
Abstract
BACKGROUND:
Long non-coding RNAs (lncRNAs) are found to involve in modulating the development of atherosclerosis (AS). But the molecular mechanism of lncRNA growth-arrest specific transcript 5 (GAS5) in AS is not fully understood.
METHODS:
QRT-PCR was performed to measure the abundances of GAS5, miR-128-3p and fibulin 2 (FBLN2). Oxidized low-density lipoprotein (ox-LDL)-treated THP-1 cells were employed as cell models of AS. The cell proliferation and apoptosis were analyzed using CCK-8 and Flow cytometry assays, respectively. Levels of all protein were examined by western blot. The interaction among GAS5, miR-128-3p and FBLN2 was confirmed via dual-luciferase reporter and RNA immunoprecipitation (RIP) assays.
RESULTS:
GAS5 was elevated and miR-128-3p was decreased in the serum of patients with AS and ox-LDL-stimulated THP-1 cells. Ox-LDL stimulation inhibited proliferation and induced apoptosis of THP-1 cells. Meanwhile, GAS5 directly targeted miR-128-3p and inversely modulated its expression. Importantly, GAS5 depletion facilitated cell proliferation and impaired apoptosis in ox-LDL-induced THP-1 cells. Additionally, GAS5 augmented FBLN2 expression through sponging miR-128-3p, and miR-128-3p facilitated proliferation and retarded apoptosis of ox-LDL-induced THP-1 cells by targeting FBLN2.
CONCLUSION:
GAS5 knockdown promoted the growth of ox-LDL-induced THP-1 cells through down-modulating FBLN2 and increasing miR-128-3p, suggesting the potential value of GAS5 for treatment of AS.
Introduction
Atherosclerosis (AS) is a common chronic inflammatory disease in cardiovascular diseases and has a high incidence and mortality worldwide [1]. The formation of AS lesions involves multiple cell types, including vascular smooth muscle cell (VSMC) accumulation, endothelial cell (EC) dysfunction and macrophage activation [2, 3]. One basic hallmark of atherosclerosis as a chronic disease is lipid accumulation, which is associated with oxidized low-density lipoprotein (ox-LDL) [4]. Ox-LDL is a pivotal factor to accelerate the development of AS and has been revealed to affect cell proliferation and apoptosis [5]. Therefore, it is necessary to find molecular targets for effective treatment of AS and clarify the potential mechanism of ox-LDL inducing the development of AS.
Another basic hallmark of atherosclerosis as a chronic disease is inflammation, which is closely involved in the consecutive events leading to the formation, progression and rupture of an atherosclerotic plaque [4]. Monocytes are important mediators of the innate immunity [6] and was considered as a major source of proinflammatory species during atherogenesis [7] because they increased quantities during inflammation [8]. In addition, monocytes take up ox-LDL via toll like receptor 4 (TLR4) and differentiated co-receptors cluster CD36, and then they differentiates to macrophages with enhanced uptake characteristics. Excessive intake of ox-LDL make they transform into foam cells, which finally form the necrotic core of the atherosclerotic plaque [9, 10]. Monocyte development or trafficking inhibition strongly affects atherosclerotic lesion formation. Combined inhibition of C-C Motif Chemokine Ligand 2 (CCL2), C-X3-C Motif Chemokine Receptor 1 (CX3CR1), and C-C motif chemokine receptor 5 (CCR5) abrogates Ly6C(hi) and Ly6C(lo) monocytosis and almost abolishes atherosclerosis in hypercholesterolemic mice [11]. The mice lack of macrophage colony-stimulating factor (op) and apolipoprotein E in both decreased atherosclerosis [12]. Furthermore, apoptosis of monocytes is a characteristic of atherosclerotic lesions, which caused accumulation of insoluble lipids and other cellular contents [13]. Monocytes apoptotic death also leads to plaque instability and rupture because the release of metalloproteinases, which degrade the extracellular matrix (ECM) proteins and neovascularization [14]. As a result, the activation of coagulation cascade, the deposition of fibrin, and the activation and recruitment of platelet combine to form a clot [15]. Therefore, prevent monocyte apoptosis is a potentially powerful therapeutic strategy for atherosclerosis treatment. Long noncoding RNAs (lncRNAs), a group of endogenous RNAs with the length more than 200 nucleotides [16], are reported as independent biomarkers for numerous complex disease processes containing AS [17]. LncRNAs are widely concerned because they are implicated in the modulation of inflammatory response, cell growth and migration of AS [18]. For example, lncRNA 430945 could facilitate proliferation and migration of VSMCs by regulating ROR2/RhoA signaling pathway in AS [19]. Silencing lncRNA H19 retarded the growth of ox-LDL stimulated VSMCs through modulating the miR-148b/WNT/β-catenin network [20]. LncRNA growth-arrest specific transcript 5 (GAS5), which includes C/D box snoRNA genes in introns, has been found to be mal-regulated in many cancers [21]. GAS5 was found to inhibit their development as a suppressor gene in laryngeal squamous cell carcinoma [22], gastric cancer [23] and cervical cancer [24]. Others like Jing et al. demonstrated that GAS5 could modulate tumourigenesis and cell metastasis in B lymphocytic leukaemia [25]. Recently, a study revealed that GAS5 was enhanced in ox-LDL-stimulated THP-1 cells and AS mice model, and its knockdown could impede the progression of AS via targeting miR-135a [26]. But the potential molecular mechanism and effect of GAS5 on the process of human AS remain not clear.
MicroRNAs (miRNA), a kind of endogenously small ncRNAs, can participate in many pathological behaviors, such as immune responses, lipoprotein metabolism and vascular function [27]. MiR-128-3p is known to be related to many cardiovascular diseases. Qing et al. indicated that miR128-3p was declined in human umbilical vein ECs treated with Laminar shear stress (LSS) and regulation of miR-128-3p facilitated monocyte adhesion [28]. In addition, Liu et al. found that miR-128 was targeted by lncRNA NEAT1 to involve in ox-LDL-stimulated inflammation in AS development [29]. The FBLN2 gene encodes the extracellular matrix protein fibulin 2, and it is mainly expressed at epithelial-mesenchymal transition sites during cardiovascular development [30]. Another study supported that the increased FBLN2 might accelerate arterial damage and promote the progression of AS in diabetics [31]. Additionally, FBLN2 regulated activator protein-1 factor (AP-1) expression, which was related to the process of AS [31, 32]. But the specific role of FBLN2 in AS is unknown.
THP-1 is a type of cells derived from the blood of patients with human monocytic leukemia. THP-1 monocytes are similar to the macrophages in morphology and differentiation characteristics [22], and THP-1 cells have been shown to have the same oxidative ability as macrophages [23]. Previous study have shown that ox-LOL stimulated macrophages RAW264.7 cells could be used as AS model [24]. In addition, ox-LOL induced THP-1 was used as AS model to study the influence of miR-135a on AS progression [15]. Therefore, in this research, ox-LOL induced THP-1 cells could be conducted as the cell model of atherosclerosis to study some pathological processes in the formation of atherosclerosis.
In this study, we assessed GAS5 expression in the serum of patients with AS as well as ox-LDL-stimulated THP-1 cells. More than that, we researched the impact of GAS5 on proliferation and apoptosis of ox-LDL-induced THP-1 cells by modulating miR-128-3p/FBLN2 axis.
Materials and methods
Tissue samples and cells
The blood samples from 38 patients with AS and 38 healthy volunteers were acquired at The Affiliated Zhangjiagang Hospital of Soochow University, and they were placed in the centrifuge tube without anticoagulant. These AS patients (50–70 years old, and the ratio of males to females is 3/2) and healthy volunteers were signed the informed consents, and those patients had not received any treatment before the blood sample was taken. Serums were obtained by centrifuging the blood samples at a speed of 3000 rpm. All the experiments in our study were permitted by the Ethics committee of The Affiliated Zhangjiagang Hospital of Soochow University.
Human monocyte macrophage line THP-1 and HEK293T cells were acquired from Procell (Wuhan, China). They were maintained in DMEM (Invitrogen, Carlsbad, CA, USA) supplemented with 10% fetal bovine serum (FBS, Invitrogen) in an incubator.
Quantitative real-time polymerase chain reaction (qRT-PCR)
The RNA from cells and serums was extracted by TRIzol® reagent (Invitrogen). For detection of GAS5 and FBLN2, the cDNA was synthesized by the PrimeScript™ RT reagent kit (Takara, Dalian, China). For miR-128-3p, the cDNA was acquired by using miRNA reverse transcription kit (Funeng, Guangzhou, China). Then, qRT-PCR was performed on a 7900HT Fast Real-Time PCR System (Applied Biosystems, Foster City, CA, USA) using SYBR Green PCR Master Mix (Applied Biosystems). GAPDH was used as the control of GAS5 and FBLN2, and miR-128-3p expression was normalized to U6. Sequence of primers: GAS5, Forward (F): 5′-AACTTGCCTGGACCAGCTTA-3′, Reverse (R): 5′-CAAGCCGACTCTCCATACCT-3′. GAPDH, F: 5′-CTCAACTACATGGTCTACATGTTCCA-3′, R: 5′-CTTCCCATTCTCAGCCTTGACT-3′. MiR-128-3p, F: 5′-GGTC ACAGTGAACCGGTC-3′, R: 5′-GTGCAGGGTCCGAGGT-3′. U6, F: 5′-TGCGGGTGCTCGCTTCGCAGC-3′, R: 5′-CCAGTGCAGGGTCCGAGGT-3′. FBLN2, F: 5′-AGGACACAGACCCCAACTCT-3′, R: 5′-CACAGGTCTCACCCTCCTTG-3′.
Ox-LDL treatment and cell transfection
THP-1 cells were stimulated with disparate concentration of ox-LDL (0, 10, 25 and 50μg/mL) (Solarbio, Beijing, China) or LDL (50μg/mL) for 24 h. MiR-128-3p mimics (miR-128-3p), miR-128-3p inhibitor (anti-miR-128-3p) and their paired NCs, small interference RNA (siRNA) targeting GAS5 (si-GAS5) and its internal control (si-NC) were synthesized by RiboBio (Guangzhou, China) The full length sequence of GAS5 or the coding sequence (CDS) of FBLN2 was inserted into the pcDNA vector (Invitrogen) to produce pcDNA-GAS5 or pcDNA-FBLN2 (FBLN2). Transfection was conducted via Lipofectamine 2000 (Invitrogen).
Cell proliferation assay
THP-1 cells (after treated with ox-LDL or blank) were uniformly tiled into 96-well plates and placed in an incubator. At the specified time points after transfection, 10μL of cell counting kit-8 (CCK-8, Solarbio) reagent was added into the cells, and cells were further incubated for 3 h. Subsequently, the absorbance of THP-1 cells was measured under a microplate reader at 450 nm.
Cell apoptosis detection assay
The apoptosis of THP-1 cells in our study was analyzed by Flow cytometry using an Annexin V FITC/PI apoptosis detection kit (Solarbio). The ox-LDL or LDL treated THP-1 cells were uniformly placed into 6-well plates. At 48 h after transfection, cells were collected and stained with FITC as well as PI for 20 min. Finally, the FACS Caliber flow cytometer (FlowCam, Shanghai, China) was used to calculate the number of apoptotic cells.
Western blot
Proteins were extracted from the transfected THP-1 cells by RIPA Buffer (Beyotime, Shanghai, China). These proteins needed to be denatured at 99°C for 6 min before being separated. Then proteins were transferred to PVDF (Solarbio) membranes. After sealed in 5% non-fat milk, the membranes were incubated at 4°C with antibodies I against CyclinD1 (1:200, Abcam, Cambridge, MA, USA), B-cell lymphoma-2 (Bcl-2, 1:2000, Abcam), BCL2-associated x (Bax, 1:2000, Abcam), Cleaved-casp-3 (1:500, Abcam), FBLN2 (1:500, Abcam) or GAPDH (1:3000, Abcam). Next, the membranes were interacted with horseradish peroxidase-conjugated antibody II (HRP, 1:5000, Abcam) for another 1 h. The bands were observed by an ECL kit (R&S, Shanghai, China).
Dual-luciferase reporter assay
The fragments of GAS5 or 3’ UTR of FBLN2 containing the predicted miR-128-3p complementary sites or corresponding mutant sites were cloned into the pmirGLO vector (Promega Corporation, Fitchburg, WI, USA) to generate reporter plasmid of wile type-GAS5 (WT-GAS5), mutant type-GAS5 (MUT-GAS5), WT-FBLN2-3′ UTR and MUT-FBLN2-3′ UTR. These plasmids were co-transfected with miR-128-3p, miR-NC, anti-miR-128-3p, anti-miR-NC, pcDNA or pcDNA-GAS5 into HEK293T cells, respectively. Finally, the luciferase activity was assessed by a dual-luciferase reporter assay kit (Promega Corporation).
RNA immunoprecipitation (RIP) assay
EZ-Magna RIP Kit (Millipore, Billerica, MA, USA) was applied to analyze the targeted relationship between GAS5 and miR-128-3p. After lysed by lysis buffer, THP-1 cells were incubated with protein A/G magnetic beads and Ago2 antibody or IgG antibody. After purification of RNAs from the magnetic beads binding complexes, the RNA was separated from the beads and stored at –80°C for qRT-PCR analysis.
Statistical analysis
All data were repeated at least three times and appeared as the mean±standard deviation (SD). The difference in two groups was analyzed by Student’s t-test analysis. The correlation was assessed by spearman’s analysis. P value less than 0.05 was considered to be significant.
Results
GAS5 was up-modulated and miR-128-3p was down-modulated in the serum of AS patients
We first investigated whether GAS5 and miR-128-3p expression levels were altered in the serum of patients with AS by qRT-PCR. As displayed in Fig. 1A-B, GAS5 expression was fortified and miR-128-3p was down-modulated in human AS patient serums (n = 38) relative to that in healthy volunteer serums (n = 38). Meanwhile, a significant inverse interplay between miR-128-3p and GAS5 was also observed in AS patient serums by spearman’s correlation coefficient analysis (Fig. 1C). These results implied that GAS5 and miR-128-3p might participate in regulating AS development.

GAS5 was up-modulated and miR-128-3p was down-modulated in the serum of AS patients. (A-B) GAS5 and miR-128-3p expression levels in the serum of 38 patients with AS and 38 healthy volunteers were detected by qRT-PCR.
GAS5 and miR-128-3p levels in THP-1 cells stimulated with ox-LDL were further examined. As shown in Fig. 2A-B, after THP-1 cells were stimulated with ox-LDL of different concentrations, GAS5 was augmented and miR-128-3p was declined in a dose-dependent manner, while there was no obvious change in the levels of GAS5 and miR-128-3p after LDL treatment compared to control. Besides, to make sure we built the model of AS cell successfully, the cells proliferation and apoptosis were assessed. CCK-8 assay revealed that the cell proliferation was markedly repressed by ox-LDL in a dose-dependent manner (Fig. 2C). It was known from the above results that 50μg/mL ox-LDL treatment had a good effect, which was used for the following experiments. Then, Flow cytometry assay showed that the apoptosis rate of THP-1 cells treated with ox-LDL was significantly enhanced (Fig. 2D). To further determine these results, we detected the levels of CyclinD1, Bcl-2, Bax and Cleaved-casp-3 using western blot. Consistent with the data above, the levels of CyclinD1 and Bcl-2 were declined, and Bax as well as Cleaved-casp-3 levels were induced in THP-1 cells following ox-LDL stimulation (Fig. 2E).
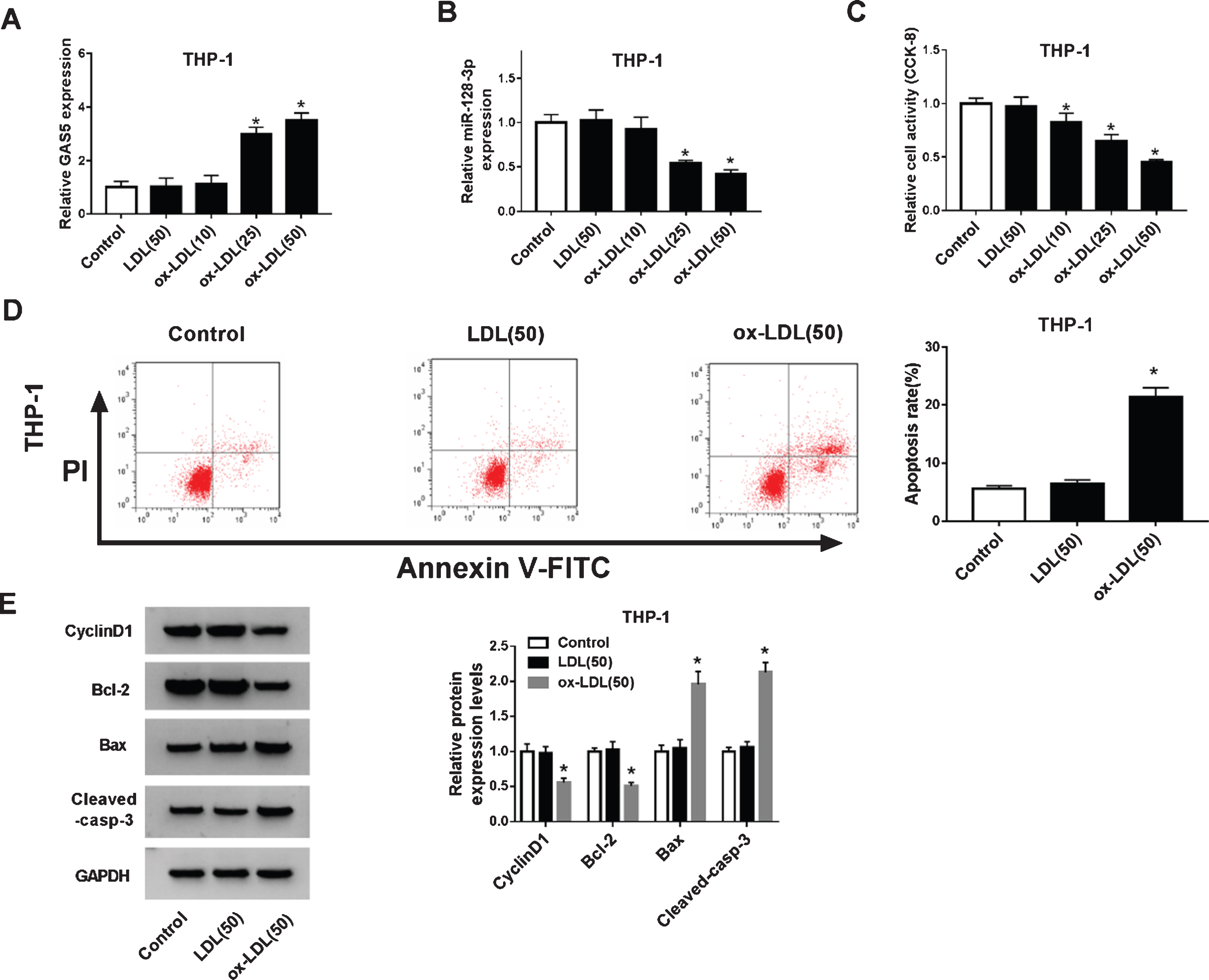
GAS5 was increased and miR-128-3p was reduced in ox-LDL-stimulated THP-1 cells.
Given that the contrary expression patterns of GAS5 and miR-128-3p in the serum of AS patients, and previous studies revealed that lncRNA could act as the competing endogenous RNA (ceRNA) of miRNA to regulate mRNA expression [33]. We found that there were binding sites between GAS5 and miR-128-3p by StarBase v2.0 (Fig. 3A), suggesting miR-128-3p might be a target miRNA of GAS5. And dual-luciferase reporter assay displayed that miR-128-3p obviously dwindled the luciferase activity of HEK293T cells in WT-GAS5 groups than that of miR-NC groups, and anti-miR-128-3p elevated the luciferase activity of WT-GAS5 in contrast to anti-miR-NC (Fig. 3B), while miR-128-3p and anti-miR-128-3p have no significant change on the luciferase activity of MUT-GAS5. PIR assay further demonstrated that GAS5 and miR-128-3p were enormously enriched in anti-Ago2 groups than that in anti-IgG groups (Fig. 3C), further supporting the targeted relationship between them. Additionally, GAS5 overexpression dwindled miR-128-3p expression and GAS5 knockdown elevated miR-128-3p expression in ox-LDL stimulated THP-1 cells (Fig. 3D-E).

GAS5 directly targeted miR-128-3p.
Next, the influence of GAS5 and miR-128-3p on cell proliferation and apoptosis of ox-LDL-treated THP-1 cells was studied. The data of qRT-PCR revealed that the promotion effect of si-GAS5 on miR-128-3p expression was attenuated by anti-miR-128-3p in ox-LDL-stimulated THP-1 cells (Fig. 4A). CCK-8 assay results showed that GAS5 knockdown promoted proliferation of ox-LDL-stimulated THP-1 cells, and the effect could alleviate by anti-miR-128-3p (Fig. 4B). And the anti-apoptotic effect of si-GAS5 on THP-1 cells could also be reversed by inhibiting miR-128-3p (Fig. 4C). The above results were also confirmed by changes in the levels of proliferation-related protein CyclinD1 and apoptosis-related proteins Bcl-2, Bax and cleaved-casp-3. As shown in Fig. 4D, the up-regulated effect on levels of CyclinD1 and Bcl-2 as well as the down-regulated impact on levels of Bax and cleaved-casp-3 by si-GAS5 in ox-LDL-stimulated THP-1 cells were weakened by anti-miR-128-3p. These data implied that GAS5 might accelerate the progression of AS by modulating miR-128-3p.

Silencing miR-128-3p overturned the effect of GAS5 depletion on proliferation and apoptosis of ox-LDL-stimulated THP-1 cells.
We searched the downstream target genes of miR-128-3p by StarBase v2.0 and found that there were binding sites between miR-128-3p and FBLN2-3’ UTR (Fig. 5A). Considering that miR-128-3p was a target miRNA of GAS5, the following dual-luciferase reporter assay was applied to assess the relationship among GAS5, miR-128-3p and FBLN2. As shown in Fig. 5B, miR-128-3p strikingly dwindled the luciferase activity of WT-FBLN2-3’ UTR, while this inhibition was overturned by up-modulating GAS5 in HEK293T cells. But no distinct change in luciferase activity was observed in MUT-FBLN2-3’ UTR group. Simultaneously, we found that FBLN2 expression at mRNA and protein levels were augmented when GAS5 was overexpressed, while this facilitation effect was counteracted by increasing miR-128-3p (Fig. 5C-D), suggesting GAS5 served as miR-128-3p sponge to fortify FBLN2 expression in ox-LDL stimulated THP-1 cells.

GAS5 up-regulated FBLN2 expression via acting miR-128-3p sponge.
Given that FBLN2 was the target gene of miR-128-3p, we further investigated whether miR-128-3p could modulate the progression of AS via targeting FBLN2. Firstly, the inhibition of FBLN2 expression by miR-128-3p could be reversed through overexpression of FBLN2 in ox-LDL-mediated THP-1 cells (Fig. 6A-B). Additionally, overexpression of miR-128-3p accelerated the cells proliferation and retarded apoptosis, whereas the effect was reversed through increasing FBLN2 (Fig. 6C-D). Congruously, miR-128-3p enhanced the levels of CyclinD1 and Bcl-2, retarded the levels of Bax and cleaved-casp-3. However, up-regulation of FBLN2 inverted the inhibition and promotion (Fig. 6E-F).

MiR-128-3p could target FBLN2 to induce proliferation and inhibit apoptosis of ox-LDL-stimulated THP-1 cells. After treated with 50μg/mL ox-LDL, THP-1 cells were transfected with miR-NC, miR-128-3p, miR-128-3p + Vector or miR-128-3p + FBLN2, respectively.
The occurrence of AS is closely associated with dyslipidemia, metabolic disorders and vascular inflammation [34]. During the development of AS, the high level of ox-LDL promoted the secretion of inflammatory cytokines [35]. Furthermore, ox-LDL stimulation could increase vascular permeability and accelerate the process of AS [36], and ox-LDL in U937 cells was found to induce foam cell formation to activate AS [37]. In spite of the effect of ox-LDL in AS was reported, the potential mechanism in AS remains to be investigated.
Accumulated evidences revealed that multiple lncRNAs are involved in regulating the progression of AS, such as down-regulation of lncRNA XIAT repressed ox-LDL-stimulated HUVEC inflammatory response and apoptosis [38]. And lncRNA DAPK-IT1 could contribute to the formation of AS via modulating cholesterol metabolism and inflammatory response of macrophages [39]. Lately, Shen et al. found that the allele of Ins/Del polymorphism (Rs145204276) in the promoter region of GAS5 related to atherosclerosis [40], and Ye et al. demonstrated that GAS5 was enriched in atherosclerotic plaques and ox-LDL-induced macrophage [41], which aroused our concern to probe the specific role of GAS5 in AS development.
In this research, GAS5 was elevated and miR-128-3p was dwindled in the serum of patients with AS. In addition, ox-LDL stimulated THP-1 cells were utilized as AS model. We examined the efficacy of ox-LDL treatment in a dose-dependent manner and results indicated that high-concentration treatment (25μg/mL, 50μg/mL) had significant effects, while the induction of ox-LDL with lower concentration (10μg/mL) and LDL stimulation had no obvious effect. In 50μg/mL ox-LDL stimulated THP-1 cells, the expression of GAS5 was upregulated and miR-128-3p was markedly down-regulated. And the proliferation ability of THP-1 cells treated with ox-LDL was inhibited, and the apoptosis rate was increased, which was consistent with previous results [42]. Coincidentally, GAS5 could serve as a sponge of miR-128-3p and inversely regulated its expression in ox-LDL-stimulated THP-1 cells. Our data also revealed that GAS5 depletion facilitated proliferation and impaired apoptosis through sponging miR-128-3p in ox-LDL-induced THP-1 cells, these data were consistent with the results of Chen et al. [19, 29]. However, Zhong et al. demonstrated that silencing MIAT could retard the growth of ox-LDL-stimulated AS cell models [42], it’s speculated that the inconsistent results were caused by different cells used in the experiments, and the specific reasons needed to be further explored.
The function of lncRNA as the ceRNA of miRNA to regulate the target mRNA expression has been widely introduced [43]. Therefore, we searched the target mRNAs of miR-128-3p and determined that FBLN2 could bind to miR-128-3p. Meanwhile, miR-128-3p accelerated the cells proliferation and hindered apoptosis, and the effect was counteracted by overexpressing FBLN2. Previous studies revealed that FBLN2 was related to arterial injury in patients with diabetes and AP-1 expression [31, 32], our results further confirmed the role of FBLN2 in ox-LOL-treated THP-1 cells. Mechanically, overexpression of GAS5 fortified FBLN2 expression by sponging miR-128-3p in AS cell model. While, our drawback is the lack of in vivo experiments, so further works are expected to verify the GAS5/miR-128-3P/FBLN2 axis in AS using animal model.
In summary, our results showed that GAS5 was augmented in the serum of patients with AS and ox-LDL-treated AS cell model. Meanwhile, our results revealed that GAS5 depletion promoted proliferation and hampered apoptosis in ox-LDL-stimulated THP-1 cells through regulating miR-128-3p/FBLN2 axis, suggesting that GAS5 has therapeutic potential in the treatment of AS.
Funding
None.
Declaration of interests
The authors have no interests to disclose.
Footnotes
Acknowledgments
None.