pro-IL-1 β (protein), (c) Expression of inflammatory cytokines (IL-1 β, IL-6 and TNF-α) (mRNA), (d) Adhesion of monocytes to cardiomyocyte monolayer, and (e) Expression VCAM-1 and ICAM-1. Bar graphs represent mean ± SD (n = 3 per group).
AT2R over-expression induced a 3–5 fold increase in pro-IL-1β and IL-1β levels (mRNA and protein (P, 0.05)), an effect not seen in cells over-expressing AT1R (Figures 3b and c).
AT1R over-expression induced a significant increase in the expression of IL-6 and TNF-α, whereas AT2R over-expression had only a modest effect on the expression of IL-6 and no effect on the expression of TNF-α (Figure 3c). Taken together, these data suggest that AT1R over-expression has a potent effect on the transcription of inflammatory genes whereas AT2R over-expression had only a small effect.
Since inflammatory cell attachment is dependent on the expression of adhesion molecules, we examined and found that ICAM-1 expression was enhanced in cells that had been transfected with AT1R cDNA (P < 0.05) (Figure 3e). In contrast, AT2R over-expression did not affect either ICAM-1 or VCAM-1 expression (Figure 3e).
In keeping with the data on adhesion molecule expression, AT1R over-expression enhanced monocyte adhesion to cardiomyocytes (P < 0.05), whereas AT2R over-expression had no such effect (P > 0.05) (Figure 3d).
Signaling in the effects of Ang II and LPS in cardiomyocytes
We observed that AT1R over-expression up-regulated the transcription of LOX-1 which translated into enhanced protein (P < 0.05) (Figure 4). AT2R over-expression, on the other hand, slightly, but significantly, resulted in a decrease in LOX-1 protein (P < 0.05) with a small decrease in LOX-1 mRNA (Figure 5). Western blotting showed that AT1R over-expression enhanced phospho-p44/42MAPK, phospho-p38MAPK, phospho-NF-κB and phospho-c-Jun expression (P < 0.05); whereas, AT2R over-expression had no significant effect on phospho-p44/42MAPK, phospho-p38MAPK, phospho-NF-κB and phospho-c-Jun expression (Figure 4).
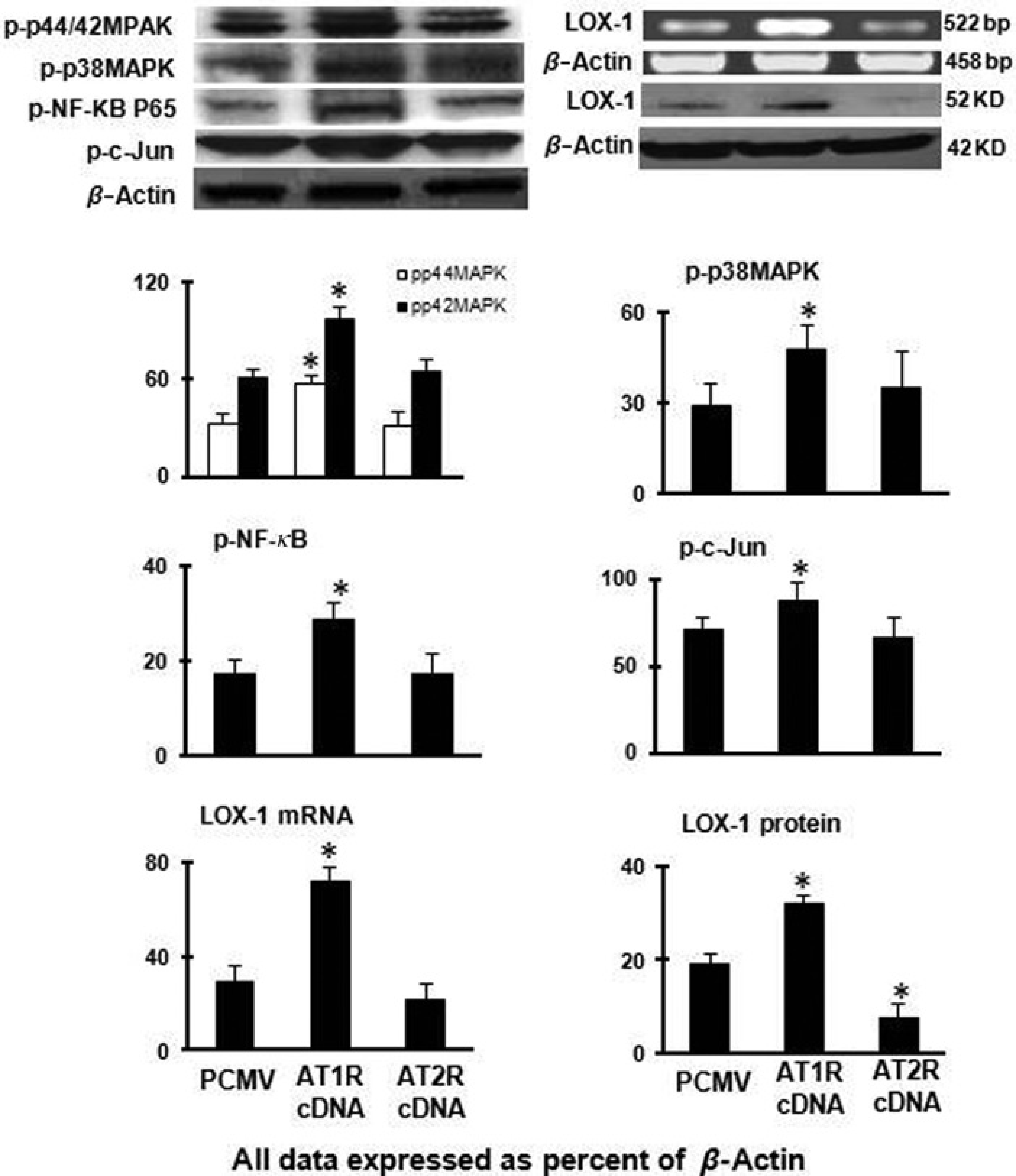
Expression of LOX-1, phospho-p44/42MAPK, phospho-p38MAPK, phospho-NF-κB and phospho-c-Jun in cells over-expressing AT1R and AT2R. Bar graphs represent mean ± SD (n = 3 per group).

(a) Treatment of in cultured cardiomyocytes over-expressing AT1R and AT2R with p44/42MAPK activation inhibitor (U0126), p38MAPK activation inhibitor (SB203580), c-Jun activation inhibitor (SP600125) and NF-κB activation blocker II (JSH-23). (b) Treatment of cultured cardiomyocytes with U0126, SB203580, SP600125 and JSH-23 followed by incubation with Ang II or LPS. Bar graphs represent mean ± SD (n = 3 per group). *P < 0.05 versus PCMV or control; #P < 0.05 versus AT1R; §P
Next, we studied the role of phospho-p44/42MAPK, phospho-p38MAPK, phospho-NF-κB and phospho-c-Jun in the up-regulation of IL-6 and TNF-κ in AT1R over-expressing cells with the use of inhibitors of each of these molecules. These inhibitors blocked the up-regulation of IL-6 and TNF-α (Figure 5a).
Next, we studied the role of p44/42MAPK, p38MAPK, NF-κB and c-Jun in the up-regulation of AT1R, AT2R and LOX-1 by Ang II. As shown in Figure 5b, Ang II-mediated up-regulation of AT1R, AT2R and LOX-1 could be inhibited by p44/42MAPK inhibitor U0126, p38MAPK inhibitor SB203580, c-Jun inhibitor SP600125, and the NF-κB inhibitor JSH-23, which suggests that the up-regulation of AT1R, AT2R and LOX-1 is, in large part, mediated by activation of MAPK/NF-κB pathway.
Interestingly, LPS-mediated up-regulation of AT1R, AT2R and LOX-1 was also inhibited by the chemical blockers of p44/42MAPK, p38MAPK, c-Jun inhibitor and NF-κB inhibitor (Figure 5b). These observations suggest that the up-regulation of AT1R, AT2R and LOX-1 by LPS shares the same signaling pathways as Ang II.
Discussion
This study shows an interplay between inflammation, Ang II receptors and LOX-1 in cultured HL-1 cardiomyocytes. We conducted these studies in cultured HL-1 cardiomyocytes, a cell line derived from atrial cardiomyocyte tumor lineage derived from AT-1 mouse, which maintains the differentiated adult cardiac phenotype and indefinite proliferation ability in vitro. 16 A number of investigators, including us, have used this cell line to study the dynamics of Ang II receptor expression and functional response to a variety of stimuli, and find this cell line as a useful model system for investigating cardiomyocyte biology.17–19
The presence of inflammation in the ischemic myocardium and increase in pro-inflammatory cytokines in the systemic circulation have been known for long time, but it is only in the recent past that inflammation in the ischemic tissues has been shown to cause expansion and extension of the area of infarct.20,21 Strategies that limit myocardial ischemic injury are almost always associated with a reduction in inflammatory response. 22 Further, a reduction in inflammatory cell number or activity results in a limitation of infarct size.20–22
Cardiac tissues have all components of RAS which becomes activated during perturbation caused by ischemia resulting in local release of Ang II. 23 Our previous studies in cultured cardiomyocytes show that Ang II is capable of inducing the expression of its own receptors AT1R and AT2R. 19 Studies in cultured endothelial cells, vascular smooth muscle cells and cardiomyocytes have shown that Ang II via AT1R up-regulates LOX-1 at gene level,24–26 and ox-LDL via LOX-1 up-regulates AT1R mRNA. 27 A novel finding in the present studies was that LOX-1 expression was diminished in cells with AT2R over-expression. Thus, it appears that AT1R and AT2R have opposing and regulatory effects on the expression of LOX-1.
Several studies implicate LOX-1 as a pro-inflammatory signal.28–30 The LOX-1 knockout mice have only limited inflammatory response to injury 31 as do animals treated with LOX-1 binding antibody. 29 The previous observation of only minimal amount of inflammation and a very small increase in AT1R expression in the ischemic hearts of LOX-1 knockout mice 31 suggests that besides being a direct pro-inflammatory molecule, LOX-1 may be an indirect modulator of inflammation via AT1R expression.
LOX-1 contributes to the ischemic injury, at least in part via the up-regulation of AT1R, 31 a well-known mediator of inflammation. However, the studies by Lu et al. 31 did not delineate the role of AT2R during inflammation. Further, results of experiments with LPS in the present study suggest that inflammation per se induces the expression of AT1R and AT2R as well as LOX-1. This study also sheds light on the role of AT2R during ischemia.
Studies in AT1R cDNA transfected HL-1 cardiomyocytes showed that these cells are capable of stimulating the synthesis of pro-inflammatory cytokines, such as IL-6 and TNF-α, and adhesion molecule ICAM-1, which result in adhesion of monocytes to cardiomyocytes. In contrast to the effect of AT1R over-expression, the over-expression of AT2R in cardiomyocytes was associated with a significant increase in pro-IL-1β and IL-1β and a small increase in IL-6, but total lack of any increase in ICAM-1 or monocyte adhesion.
The precise contribution of pro-IL-1β and IL-1β in response to AT2R over-expression to the inflammatory process is unclear, but some reports suggest that IL-1β has an important role in the regulation of apoptosis. 32 Ing et al. 33 showed that cytokine-mediated induction of apoptosis in neonatal rat cardiac myocytes could be attributed entirely to IL-1β. In addition, these authors showed that the pro-apoptotic effect of IL-1β could be blocked by the inhibitors of NOS activity. 33 Siragy 34 have also suggested that AT2R is involved in the development of apoptosis via release of NO. We propose that the up-regulation of AT2R in response to Ang II and LPS participates in the development of apoptosis in the ischemic tissues related, at least in part, to the release of pro-IL-1β and IL-1β.
These observations taken together imply that the expression/activation of AT1R in response to Ang II and inflammatory milieu may serve to enhance the inflammatory response, whereas AT2R expression/activation may serve to initiate apoptosis and reduce LOX-1 transcription.
We studied intracellular signaling in the effects of Ang II, and observed an important role of MAPK/NF-κB pathway in the transcription of AT1R and AT2R as well as LOX-1. The MAPK/NF-κB pathway was also activated in cells treated with LPS resulting in the transcription of AT1R and AT2R as well as LOX-1. The central role of this pathway became evident in experiments with specific inhibitors of MAPKs, c-Jun and NF-κB that blocked the transcription of AT1R and AT2R as well as LOX-1 in response to both Ang II and LPS. MAPKs, c-Jun and NF-κB are activated during the redox state. It is well known that the ischemic myocardium generates significant amount of ROS and induces peroxidation of lipids in the cell membranes. 31
It is noteworthy that in cells with forced over-expression of AT2R, there was no activation of MAPK/NF-κB pathway, no change in ICAM-1 and VCAM-1 expression and no monocyte adhesion; however, there was a significant decrease in LOX-1 transcription. Down-regulation of LOX-1 and absence of monocyte adhesion with AT2R up-regulation may be inter-twined. Even though there was a small increase in IL-6 and TNF-α expression in AT2R over-expressing cells, this increase was not associated with activation of MAPK/NF-κB signaling. These data can be interpreted to suggest that Ang II and the inflammation mediators like LPS activate MAPK/NF-κB pathway to induce AT1R and LOX-1 transcription in a positive feedback manner. On the other hand, Ang II- and LPS-induced AT2R transcription may serve to decrease the inflammatory response via inhibition of LOX-1.
In summary, we show that Ang II and pro-inflammatory stimuli up-regulate AT1R and LOX-1 in a positive feedback manner leading to a self-perpetuating inflammatory milieu in the injured tissues. While AT2R is also up-regulated in response to Ang II and LPS, it mainly serves to activate apoptosis-related signals, and to inhibit LOX-1 transcription.
Footnotes
Acknowledgements
This study was supported in part by funds from the Department of Veterans Affairs, Veterans Health Administration, Office of Research and Development, Biomedical Laboratory Research and Development, Washington, DC.