
Abstract
Abdominal aortic aneurysms (AAAs) comprise the tenth leading cause of death in Caucasian males 65 to 74 years of age and accounted for nearly 16,000 deaths overall in 2000. Therefore, understanding the pathophysiology of AAAs is an important undertaking. Clinically, multiple risk factors are associated with the development of AAAs, including increasing age, positive smoking history, and hypertension. Male gender is also a well-established risk factor for the development of an AAA, with a 4:1 male to female ratio. The reason for this gender disparity is unknown. The pathogenesis of AAAs formation is complex and multifactorial. Histologically, AAAs are characterized by early chemokine-driven leukocyte infiltration into the aortic wall. Subsequent destruction of elastin and collagen in the media and adventitia ensues owing to excessive local production of matrix-degrading enzymes and is accompanied by smooth muscle cell loss and thinning of the aortic wall. At present, no medical therapies are available to treat patients with aortic aneurysms, using only the crude measurement of aortic diameter as a threshold for which patients must undergo life-threatening and costly surgery. Defining the early mechanisms underlying gender-related differences in AAA formation is critical as understanding differences in disease patterns based on gender may allow us to develop new translational approaches to the prevention and treatment of patients with aortic aneurysms.
The pathogenesis of AAAs involves a complex series of events that have not been fully elucidated yet as most studies of AAAs primarily involve cohort studies from the end stage of the disease process. Characteristics of aneurysm development include degradation of elastin and collagen in the media and adventitia, release of cytokines and chemokines, and an oxidative burst resulting from a catalytic process that occurs following infiltration of inflammatory cells into the aortic wall.9–13
Clinical Impact of Gender on AAA
Clinical risk factors for AAA, including advanced age, family history, smoking history, atherosclerosis, hypertension, and chronic obstructive pulmonary disease, are equally distributed between males and females.5,14,15 In contrast, females seem to be protected from developing AAAs. Lederle and colleagues documented that female gender was considered a negative risk factor for developing a small AAA (odds ratio [OR] 0.47, 95% confidence interval [CI] 0.32–0.69 for AD 3.0 to 3.9 cm; OR 0.18, CI 0.07–0.48 for AD > 4.0 cm). 5 In contrast, the prevalence of AAAs among males greater than 50 years old is four to five times greater than that among females of the same age.15,16 As men age, the prevalence of AAAs increases, reaching a peak prevalence of 5.9% at 80 to 85 years and then decreasing afterward. In women, the prevalence increases to 4.5% after age 90 years. 17 After analyzing population-based screening studies, Bengtsson and colleagues demonstrated that AAAs develop 10 to 15 years later in women. 17 Speculation as to why this occurs may be due to decreased circulating levels of estrogen in postmenopausal women, as has been suggested by investigational studies in rodents. 18
Observations such as these have diminished the enthusiasm for screening women for AAAs, even those with a significant number of risk factors for developing an AAA. Currently, the US Preventive Task Force provides a grade D recommendation for screening for AAA among women owing to the low prevalence of the disease. 19 However, this recommendation conflicts with that of the Society for Vascular Surgery and the Society for Vascular Medicine and Biology, which recommend screening for women aged 60 to 85 years with cardiovascular risk factors or women more than 50 years old with a family history of AAA. 20 Derubertis and colleagues recently documented that women of advanced age (≥ 65 years) with a history of smoking or heart disease should be considered for AAA screening. 21
Regarding different thresholds for AAA repair among gender, the 2005 American College of Cardiology/American Heart Association (ACC/AHA) practice guidelines recommend an AAA diameter of 5.5 cm as a lower limit, with no mention of variation between sexes.
1
However, numerous studies have documented that females consistently suffer significantly higher mortality rates compared with males following elective abdominal aortic aneurysm (eAAA) repair (Figure 1), ruptured abdominal aortic aneurysm (rAAA) repair, and EVAR.4,16,22–24 Methods suggested to decrease mortality rates in females include referral to high-volume hospitals
23
or decreasing the diameter threshold for repair.3,25 According to Dimick and colleagues, there were significant decreases in mortality rates for women undergoing AAA repair when they were admitted to a high-volume hospital versus a low-volume hospital (Table 1).
23
In addition, numerous retrospective studies have suggested an almost paternalistic approach by caregivers for women with AAAs as females were less likely than males to undergo eAAA repair, rAAA, or EVAR.4,16,22 Anatomic contraindications, such as small iliac arteries or a small aortic bifurcation diameter, and increased intraoperative morbidity, such as iliac artery occlusions, dissections, and ruptures, are factors that occur more frequently in females and may preclude women from undergoing EVAR, with its attendant lower short-term mortality.
26
Comparison of in-hospital mortality rates for elective open abdominal aortic aneurysm repair documenting increased mortality in females compared with males. *p < .05.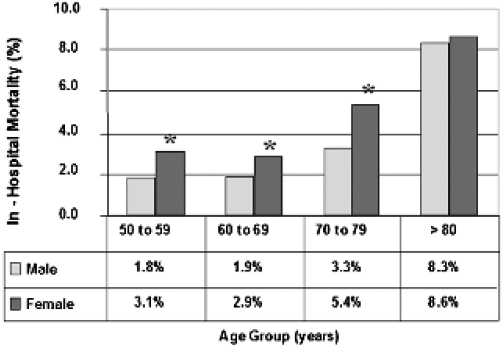
Unadjusted In-Hospital Mortality Rates for Patients Undergoing Repair of Intact and Ruptured Abdominal Aortic Aneurysm in the United States, 1996 to 1997

Adapted from Dimick JB et al. 23
p < .05 of chi-square test.
A lower threshold for eAAA repair in women with an AD of 4.5 to 5.0 cm, which is in contradiction to the 2005 ACC/AHA practice guidelines, has been recommended by the American Association for Vascular Surgery and the Society for Vascular Surgery. 3 Evidence to support this recommendation was primarily based on a study by Brown and colleagues, which reviewed the risks of small aneurysm rupture in patients undergoing ultrasound surveillance. The study documented that female sex was significantly associated with aneurysm rupture (hazard ratio 4.50, CI 1.98–10.2, p = .001) and that mean AD at rupture was 5 cm in women versus 6 cm in men. 25 The reasons for these differences may be that aneurysms of the same size in both sexes represent more advanced disease in the females, who generally have smaller initial ADs.27,28 An alternative hypothesis is that although AAAs occur less frequently in females, women have faster growth rates of their aorta compared with males secondary to increased inflammation in the aortic wall once they have a small AAA.29,30
Other disease states are associated with an increased incidence of aortic aneurysms, such as polycystic kidney disease (PKD), in which the severity of the disease is associated with alterations in gonadal hormones. A recent study by Smith and colleagues suggested that testosterone is associated with a decline in renal function, whereas estrogen was protective in mice with juvenile cystic disease. 31 A link between steroid hormones and matrix metalloproteinase (MMP)-9 and MMP-1 in patients with PKD has been established. 32 A link between Turner syndrome, a chromosomal deletion leaving patients with an XO sex-linked chromosome pair, and aortic disease is also well documented. 33 Observations such as these have led some to suggest that “males and females have different patterns of illness…. Understanding the basis of these gender-based differences is important to developing new approaches to prevention, diagnosis, and treatment.’' 34
Increasing Incidence of AAAs in Males in Rodent Experimental Models
The mechanism(s) by which aneurysms are initiated and the series of events by which an AAA forms are poorly understood. 35 Although the specific etiology is unclear, AAAs are initiated by some unknown aortic wall injury. In a conducive genetic and environmental background, recruitment of leukocytes into the aortic wall media and adventitia by chemokines appears to be an early and critical event. These chemokines may be secreted by a number of cells, including leukocytes and smooth muscle cells (SMCs). Subsequent leukocyte infiltration into the aortic wall is associated with the production of many proinflammatory cytokines as part of the innate inflammatory response. During this stage, a number of matrix-degrading enzymes are released, leading to aortic wall elastin and collagen degradation. With continued aortic wall thinning secondary to SMC apoptosis 36 and elastin fragmentation, AAA formation occurs.
Three published reports have directly compared male and female animals in experimental AAA models and have suggested that, similar to humans, females are protected from AAA formation.18,37,38 Daugherty and colleagues documented in angiotensin (AT)II–induced AAAs in apolipoprotein E−/- and low-density lipoprotein (LDL) receptor−/- mice that males are more susceptible to aneurysm development than females. 39 Interestingly, in the initial report using this model describing suprarenal aortic aneurysm formation, female mice were used exclusively, and only 20% and 33% of mice developed an AAA. 38 In subsequent studies, male mice have been used primarily and develop an AAA approximately 80% of the time. 38
In the elastase-induced rodent aortic aneurysm model, a report from our laboratory documented that males form AAAs more often than females and that this was associated with an increase in the number of MMP-9-secreting macrophages in the aortic wall. 18 The mean increase in AD 14 days after elastase perfusion in male aortas was 200 6 38%, whereas female aortas had an increase of only 69 6 27% (p = .023). The incidence of AAAs, defined as a 100% increase in AD from baseline, was 82% in male rats compared with 29% in female rats (p = .023). A follow-up study in the rat elastase model in males using microarrays also suggested that early (day 3) during AAA development, the androgen receptor and the estrogen-related α receptor are down-regulated 50-fold and 126-fold, respectively, compared with control animals. 40 A number of other cytokines, including interleukin (IL)-1, were also found to be downregulated in females compared with males (Table 2). 41
Superarray Data from Male and Female Elastase-Perfused Abdominal Aortic Aneurysms Documenting Downregulation of a Number of Gene Families in Females Compared with Males

Adapted from Sinha I et al. 41
ND = expression was present in males but not detectable in females.
Recent experiments in our laboratory have confirmed that the phenotypic changes that occur in rats also occur in mice in the elastase-induced AAA model. In these experiments, male and female mice underwent isolated elastase or heat-inactived elastase aortic perfusion. The results were similar to those in rats in that male mice form larger AAAs with increased incidence compared with female mice (Figure 2A). In addition, MMP-9 and MMP-2 activity as determined by zymography is increased in the mouse aortic wall in males compared with females (Figure 2B).
A, Bar graph representing greater increase in aortic diameters (ADs) of male mouse aortas versus female mouse aortas at 14 days following elastase perfusion. The y axis indicates percent enlargement of AD, calculated as follows: {[(post AD – pre-AD)/pre-AD]*100}. B, Bar graph representing matrix metalloproteinase (MMP)-9 and MMP-2 activity levels analyzed via zymography in male and female mouse aortas harvested at 14 days following elastase perfusion. There was a significantly greater amount of MMP-9 activity in male elastase-perfused aortas versus female elastase-perfused aortas (p = .016). There was no significant difference in MMP-2 activity levels between the two groups. The γ-axis represents integrated optical density (IOD) of zymogen activity levels corrected for total protein. FC = female control-perfused aortas; FE = female elastase-perfused aortas; MC = male control-perfused aortas; ME = male elastase-perfused aorta; ns = not significant.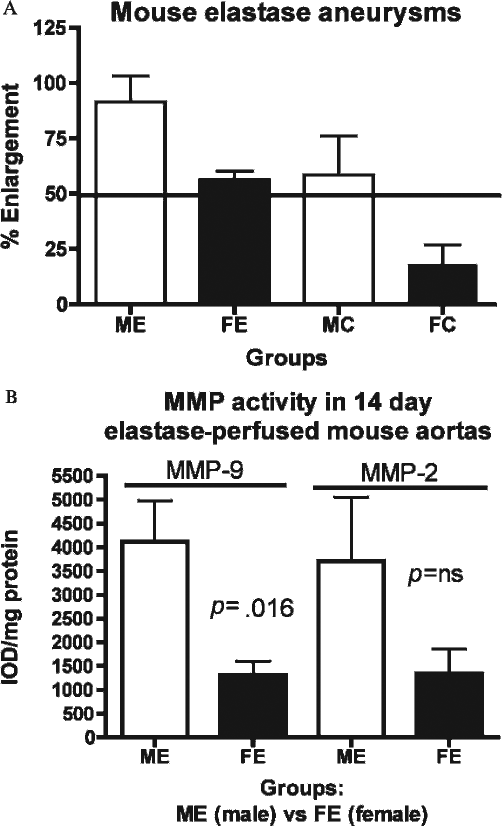
Recruitment of Leukocytes Is Critical during Early AAA Formation
The pathogenesis of AAAs involves a complex series of events, characterized ultimately by the degradation of elastin and collagen in the media and adventitia through a catalytic process that occurs following infiltration of inflammatory cells into the aortic wall.10,12,42–45 The macrophage, in particular, has been implicated as critical in the pathogenesis of AAAs. 46 The importance of the neutrophil during AAA formation, although not as well studied, has also been established in humans47–49 and, recently, in a mouse model of aneurysm formation. 50
The broad context for another study from our laboratory using a neutrophil depletion strategy prior to AAA formation was to better define the role of the neutrophil in the sequence of events leading to AAA formation. 50 Briefly, control, rabbit serum–treated (n = 27), or antineutrophil antibody–treated (anti–polymorphonuclear neutrophil [PMN], n = 25) C57BL/6 mice underwent aortic elastase perfusion to induce experimental aneurysms. Control mice 14 days after elastase perfusion exhibited a mean AD increase of 104 ± 14% (p < .001) and 67% developed AAAs, whereas anti-PMN-treated mice exhibited a mean AD increase of only 42 ± 33%, with 8% developing AAAs. Tissue neutrophils and macrophages 14 days after elastase perfusion were significantly decreased in the anti-PMN group compared with controls. Earlier time points (4 and 7 days) reflected these differences as well.
The mechanisms by which neutrophils and macrophages are recruited into the aortic wall during aneurysm formation are critical early events during AAA formation 51 and appear to be mediated by two primary mechanisms: (1) soluble mediators, including chemokines and cytokines, and (2) cell-specific receptors, such as the selectins. Multiple chemokines and cytokines have been studied in both human and experimental AAAs. The CC chemokines appear to be critical during AAA formation and have previously been shown to be important intermediaries during the initial inflammatory process, leading to AAA formation.52,53 Each of these chemokines, although not completely selective, tends to recruit a particular cell type to sites of inflammation. For example, macrophage inflammatory protein (MIP)-2 and keratinocyte chemokine (KC) (human IL-8) are primarily associated with recruitment of neutrophils, whereas MIP-1α and monocyte chemotractant protein (MCP)-1 (C-C chemokine ligand 2) are involved in the recruitment of macrophages.52,53 MIP-1α has been shown to be upregulated in macrophages in response to leukotriene LTD4 in hyperlipidemic apolipoprotein E−/- and LDL receptor−/- mice that form AAAs. 54 MCP-1 is essential for monocyte recruitment in vivo in several different inflammatory models55–57 and is upregulated in both mouse and human AAAs.58,59 The importance of MCP-1, in particular, during AAA formation is also supported by the observation that mice deficient in the CCR-2 receptor fail to develop aneurysms in the ATII-induced apolipoprotein E−/-mice. 60 This observation is associated with a marked decrease in monocyte-mediated inflammation in the aortic wall and an increase in IL-1β expression.
In separate experiments in mice using the elastase perfusion AAA model, the temporal course of MCP-1 expression and protein levels was defined. 58 Mouse AD (mean ± SEM) increased to aneurysmal proportions 14 days after elastase perfusion (from 0.51 ± 0.03 mm to 1.34 ± 0.32 mm; 163% increase; p < .05). Aortic wall messenger ribonucleic acid (mRNA) expression increased 28-fold for MCP-1 on day 4, with maximal production of chemokine protein on day 7. These data suggest that increased aortic wall expression of MCP-1 occurs early in development of elastase-induced AAAs before the onset of the chronic inflammatory response. The source of MCP-1 was aortic SMCs, providing an important link between enzymatic injury, leukocyte recruitment, and aneurysmal degeneration of the aortic wall.
Another group of soluble mediators involved early in the cascade of events that occurs during AAA formation is cytokines, which serve as cytotoxic mediators damaging SMCs in the aortic wall. Two well-studied cytokines, tumor necrosis factor α (TNF-α) and IL-1β, may play a central role in the gender disparity seen during AAA formation. Importantly, increased serum and tissue levels of TNF-α and IL-1β have been documented in patients with aortic aneurysms.61,62 These two cytokines are particularly relevant during AAA formation as they both inhibit type I and III collagen synthesis.63,64 In general, although often cited as having similar effects in inflammatory and septic models, a recent study has demonstrated enhanced TNF-α expression without corresponding changes in IL-1β in small AAAs. 65 Temporal and concentration differences in the expression of these two cytokines may be critical during the observed differential phenotypic expression of AAA between males and females. The expression of MMPs and tissue inhibitors of metalloproteinases is also controlled in part at the transcriptional level by cytokines.
In addition to the multiple potential soluble mediators of early leukocyte recruitment during AAA formation, various cell-based receptors are also critical during this process. One group of adhesion molecules that may be relevant is the selectins. The selectins are a family of three adhesion molecules: (1) E-selectin on the surface of activated endothelial cells, (2) P-selectin on the surface of activated platelets and endothelial cells, and (3) L-selectin constitutively expressed on the surface of most leukocytes (PMNs and macrophages).66,67 The primary function of selectins is to promote leukocyte capture to sites of inflammation. Without selectins, inflammatory cell recruitment is significantly diminished.68–72
The critical role of L-selectin in AAA formation in male rodents has been studied extensively in our laboratory. 73 Briefly, male rat abdominal aortas were perfused with saline or elastase and studied on postperfusion days 1, 2, 4, 7, and 14. L-selectin expression and protein levels in aortic tissue were determined by polymerase chain reaction and Western blot, respectively. Elastase-perfused ADs were significantly increased compared with control aortas at all time points except day 1 (p < .05). L-selectin mRNA expression in elastase-perfused aortas was 18 (p = .018), 17 (p <.001), and 8 (p = .02) times greater than in control aortas at days 1, 2, and 4, respectively. Western blot demonstrated a significant 69% increase in L-selectin protein at day 7 in elastase-perfused aortas compared with saline-perfused aortas (p = .005). Subsequent experiments involved studies on post–elastase perfusion days 4, 7, and 14 of aortas from C57Bl wild-type (n = 21) and L-selectin knockout (LKO) mice (n = 19). LKO mice had significantly smaller ADs at day 14 compared with wild-type mice. PMN counts were significantly greater in elastase-perfused wild-type mouse aortas compared with LKO mouse aortas at day 4 postperfusion (12.8 vs 4.8 PMNs/high-power field [HPF], p = .02). Macrophage counts were significantly greater at all time points in elastase-perfused wild-type mouse aortas compared with elastase-perfused LKO mouse aortas, with a maximum difference at day 7 postperfusion (13.3 vs 0.5 macrophages/HPF, p < .001)
In separate experiments from our laboratory to document phenotypic and mechanistic changes in AAA formation, male wild-type (n = 17) and P-selectin knockout (PKO)−/- mice (n = 10) underwent elastase perfusion, and ADs were determined postperfusion day 14.
74
The results document that, even to a greater extent than L-selectin−/- mice, the absence of P-selectin inhibits AAA formation (p < .001) (Figure 3).
Wild-type (WT) mice had significantly larger aortic diameters compared with P-selectin knockout (PKO) mice at day 14 following elastase perfusion (116% vs 38%, p < .001). In addition, aortic aneurysm incidence was 52% in WT mice and 0% (p = .01) in PKO mice. An aortic aneurysm was defined as a 100% or greater increase in AD from preperfusion measurement.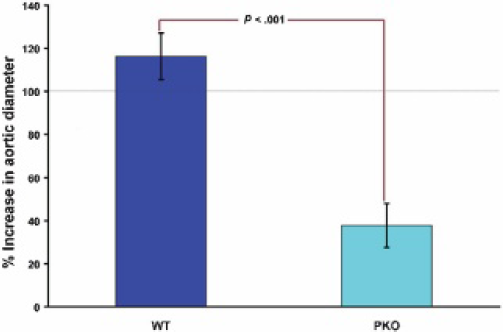
Manipulation of Gonadal Hormones Alters Leukocyte Trafficking and Aortic Wall Degradation
Although few studies have examined the role of pharmacologic or surgical alteration of gonadal hormones on leukocyte recruitment during AAA formation, a protective role for estrogen and its derivatives in AAA formation receives indirect support from a number of studies. A study in the AT-II-induced AAA model suggested that 17β-estradiol implanted subcutaneously attenuates aneurysm development. 75 Inhibition of proinflammatory genes, including MCP1, was proposed as the mechanism responsible for this effect. It has been suggested that orchidectomy, not ovariectomy, is capable of altering gender-specific rates of AAA formation. 76
Our laboratory confirmed the inhibitory effects of estradiol in the aortic elastase-perfusion rodent model. 18 Male rats were randomized to implantation of an estrogen pellet or sham implantation (n = 13, each). Rat aortas from both groups were subjected to elastase perfusion 5 days later. The aortic size was then determined 14 days following elastase perfusion, and the aorta was excised for study. By day 14, male rats receiving estradiol had significantly smaller aneurysms (241 ± 57%) compared with sham rats (538 ± 105%, p = .023). 18 ED-1-positive cell counts (mononuclear leukocyte lineage) were 1.8 ± 0.3 cells/HPF in males receiving estradiol versus 5.2 ± 0.5 cells/HPF in male sham rats (p = .0006).
Multiple observations from the trauma and sepsis literature suggest that gonadal hormones regulate leukocyte trafficking. Knoferl and others developed the hypothesis that leukocyte immune responses, through multiple soluble mediators, are depressed in males and enhanced in females following trauma and hemorrhage.77–82 Other models have also been used to examine the role of estrogen on leukocyte migration. In rat carotid arteries following acute balloon injury, a large number of leukocytes (neutrophils and macrophages) invade the carotid artery within 24 hours following injury. This response is markedly blunted by exogenous estrogen. Following ovariectomy in rats, estrogen therapy is able to negatively modulate the proinflammatory response by altering expression and protein levels of a number of chemokines and cytokines, including MCP-1, IL-1β, and TNF-α. Inhibition of these chemokines and cytokines by estrogen in oopherectomized animals translated into decreased migration of neutrophils in a chemotaxis assay.83–85 Estrogen also has a direct inhibitory effect on macrophage recruitment, through its effects on MCP-1.86–89 Furthermore, increased estrogen levels in women, including those on estrogen replacement therapy, correlated with reductions in circulating MCP-1 levels.
Gonadal hormones may also affect AAA formation through their effects on gonadal receptors. Mutliple genetic studies have suggested a role for the estrogen receptors in influencing cardiovascular disease. Although not examined in detail experimentally in the development of AAAs, vascular SMCs express functional estrogen receptor (ER)-α and -β. ER-α polymorphisms are associated with premature coronary artery disease in men 90 and postmenopausal women. 91 Similarly, a polymorphism in the ER-β gene has been documented in postmenopausal Japanese women with hypertension, 92 a known risk factor for aneurysm development.
Estrogen may also affect AAA development by directly inhibiting macrophage and SMC production of MMPs. For example, estrogen treatment of U937 cells decreases MMP-2 production directly. 93 In a study by Potier and colleagues, estrogen and progesterone differentially regulated MMP-9 and MMP-2 expression and activity in aortic arch–derived SMCs isolated from atherosclerosis-susceptible and – resistant mice. 94 Animals treated with estradiol also have decreased vascular SMC contractility and proliferation. 95
A third possible mechanism by which gender might alter AAA formation is through the effect of gonadal hormones on aortic wall extracellular matrix (ECM) proteins. In particular, the influence of sex hormones on aortic wall elastin and collagen degradation has been studied. Fischer and Swain examined the effects of castration in the presence of testosterone and estradiol in male rats and concluded that (1) estradiol, in the presence or absence of testosterone, alters the proportions of collagen and elastin so that the vessel is more distensible, and (2) testosterone has the opposite, but markedly less, effect of estradiol on vascular tissue. 96 These studies were extended by Simpson and Cardeilhac, who, following the administration of β-amino-propionitrile (BAPN) in turkeys, demonstrated that 65% of BAPN-treated male turkeys died of dissecting aneurysms, whereas only 21% of females died of the same disease. Ultrastructural alterations in aortic collagen and elastin occurred more frequently in males. 97 Potier and colleagues also suggested that estrogen differentially regulates collagen in atherosclerosis-susceptible and atherosclerosis-resistant mice. 94 In postmenopausal women, phytoestrogens result in decreased aortic stiffness. 98
Summary
Despite human epidemiologic and animal investigational data strongly supporting a gender bias in the development of AAAs, little is known about the specific mechanisms driving this disparity. This review provides data supporting the concept that gonadal hormones and their receptors work through multiple mechanisms, including (1) inhibiting leukocyte recruitment and aortic wall inflammation, (2) altering MMP expression or activity, or (3) effecting levels of ECM proteins elastin and collagen, ultimately leading to gender-based differences in AAA formation. Clearly, leukocyte recruitment plays a critical role during early AAA formation, and data suggest that this is influenced by gonadal hormones. Improved understanding of leukocyte trafficking during AAA formation will help better elucidate the pathophysiologic mechanism of aneurysm formation in men and women and may ultimately be expected to translate into novel therapies by which we might treat the large number of patients with small AAAs.