
Abstract
Open vascular repair of ischemic myocardium and aortic aneurysms results in a systemic inflammatory response that influences the mortality and morbidity of these procedures. Recent studies in animal models of complex vascular reconstruction indicate that the activity of poly(ADP-ribose) polymerase (PARP) may influence the mortality and morbidity of these kinds of reconstructions. PARP's activity, localized to nuclei and mitochondria, is stimulated by deoxyribonucleic acid (DNA) strand breaks. Activation of PARP results in synthesis of poly(ADP-ribose) sugar moieties, whose primary role is to protect DNA from degradation during cytotoxic stress. Paradoxically, when stressful conditions similar to those experienced during vascular reconstructions result in overactivation of PARP, depletion of cellular levels of adenosine triphosphate and nicotinamide adenine dinucleotide can result in exacerbation of tissue injury. Herein we review the role of PARP in inflammation and its relevance to cardiovascular reconstructions.
Keywords
Although endovascular 1–3 and minimally invasive techniques 4,5 now provide the mainstay of cardiovascular reconstructions, open surgical techniques remain necessary to treat extremely complex conditions. In these instances, some degree of acute ischemia-reperfusion injury is superimposed on the patient's general medical condition. This acute ischemia-reperfusion injury is associated with the development of a severe systemic inflammatory response during and after the vascular reconstructions. 6–9 These inflammatory responses are believed to influence outcomes following cardiovascular repair. 8,10,11 Recent work using animal models of myocardial and aortic ischemia-reperfusion has indicated that the activity of poly(ADP-ribose) polymerase (PARP) may be up-regulated in response to ischemia-reperfusion. The purpose of this article is to review the biology of PARP and discuss its relevance to vascular reconstructions.
Basic Facts
PARP is present in eukaryotes, where its primary structure is highly conserved. The catalytic domain of this enzyme contains the so-called PARP signature sequence, a 50–amino acid block showing 100% homology between vertebrate species. PARP functions as a deoxyribonucleic acid (DNA) damage sensor and signaling molecule, binding to both single- and double-stranded DNA breaks. After binding to damaged DNA, PARP catalyzes the cleavage of oxidized nicotinamide adenine dinucleotide (NAD+) into nicotinamide and adenosine diphosphate (ADP) ribose (Figure 1). It uses the latter to synthesize branched nucleic acid–like polymers covalently attached to nuclear acceptor proteins. These PARPs act to stabilize DNA and modulate the activity of nuclear acceptor proteins. In vivo, PARP-1 is the most abundant poly(ADP ribosylated) protein, and auto-poly(ADP ribosylation) represents a major mechanism to provide negative feedback, thereby decreasing PARP activity. Histones are another major acceptor protein of poly(ADP ribose). Poly(ADP ribosylation) confers negative charge to histones, leading to electrostatic repulsion between DNA and histones. This process has been implicated in chromatin remodeling, DNA repair, and transcriptional regulation. Several transcription factors, 12 signaling molecules, 13 and DNA replication factors 14 have been shown to become poly-ADP ribosylated by PARP.
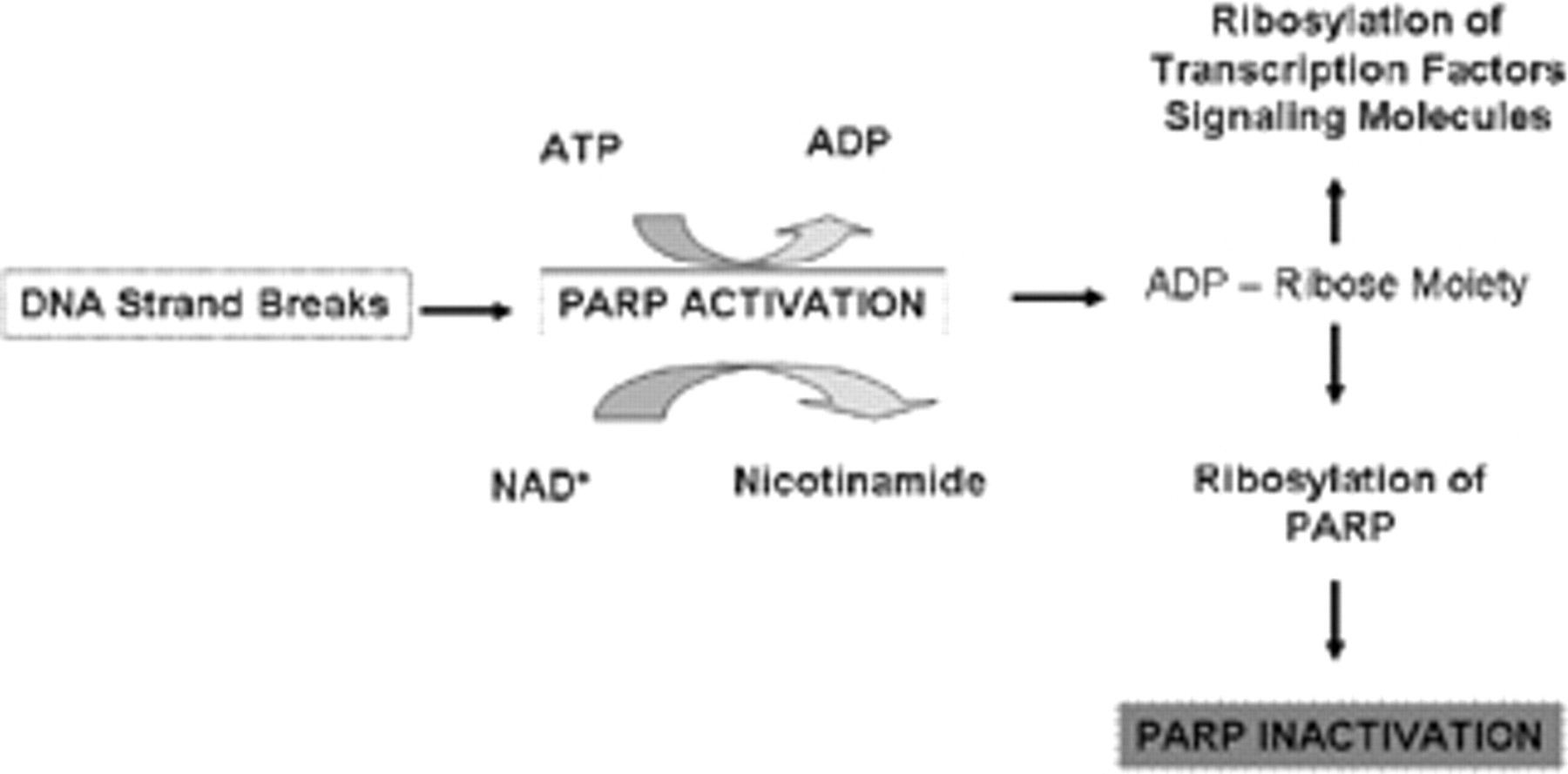
Poly(ADP-ribose) polymerase (PARP) activation leads to depletion of intracellular adenosine triphosphate (ATP) and nicotinamide adenine dinucleotide (NAD) to create adenosine disphosphate (ADP)-ribose polymers, which inactivate PARP and modulate the activity of transcription factors and signaling molecules. DNA = deoxyribonucleic acid.
Pathophysiology of PARP Activation
It does indeed seem paradoxical that a molecule designed to preserve genomic integrity has cytotoxic potential. This paradox is clearly related to the degree of PARP activation (Figure 2). In the setting of severe prolonged PARP activation, there is a substantial depletion of NAD+ and adenosine triphosphate (ATP), leading to cell death and necrosis. Under moderate levels of PARP activation, cellular levels of ATP and NAD+ are relatively well preserved, thereby allowing activation of transcription factors and caspases, which cleaves and inactivates PARP. Because of this fine control and modulation of transcription, several studies of PARP activation or inhibition have been undertaken in models of systemic stress, inflammation, and reperfusion. These studies have been facilitated largely by development of pharmacologic inhibitors of PARP.
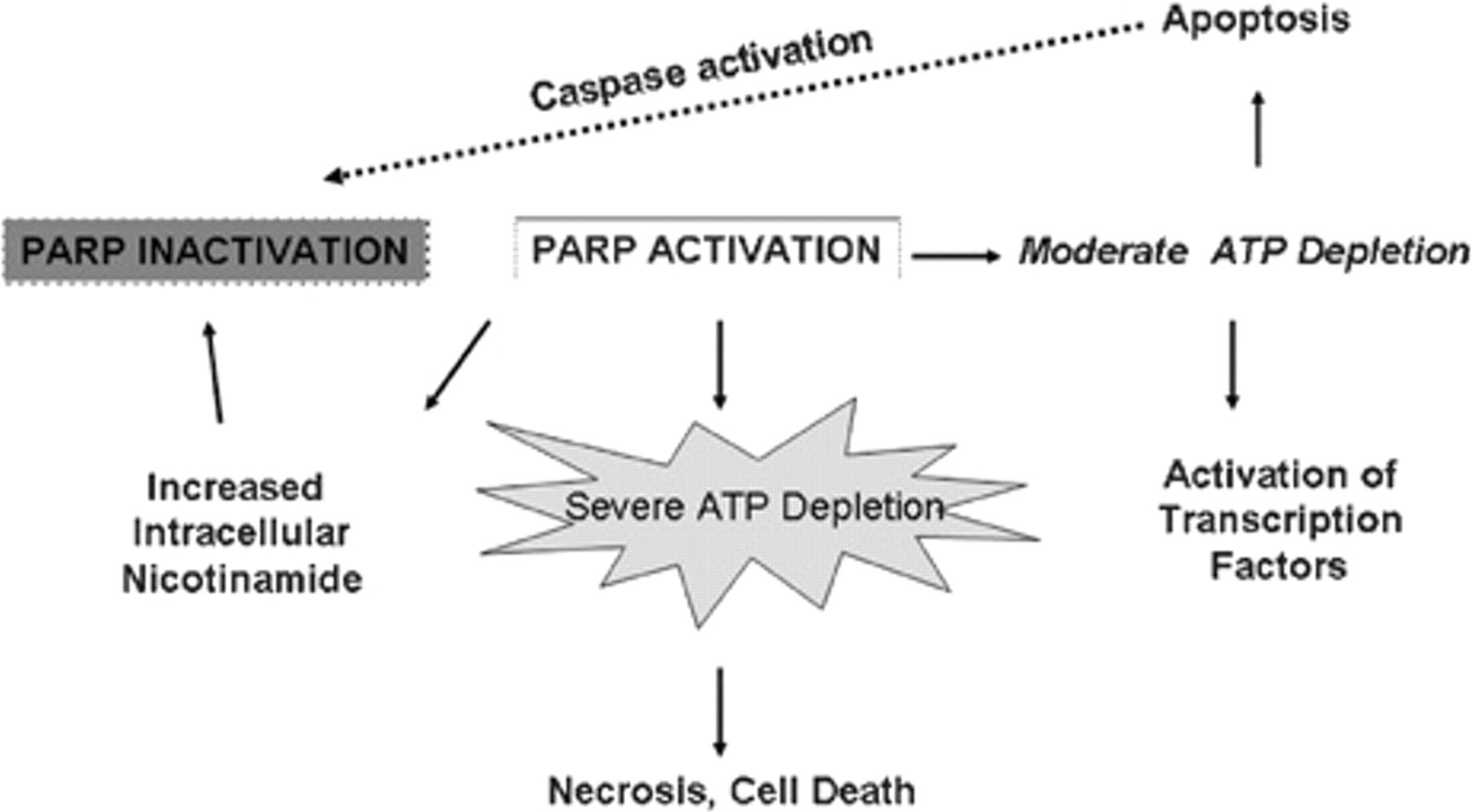
Consequences of poly(ADP-ribose) polymerase (PARP) activation. Depending on the degree of PARP activation, tissue necrosis or apoptosis may occur. Coincident with activation of apoptotic pathways, PARP itself is inactivated by the proteolytic action of caspases 3 and 7. ATP = adenosine triphosphate.
PARP Inhibitors
Given that nicotinamide is an endogenous inhibitor of PARP, its structure has served as a prototype for many inhibitors, 15 such as 3-aminobenzamide. 16 These substances inhibit PARP with low potency and minimal specificity and have limited cellular uptake. Most PARP inhibitor compounds fall into categories of monoaryl amides and bi-, tri-, or tetracylic lactams. Most PARP inhibitors block NAD+ binding to the catalytic domain of the enzyme. The catalytic domain is highly preserved among PARP isoforms; thus, it is likely that potent competitive inhibitors of PARP will inhibit the catalytic activity of all PARP isoforms. PJ34 is a tricyclic structure that has marked in vivo activity and is bioavailable when orally administered. 17 In addition to competitive inhibitors, calcium chelators have been shown to protect against oxidant-mediated PARP activation because PARP activation is dependent on increased levels of intracellular calcium. 18
PARP, Inflammation, and Shock
Recent studies have clearly demonstrated a role for PARP activation in local inflammation induced by typical stimuli such as carrageen-induced paw edema (model of arthritis) 19 and pleurisy (carrageen-induced pleural effusion). 20 Activation of the PARP pathway has been demonstrated in the pathophysiology of endotoxic shock by demonstrating increased DNA strand breakage, decreased intracellular NAD+, and ATP levels in mitochondria from peritoneal macrophages in rats subjected to endotoxic shock. 21 In a murine model of hemorrhagic shock, PARP activation seems to play a major role. PARP suppression was not associated with modulation of the nitric oxide or signaling via interleukin-6, which were found to be increased in wild-type and genetically deleted PARP-1−/−mice. 22
PARP and Vascular Reconstructions
In a canine model of cardiopulmonary bypass, PARP inhibition preserved myocardial and pulmonary function after 60 minutes of hypothermic arrest. 23 This experimental finding has considerable relevance to vascular reconstructions since cardiopulmonary bypass remains a useful tool in managing a selected group of complex thoracoabdominal aneurysms. 24 PARP inhibition is also effective in ameliorating regional myocardial dysfunction and decreasing infarct size in a porcine model of coronary occlusion and reperfusion. 25 The effect of PARP inhibition on spinal cord dysfunction has been evaluated in a clinically relevant murine model of spinal cord injury. 26 Administration of PJ34 prior to ischemia and immediately after reperfusion completely reversed the dense neurologic deficit observed in untreated mice. 27 These studies provided histologic evidence of PARP activation in the anterior spinal column of untreated mice and absent activation in PJ34-treated animals. Organ-specific analysis using this model demonstrated complete amelioration of renal injury following thoracic aortic ischemia-reperfusion in PJ34-treated mice. 28 A global interrogation of lung, hepatic, and renal tissue in addition to operative mortality revealed a marked improvement in murine survival and diminished systemic inflammation following ischemia-reperfusion in PJ34-treated mice subjected to thoracic aortic ischemia-reperfusion. 29
Conclusions
A major concern regarding the use of PARP inhibitors in clinical scenarios relates to the general role of PARP in DNA repair. It is possible that by blocking PARP activation, DNA repair may be compromised, thereby sensitizing cells to malignant transformation. Although PARP-deficient mice have been found to be more sensitive than wild-type mice to ionizing radiation or nonfunctional alkylating agents, 30,31 there is no evidence to suggest a spontaneous increase in malignancy. Furthermore, therapeutic PARP inhibition associated with vascular reconstructions would only be transient, not permanent, as found in PARP−/− mice. Thus, therapeutic PARP inhibition for vascular reconstructions and inflammatory disease processes in humans holds great promise.
Footnotes
Support was received from the Department of Surgery, Division of Vascular and Endovascular Surgery, Massachusetts General Hospital, the Pacific Vascular Research Foundation, and the American Diabetes Association