
Abstract
There is increasing concern that HIV treatment failure may result from inadequate central nervous system (CNS) penetration of antiretroviral drugs, allowing compartmentalized viral replication and development of resistance. We discuss a patient who maintained a suppressed plasma viral load for four years on antiretroviral therapy (ART) before developing HIV encephalitis with a cerebrospinal fluid (CSF) HIV viral load of 861 copies/mL and newly detectable plasma viral load of 68 copies/mL. Identification of major resistance mutations to his combination therapy supported concerns that resistant HIV had developed within the CNS. His ART was changed to optimize CNS penetration, leading to maintained clinical improvement. Imaging presented demonstrates corresponding radiological improvement. The report illustrates the need to exclude CNS viral rebound or incomplete suppression in HIV patients with neurological symptoms, and suggests that the extent of this emerging problem is only beginning to be recognized as the implications of long-term peripheral HIV suppression unfold.
INTRODUCTION
Poor central penetration of antiretroviral therapy (ART) may allow compartmentalized HIV replication within the central nervous system (CNS) despite suppressed plasma HIV copy number. We present a case of primary HIV encephalitis due to uncontrolled CNS HIV replication, to illustrate the risk of CNS viral rebound or incomplete suppression, and the potential for treatment through tailored ART to maximize CNS penetration.
CASE
A 39-year-old man was diagnosed with HIV following presentation with left-sided weakness due to cerebral toxoplasmosis. After successful toxoplasmosis treatment he commenced ART of tenofovir, emtricitabine and lopinavir–ritonavir. His viral load subsequently fell to <50 copies/mL with a sustained increase in CD4 count from a nadir of 82 cells/μL to around 200 cells/μL. Four years later, left-sided weakness progressively recurred, associated with vertigo, nausea, headache and fevers. Examination confirmed left-sided pyramidal weakness with ataxia but found no sensory deficit or cognitive impairment. His CD4 count was stable at 199 cells/mm3 (10%). His plasma HIV viral load was detectable for the first time (68 copies/mL). Other routine laboratory tests were within normal limits. He had fully adhered to ART and prophylactic co-trimoxazole, as demonstrated by consistent viral load suppression, in addition to pharmacy records indicating appropriate resupplying of medication, and standard clinical care questioning at clinic visits.
Magnetic resonance imaging (MRI) of the brain demonstrated extensive bilateral signal alteration, involving the right parietal cortex and extending into the brainstem and cerebellum (Figure 1a). These changes were not present in earlier imaging on completion of his toxoplasma treatment. Unchanged right motor cortex lesions remained from this time. Lumbar puncture had a normal opening pressure (<20 cmH2O) and confirmed a lymphocytic meningoencephalitis with white cell count 58/mm3 (100% lymphocytes), protein 1.03 g/L and glucose 3.3 mmol/L (serum glucose 6.8 mmol/L, ratio to plasma 48%).
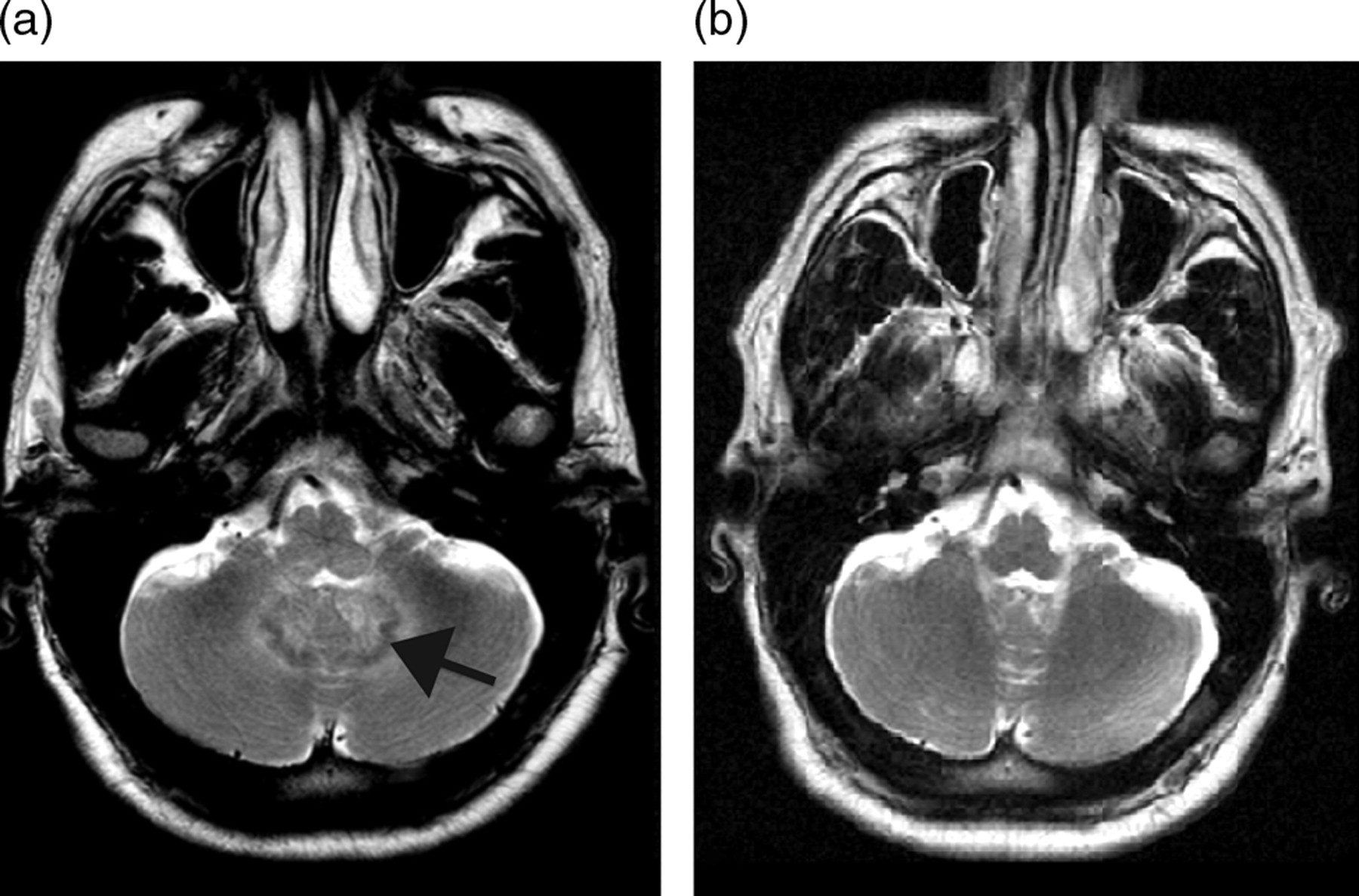
Axial T2-weighted MRI at the level of the upper medulla. (a) Baseline scan demonstrates confluent high signal delineating inflammatory change about the dentate nuclei (arrow). Similar extensive T2 high signal was also present throughout the brainstem, capsular and subcortical white matter (not shown). (b) Six months after appropriate ART treatment, MRI demonstrates resolution of signal change around the dentate nuclei. Similar improvement was seen throughout the brainstem and supratentorial white matter (not shown). MRI = magnetic resonance imaging; ART = antiretroviral therapy
Bacterial, fungal and mycobacterial cultures were negative, as was cryptococcal antigen. Polymerase chain reaction was negative for neurotropic viruses including JC virus. Cerebrospinal fluid (CSF) flow cytometry was normal with no evidence of lymphoma. In fact, the only positive finding was of a CSF HIV viral load of 861 copies/mL. Subsequently, major resistance mutations M184V, I54V and V82A were identified in both plasma and CSF, confirming resistance to emtricitabine and lopinavir.
Plasma and CSF HIV were measured with the same HIV quantification assay (Roche COBAS Ampliprep/COBAS TaqMan HIV-1 version 1 assay, designated TaqMan). Viral tropism was measured with an in-house genotypic test, using an appropriate archived blood sample, which revealed CCR5-tropic virus. Since standard tropism tests require a viral load exceeding 1000 copies/mL for an interpretable result, the most recent samples were not tested.
His ART was tailored to try to maximize CNS penetration by prescribing zidovudine, abacavir and maraviroc. Over the following 12 months of follow-up he sustained a marked improvement. Of note, repeat MRI at six month demonstrated a marked reduction in inflammatory changes (Figure 1b).
DISCUSSION
HIV patients taking ART can now expect to live with peripherally suppressed viral loads for many years. However, under-treatment of compartmentalized CNS HIV replication risks neurological injury and neglects a potential breeding ground for resistance, highlighted by a recent case series of patients with neurological symptoms and active CSF HIV replication, despite suppressed viraemia. 1 Another cross-sectional study of 69 neurologically asymptomatic HIV-positive patients found CSF HIV (>50 copies/mL) in 10% of those with undetectable viraemia, suggesting that this phenomenon may be more common than previously thought. 2 Conventionally, resistance is held to develop in the plasma, but poor CNS drug penetration allows an opportunity for low-level viral replication.
HIV infection damages the CNS through several related pathways. Peripheral immunosuppression permits opportunistic infections and malignancy. Synergistic activity between HIV and the JC virus may cause demyelination in progressive multifocal leucoencephalopathy. 3 HIV is directly virulent to neurons via primary inflammatory processes involving degeneration of synaptic, dendritic and neuronal pathways. Chemokines can exacerbate the impact of viral proteins such as gp120. 4 Once damaged, neurocognitive function is often incompletely regained despite the initiation of ART.
Our patient's broad clinical differential diagnosis included tuberculosis, toxoplasmosis, CNS lymphoma or an opportunistic viral encephalitis; however, neither imaging nor extensive laboratory studies supported these diagnoses. The CSF HIV viral load of 861 copies/mL, much exceeding the plasma value of 68 copies/mL, suggested HIV encephalitis, raising the possibility of viral resistance.
Divergence between plasma and CSF HIV genomic populations is documented; 5 thus, CSF may incubate resistant strains not apparent in plasma. Non-human primate evidence suggests poorly penetrating ART leads to ongoing CNS inflammation and persistence of viral DNA. 6 In our patient identical resistance mutations were identified in plasma and CSF, suggesting that HIV incubated within the CSF had re-seeded the previously suppressed plasma compartment, risking systemic treatment failure. This is supported by the clinical setting of a neurological prodrome preceding the occurrence of detectable plasma virus.
We acknowledge that the origin of the mutant virus cannot be definitively demonstrated retrospectively; indeed, arguably mutant virus developing in the plasma might have amplified more rapidly within the CSF; however, the patient's lengthy neurological symptoms, with sustained suppressed peripheral HIV viral load, suggest a central origin. Moreover, development of resistant virus in the plasma with disproportionate amplification within the CSF suggests the same underlying concern: poor central penetration of ART permitting viral amplification within the CSF.
Initial human studies suggest potential benefit from regimens tailored to maximize CSF penetration. 7 Furthermore, classification of antiretroviral drugs by CNS efficacy is beginning to permit choice between evidence-based regimens to optimize CNS treatment. For example, measurements of CSF drug concentrations rank zidovudine and abacavir as antiretroviral drugs more likely to be successful in tackling CNS disease; 7 likewise, initial studies suggest that maraviroc delivers therapeutic CSF drug concentrations. 8,9 Results of currently recruiting prospective comparisons between different ART regimens in neurocognitive HIV disease are awaited. 10
In summary, under-treating HIV by using poorly penetrating HIV medicines may compromise long-term neurological outcomes and risks development of resistant HIV strains. Thus, CNS viral rebound or incomplete suppression should be excluded and treated in all HIV patients with neurological symptoms. In addition, better knowledge of ART CSF penetration is needed by physicians to meet this new challenge in the management of the increasingly large number of individuals sustaining long-term peripheral HIV suppression.