
Abstract
OBJECTIVE:
Acute myeloid leukemia (AML) is a hematological malignancy. This study was attempted to uncover the effects of long noncoding RNA taurine-upregulated gene1 (TUG1) on the viability and apoptosis of AML cells.
METHODS:
QRT-PCR was implemented to examine the expression of TUG1, miR-221-3p and KIT in AML. The correlation between TUG1 and clinicopathological features of AML patients was evaluated. The effect of TUG1 on AML cells were studied by RNA interference approach. AML cells were transfected with miR-221-3p mimic and miR-221-3p inhibitor, respectively. Then the viability and apoptosis of AML cells were examined by MTT and flow cytometry assay, respectively. Additionally, dual-luciferase reporter assay was used to confirm the interactions among TUG1, miR-221-3p and KIT. Western blot was applied to analyze protein expression of KIT.
RESULTS:
The expression of TUG1 and KIT was up-regulated in AML, but miR-221-3p was down-regulated. TUG1 expression had obviously correlation with World Health Organization (WHO) grade in AML patients. The functional experiment stated that TUG1 silencing suppressed the viability and accelerated the apoptosis of AML cells. Moreover, the mechanical experiment demonstrated that TUG1 and KIT were both targeted by miR-221-3p with the complementary binding sites at 3’UTR. Up-regulation of miR-221-3p inhibited the protein expression of KIT. Furthermore, in the feedback experiment, miR-221-3p inhibition or KIT overexpression reversed the repression of tumor behavior induced by TUG1 silencing.
CONCLUSIONS:
TUG1 silencing retarded viability and promoted apoptosis of AML cells via regulating miR-221-3p/KIT axis, providing a potential therapeutic target for AML.
Introduction
Acute myeloid leukemia (AML) is an aggressive heterogeneous hematological malignancy [1]. Criterion of AML is more than 20% myeloblasts in blood (or marrow) with myeloid lineage [2]. AML results from aggregation of aberrant myeloblasts, often occurs in bone marrow, guiding to hematopoietic insufficiency and death [3]. Although hematopoietic stem cell transplants and chemotherapy have improved the prognosis of AML patient, overall long-term survival remains insufficient [4]. So far, the occurrence of AML is commonly the result of the mutual modulation of various genes [5, 6]. Thus, it is indispensable to explore the key molecules for AML treatment.
Long noncoding RNAs (lncRNAs) participate in processes of diverse hematological malignancies, such as acute lymphoblastic leukemia [7], multiple myeloma [8] and AML [9]. Long noncoding RNA taurine-upregulated gene1 (TUG1), a critical oncogenic lncRNAs of human [10], has been proved to take part in hematological cancers. For instance, TUG1 is up-regulated in multiple myeloma patients and serves as a relevant biomarker [11]. TUG1 accelerates cell proliferation and retards cell apoptosis via decreasing miR-29b-3p in multiple myeloma [12]. Interestingly, previous studies have demonstrated that TUG1 expression is increased in AML [13, 14]. Qin et al. have reported that TUG1 is associated with poor prognosis and exhibits oncogenic activity in AML [15]. Li et al. and Wang et al. have shown that TUG1 elevates cell viability and hampers cell apoptosis in AML [16, 17]. Li et al. have displayed that knockdown of TUG1 markedly reduces the viability and metastasis of AML cells [18]. Hence, TUG1 may have a promoting effect on the progression of AML. However, the underlying mechanism of TUG1 in AML is not fully revealed.
As regulative genes, microRNAs (miRNAs) are proved to involve in the development of hematological tumors including AML [19, 20]. MiR-125a expression is inhibited in AML and its overexpression suppresses AML cell proliferation and facilitates apoptosis [21]. MiR-370 acts as a tumor suppressor in AML via repressing FoxM1 [22]. Interestingly, miR-221 has been emerged as a critical regulator of multiple tumors. For example, miR-221-3p restrains cell proliferation and migration by targeting ARF4 in epithelial ovarian cancer [23]. MiR-221-3p suppresses cell proliferation and induces apoptosis in medulloblastoma by inhibiting EIF5A2 [24]. MiR-221 overexpression alleviates glucocorticoid resistance in MLL-AF4 acute lymphocytic leukemia [25]. Importantly, miR-221 expression is inhibited in AML with deranged core-binding factor subunits [26]. In addition, Guo et al. have reported that miR-221 is a target of TUG1, which is negatively regulated by TUG1 in non-small cell lung cancer cells [27]. However, whether TUG1 can regulate the progression of AML via binding to miR-221-3p remains undefined.
KIT proto-oncogene receptor tyrosine kinase (KIT or c-KIT) participates in hematopoiesis and is related to the hematological malignancy [28, 29]. Previous researches have demonstrated that there is a high expression of KIT in AML [30, 31]. Liu et al. have displayed that overexpression of KIT promotes the proliferation of AML cells [32]. In addition, accruing reports have indicated that miRNAs are involved in the process of AML by regulating KIT. For instances, miR-137 inhibits KIT expression to repress malignant progression of AML [33]. MiR-193a inhibits growth and elevates apoptosis of AML cells by repressing KIT [34]. Notably, KIT has also been confirmed as a target of miR-221 in erythroleukemic cells [35], melanomas cells [36], and gastrointestinal stromal tumor cells [37]. Therefore, we proposed that whether miR-221-3p can regulate the progression of AML by interacting with KIT.
Herein, we detected the expression of TUG1, miR-221-3p and KIT. Then, we explored the function roles of TUG1 on viability and apoptosis of AML cells. Moreover, we verified whether miR-221-3p is a target of TUG1. The relationship between miR-221-3p and KIT was also confirmed. Our study may uncover a potential therapeutic target for AML.
Materials and methods
Patients and tissue samples
In total 59 bone marrow tissues of AML patients (27 males and 32 females) were collected from the Pediatric Intensive Care Unit (PICU) of the Liaocheng Second People’s Hospital between March 2017 and October 2018. Patients had received neither radiotherapy nor chemotherapy prior to resection. In addition, 59 normal bone marrow (NBM) tissues from healthy donors were collected. AML tumor tissues and NBM tissues were stored in liquid nitrogen. We separated our patient cohort according to the median TUG1 expression into high TUG1 (n = 30) and low TUG1 groups (n = 29). This study was permitted by our hospital ethics committee, and informed consents were obtained from each patient and their guardian.
Cell culture
Human AML cell lines (HL-60, THP-1, MV-4-11 and AML-193) and human bone marrow progenitor cells CD34+ were obtained from the American Type Culture Collection (Manassas, VA, USA). Cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM, Invitrogen, Carlsbad, CA, USA) with 10% fetal bovine serum (FBS, Invitrogen) at 37°C containing 5% CO2.
Cell transfection
The siRNA negative control (si-NC), si-TUG1-1, si-TUG1-2, miR-NC, miR-221-3p mimics, miR-221-3p inhibitor, pcDNA3.1 (pcDNA)-NC and pcDNA-KIT were purchased from Shanghai Genepharma (Shanghai, China). HL-60 and THP-1 cells grown to 85% confluence were transfected or co-transfected with these above agents using Lipofectamine 3000 (Invitrogen) according to the guidelines.
Quantitative real time polymerase chain reaction (qRT-PCR)
Total RNA was extracted from tissues and cells using the TRIzol reagent (Invitrogen). Then, cDNA samples were gained through reverse transcription using PrimeScript RT Reagent Kit (TaKaRa, Japan). Next, qRT-PCR was conducted on 7500 Real-time PCR System (Applied Biosystems, Waltham, MA, USA). Relative expression was calculated by the 2-ΔΔCt method. GAPDH, U6 and β-actin were used for the normalization of TUG1, miR-221-3p and KIT, respectively. The primer sequences were shown in Table 1.
Primers sequences
Primers sequences
Total proteins were extracted from HL-60 and THP-1 cells, and then transferred into SDS-PAGE. Separated protein was transferred onto polyvinylidene fluoride membranes, blocked with 5% skimmed milk, and incubated at 4°C overnight with primary anti-KIT antibody (1 : 1000, SAB4501647MSDS, Sigma, St. Louis, MO, USA) or β-actin (1 : 4000, SAB2701711MSDS, Sigma). Afterwards, the membranes were subjected to HRP-labeled goat anti-rabbit IgG (1 : 5000, 12-348MSDS, Sigma) secondary antibody at 25°C for 1 h. The immunoblots were measured through ECL system, and quantified by ImageLab software (Bio-Rad, Hercules, CA, USA).
Dual-luciferase reporter assay
The potential binding sites of TUG1 and miR-221-3p or KIT and miR-221-3p were predicted by Starbase or TargetScan, respectively. TUG1 and KIT with WT or MUT miR-221-3p-binding sites were generated and fused to the psiCHECK-2 vectors (YouBio, Hunan, China). HL-60 and THP-1 cells were co-transfected with above luciferase vectors and miR-NC or miR-221-3p mimics using Lipofectamine 3000 (Invitrogen).
MTT assay
The HL-60 and THP-1 cells were seeded into 96-well plates (2×103 cells/well) and cultured with 5% CO2 at 37°C for 0, 24, 48, 72, 96 h, respectively. Cell viability was measured using the MTT cell proliferation assay kit (Sigma) according to the guidelines.
Flow cytometer
The HL-60 and THP-1 cells were trypsinized and washed with phosphate buffered saline twice. Then, HL-60 and THP-1 cells were stained by using Annexin V-FITC and propidium iodide (Invitrogen) for 15 min in a dark room. Subsequently, the apoptotic cells were observed by MUSE™ flow cytometer (Beckman, Miami, FL, USA).
Statistical analysis
Data statistical analysis was performed using GraphPad Prism 7.0 (GraphPad, San Diego, CA, USA). Data were presented as mean±standard deviation. The differences between two groups or among multiple groups were assessed by Student’s t-test or one-way ANOVA followed by Tukey’s post-hoc test. The correlation significance was evaluated by Pearson correlation analysis. Differences were considered statistically significant at P < 0.05.
Results
TUG1 expression was enhanced in AML tumor tissues
As shown in Fig. 1A, the TUG1 expression in 70 NBM tissues was found to be lower than those in 173 AML tumor tissues by using GEPIA web tool. To demonstrate whether TUG1 is differently expressed in AML tumor tissues and NBM tissues, qRT-PCR was implemented. Results revealed that TUG1 expression was markedly increased in AML tumor tissues compared with the NBM tissues (P < 0.001, Fig. 1B). And its expression in AML patients in TNM III/IV was obviously higher than that in TNM I/II (P < 0.001, Fig. 1C). In addition, the TUG1 expression was visibly enhanced in HL-60, THP-1, MV-4-11 and AML-193 cells by contrast to the CD34+ cells (P < 0.01, Fig. 1D). As shown in Table 2, TUG1 expression had dramatically correlation with World Health Organization (WHO) grade in AML patients (P < 0.01).

TUG1 expression was enhanced in acute myeloid leukemia (AML) tumor tissues. (A) TUG1 expression in 173 AML tumor tissues and 70 normal bone marrow (NBM) tissues was analyzed by GEPIA; (B) The expression of TUG1 in AML tumor tissues and NBM tissues was measured by qRT-PCR; (C) Relative expression of TUG1 in AML patients at the TNM I/II and TNM III/IV; (D) QRT-PCR was performed to detect the expression of TUG1 in CD34+, HL-60, THP-1, MV-4-11 and AML-193 cells. **P < 0.01 vs. CD34+.
Correlation between TUG1 expression and clinicopathological features in acute myeloid leukemia patients
Note: **P < 0.01. WHO, World Health Organization.
We used RNA interference approach to explore whether TUG1 silencing affects the tumorigenesis of AML cells. QRT-PCR displayed that transfection of si-TUG1-1 and si-TUG1-2 could considerably decline TUG1 expression in HL-60 and THP-1 cells (P < 0.01, Fig. 2A). The si-TUG1-1 was used for subsequent assays. After that, MTT assay observed that si-TUG1-1 significantly attenuated the cell viability of HL-60 and THP-1 cells at 96 h post-culturing (P < 0.01, Fig. 2B). In addition, we investigated the effect of si-TUG1-1 on apoptosis by flow cytometry. The apoptosis rate of HL-60 and THP-1 cells was obviously elevated by TUG1 knockdown (P < 0.01, Fig. 2C).
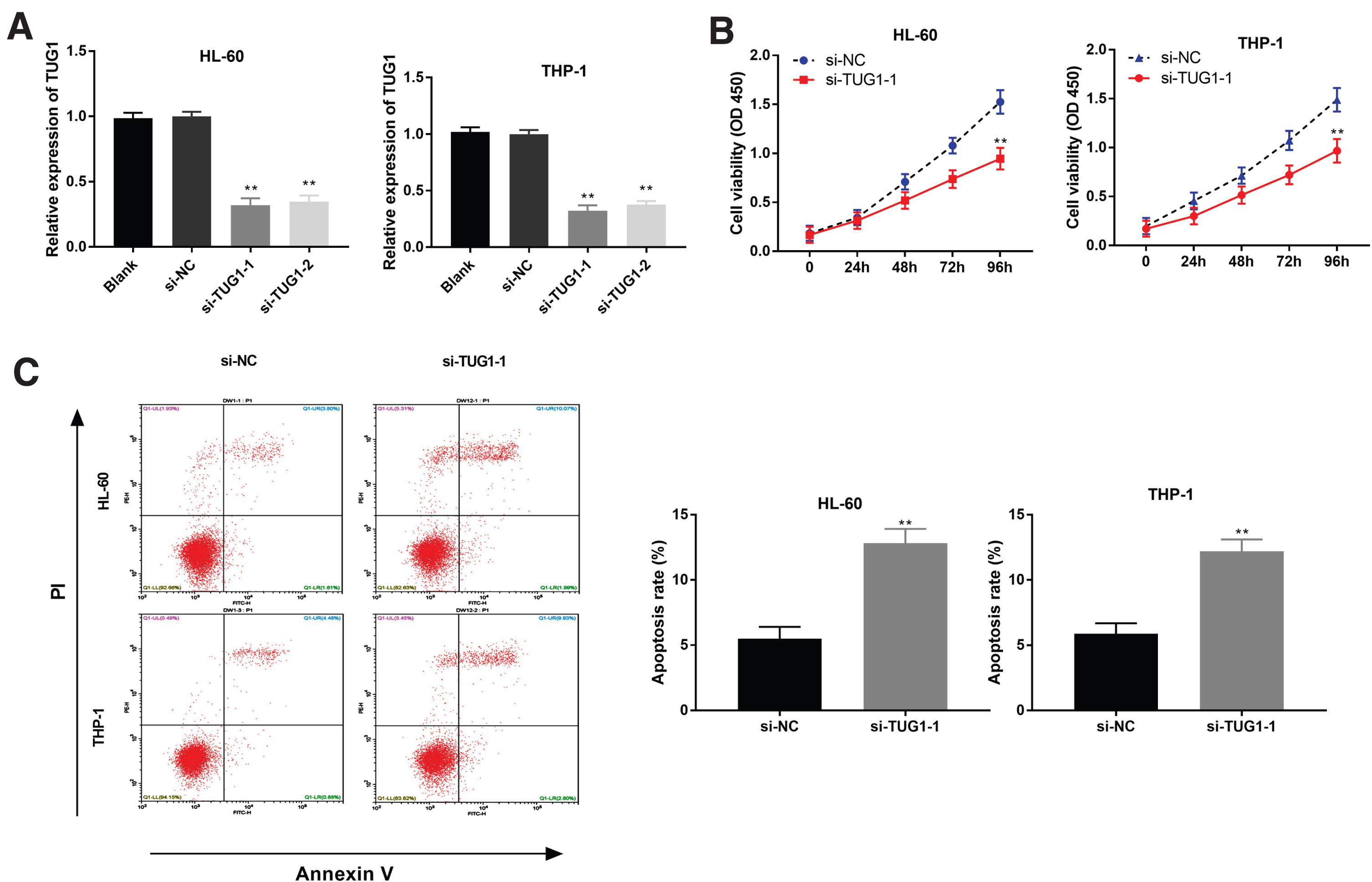
TUG1 silencing repressed the viability and induced apoptosis of acute myeloid leukemia (AML) cells. (A) The transfection efficiency of si-NC, si-TUG1-1 and si-TUG1-2 in HL-60 and THP-1 cells was measured by qRT-PCR. **P < 0.01 vs. Blank; (B) The viability of HL-60 and THP-1 cells was measured by MTT assay; (C) The apoptosis rate of HL-60 and THP-1 cells was determined by flow cytometry. **P < 0.01 vs. si-NC.
According to searching in StarBase, miR-221-3p was found to has the putative binding sites with TUG1 (Fig. 3A). As shown in Fig. 3B, TUG1 down-regulation could markedly enhance miR-221-3p expression in HL-60 and THP-1 cells (P < 0.01). Then, dual-luciferase reporter assay confirmed that miR-221-3p overexpression obviously reduced the relative luciferase activity of the reporter with TUG1 WT in HL-60 and THP-1 cells (P < 0.01, Fig. 3C). Notably, a considerably inhibition of miR-221-3p expression was appeared in tumor tissues of AML patients compared with NBM tissues (P < 0.01, Fig. 3D). Furthermore, there was a negative correlation between TUG1 and miR-221-3p expression in AML tumor tissues (N = 59, r = –0.1337, P = 0.3128, Fig. 3E). Interestingly, miR-221-3p expression was markedly decreased in HL-60, THP-1, MV-4-11 and AML-193 cells compared with the CD34+ cells (P < 0.01, Fig. 3F).

MiR-221-3p served as a target of TUG1. (A) Starbase showed the predicted binding site between TUG1 and miR-221-3p; (B) The expression of miR-221-3p was high-expressed by the transfection of si-TUG1-1 in HL-60 and THP-1 cells. **P < 0.01 vs. Blank; (C) Relative luciferase activity in HL-60 and THP-1 cells was measured by dual-luciferase reporter assay. **P < 0.01 vs. miR-NC; (D) QRT-PCR was performed to confirm the expression of miR-221-3p in tumor tissues of acute myeloid leukemia (AML) patients and normal bone marrow (NBM) tissues; (E) The expression of TUG1 was negatively correlated with miR-221-3p; (F) The expression of miR-221-3p in CD34+, HL-60, THP-1, MV-4-11 and AML-193 cells was examined by qRT-PCR. **P< 0.01 vs. CD34+.
To determine the biological function of miR-221-3p in AML, miR-221-3p was enhanced and antagonized by the transfection of miR-221-3p mimics and miR-221-3p inhibitor (P < 0.01, Fig. 4A). MiR-221-3p strikingly inhibited the viability of HL-60 and THP-1 cells at 72 and 96 h post-culturing (P < 0.01, Fig. 4B). Additionally, the apoptosis rate of HL-60 and THP-1 cells was visibly elevated by miR-221-3p overexpression (P < 0.01, Fig. 4C).
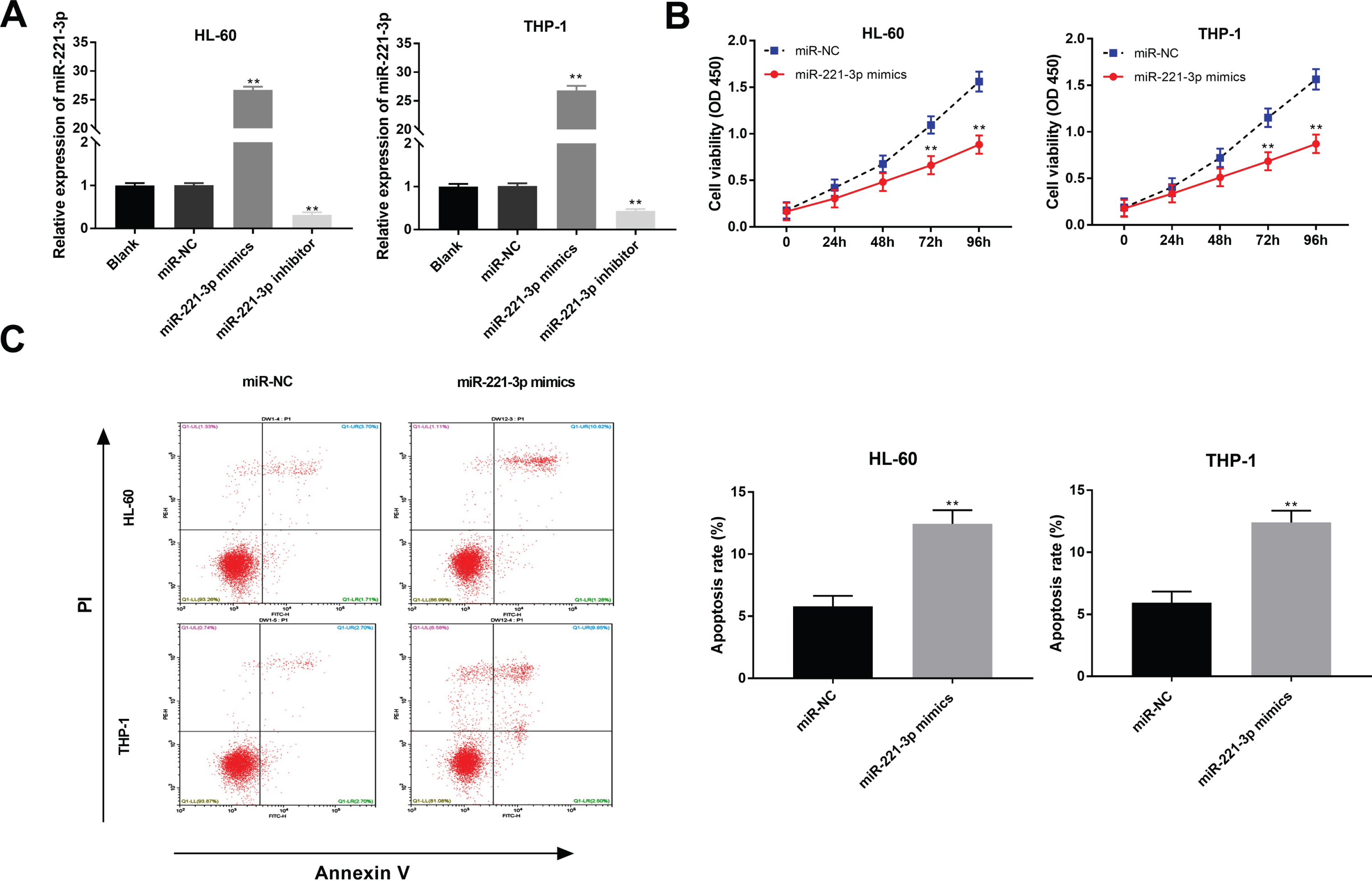
MiR-221-3p decreased the viability and induced apoptosis of acute myeloid leukemia (AML) cells. (A) The transfection efficiency of miR-NC, miR-221-3p mimics and miR-221-3p inhibitor was demonstrated using qRT-PCR in HL-60 and THP-1 cells. **P < 0.01 vs. Blank; (B) MTT assay were performed after transfected with miR-221-3p mimics or miR-NC in HL-60 and THP-1 cells; (C) The effect of miR-221-3p overexpression on the apoptosis rate of HL-60 and THP-1 cells was assessed. **P < 0.01 vs. miR-NC.
To explore the molecular mechanism of miR-221-3p in AML, TargetScan was used to predict the target genes of miR-221-3p, and miR-221-3p was found to directly target the 3’UTR of KIT (Fig. 5A). MiR-221-3p mimics obviously declined the luciferase activity of WT KIT reporter vector in HL-60 and THP-1 cells (P < 0.01, Fig. 5B). Moreover, GEPIA web tool displayed that the KIT expression in 173 AML tumor tissues was higher than that in 70 NBM tissues (Fig. 5C). Interestingly, KIT expression was obviously elevated in tumor tissues of AML patients (P < 0.001, Fig. 5D). A negative correlation between miR-221-3p and KIT expression in AML tumor tissues was exhibited (N = 59, r = –0.3607, P = 0.0050, Fig. 5E). Besides, KIT expression was positively correlated with TUG1 expression in AML tumor tissues (N = 59, r = 0.6339, P < 0.001, Fig. 5F). By contrast to the CD34+ cells, KIT expression was clearly enhanced in HL-60, THP-1, MV-4-11 and AML-193 cells (P < 0.01, Fig. 5G). Furthermore, miR-221-3p overexpression could inhibit the protein expression of KIT in HL-60 and THP-1 cells (P < 0.01, Fig. 5H).
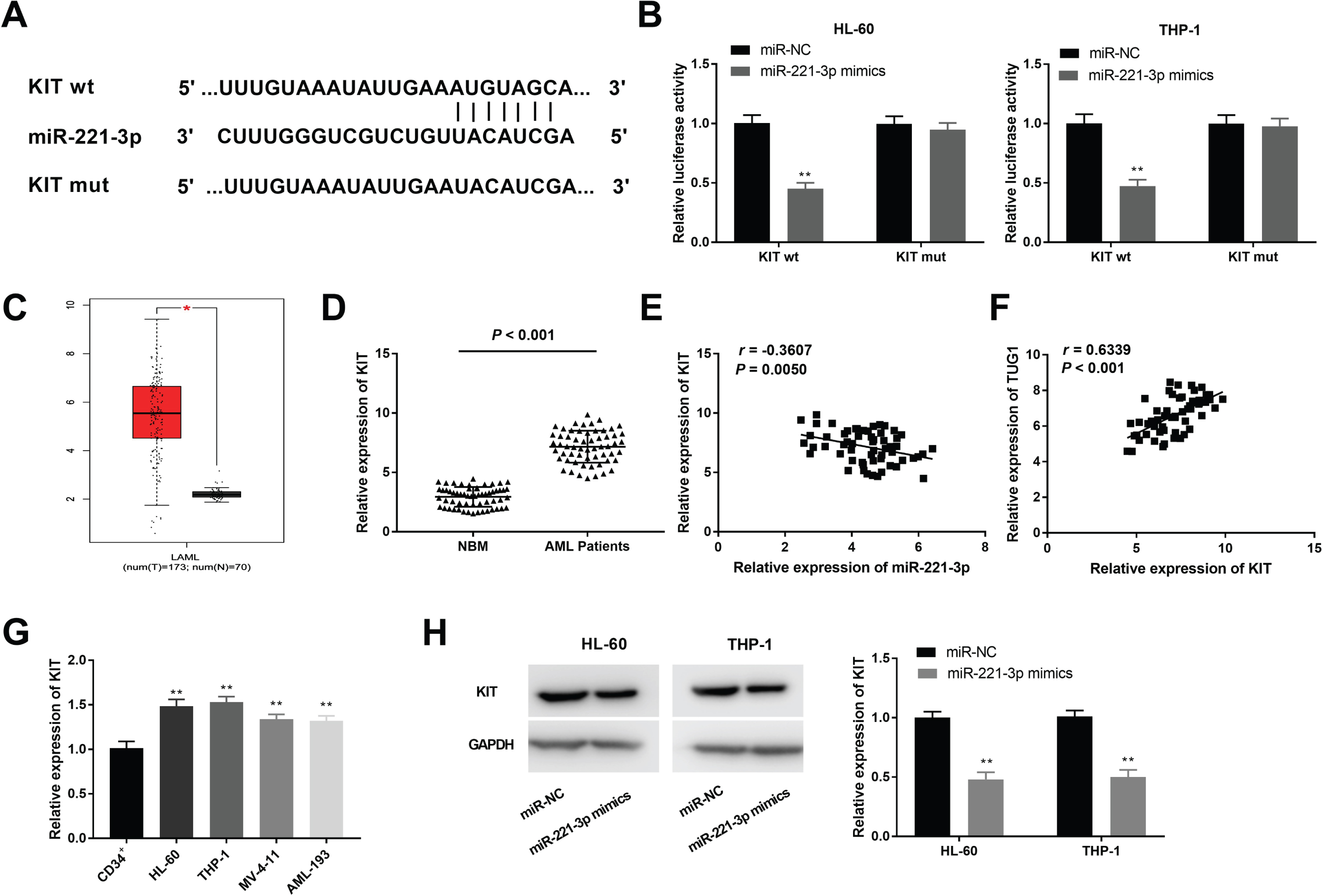
MiR-221-3p modulated the KIT expression. (A) TargetScan exhibited the predicted binding site between KIT and miR-221-3p; (B) Dual-luciferase reporter assay was performed to measure the relative luciferase activity in HL-60 and THP-1 cells. **P < 0.01 vs. miR-NC; (C) KIT expression in 173 acute myeloid leukemia (AML) tumor tissues and 70 normal bone marrow (NBM) tissues was analyzed by GEPIA. (D) QRT-PCR was used to detect the expression of KIT in tumor tissues of AML patients and NBM tissues. (E) The expression of KIT was negatively correlated with miR-221-3p; (F) The expression of KIT was positively correlated with TUG1; (G) QRT-PCR was performed to evaluate the expression of KIT in CD34+, HL-60, THP-1, MV-4-11 and AML-193 cells. **P < 0.01 vs. CD34+; (H) The protein expression of KIT in HL-60 and THP-1 cells was measured by western blot. **P < 0.01 vs. miR-NC.
To further verify the molecular mechanism by which TUG1 silencing inhibited progression of AML, feedback experiment was performed using MTT and flow cytometry assay. As illustrated in Fig. 6A and B, TUG1 knockdown could visibly reduce the viability, while promote the apoptosis of HL-60 cells (P < 0.01). MiR-221-3p inhibition or KIT overexpression could not only notably reverse the inhibitory effect of si-TUG1-1 on the viability of HL-60 cells (P < 0.01), but also markedly mitigated the promoting effect of si-TUG1-1 on the apoptosis of HL-60 cells (P < 0.05).

TUG1 inhibition attenuated viability and promoted apoptosis via regulating miR-221-3p/KIT axis in acute myeloid leukemia (AML) cells. (A) Overexpression of miR-221-3p or inhibition of KIT reversed the inhibitory effect of TUG1 knockdown on viability of HL-60 cells. (B) The promoting effect of TUG1 silencing on apoptosis of HL-60 cells was mitigated by miR-221-3p overexpression or TUG1 knockdown. **P < 0.01 vs. si-NC; #P < 0.05, # #P < 0.01 vs. si-TUG1-1.
Recent researches have demonstrated that enhanced expression of lncRNAs such as lncRNA SNHG5 [38], lncRNA HOTTIP [39] and lncRNA ANRIL [40] is uncovered in bone marrow tissues of AML patients and AML cells. In our study, TUG1 expression was high-expressed in both bone marrow tissues of AML patients and AML cells, and had obviously correlation with WHO grade in AML patients, which was similar with some lncRNAs in AML. For example, enhanced expression of lncRNA LINP1 is observed in AML patients, and is related to histological grade [41]. LINC00152 is up-regulated in AML specimens and cells, and predicts poor outcome [42]. LncRNA MVIH expression is dramatically elevated in AML patients with poor risk compared to AML patients with intermediate and better risk [43]. Above all, we speculate that TUG1 may be an onco-lncRNA for AML. Previous studies have indicated that TUG1 has a vital function in AML. For instance, TUG1 induces cell proliferation, and restrains cell apoptosis in AML by targeting aurora kinase A [17]. TUG1 knockdown increases adriamycin cytotoxicity and apoptosis rate of AML cells [44]. TUG1 silencing decreases the IC50 of adriamycin, and promotes adriamycin-induced apoptosis in AML cells by miR-34a/EZH2 axis [13]. TUG1 facilitates the cell viability and metastasis by targeting miR-370-3p/MAPK1/ERK in AML [18]. In this study, TUG1 silencing attenuated the cell viability, and promoted cell apoptosis of AML. The role of TUG1 was similar with above studies, indicating that TUG1 may be a potential therapeutic target for AML.
LncRNAs can act as a molecular sponge or ceRNA in modulating miRNA in the promotion of AML [45, 46]. Increasing evidences have verified that TUG1 serves as an onco-lncRNA by targeting miRNAs. For instance, TUG1 enhances tumorgenesis of pancreatic cancer by targeting miR-29c [47]. TUG1 accelerates cell proliferation and metastasis in gallbladder carcinoma through negatively modulating miR-300 [48]. Here, miR-221-3p contained complementary binding sites with TUG1, and we assume that TUG1 regulates AML according to similar mechanisms above. Numerous documents have proved that miR-221 exerts tumor suppressor effect on multiple cancers. MiR-221-3p expression is reduced in patients with triple negative breast cancer and correlated with poor prognosis [49]. MiR-221-3p restrains cell proliferation and migration by targeting ARF4 in epithelial ovarian cancer [23]. MiR-221 retards the progression of chronic myeloid leukemia by improving imatinib sensitivity [50]. Here, miR-221-3p expression was high-expressed and inversely related to TUG1 expression in AML, suggesting that TUG1 may exert cancer-promoting effect on AML cells via inhibiting miR-221-3p expression. We also observed that miR-221-3p attenuated the proliferation and induced apoptosis of AML cells. Additionally, miR-221-3p knockdown apparently reversed the effect that TUG1 silencing exerted. Taken together, TUG1 silencing may inhibit the viability and increase the apoptosis of AML cells by enhancing miR-221-3p expression.
KIT has been found to be an oncogenic gene, and KIT expression is elevated in different cancers, including lung cancer [51], gastrointestinal stromal tumor [52] and AML [32]. Similarly, KIT expression was obviously increased in AML in this study. Mounting researches have verified that KIT usually exerts its role in mediating the AML progression by interacting with miRNAs. MiR-193b hampers cell proliferation by targeting KIT in AML [53]. MiR-137 inhibits KIT expression to attenuate proliferation and induce apoptosis of AML cells [33]. Notably, lncRNA HOTAIR plays an oncogenic role via decreasing miR-193a and further regulating KIT in AML cells [54]. Here, KIT was a target of miR-221-3p, the expression of KIT and miR-221-3p was inversely correlated. We suspect that miR-221-3p involves in the development of AML by targeting KIT. Additionally, the KIT expression was positively related to TUG1 in AML. Considering the interaction between TUG1 and miR-221-3p, we hypothesize that TUG1 may elevate KIT expression by inhibiting miR-221-3p in AML. Here, feedback approach showed that KIT overexpression mitigated the inhibitory effect on cell viability, as well as weakened the promoting effect on cell apoptosis caused by TUG1 silencing in AML. To sum up, we suggest that TUG1 silencing inhibits viability and elevates apoptosis of AML cells by regulating miR-221-3p/KIT axis.
In conclusion, we found that TUG1 expression was up-regulated in AML patients and AML cells. Silencing of TUG1 inhibited KIT expression by targeting miR-221-3p. TUG silencing may reduce viability and promote apoptosis of AML cells by regulating miR-221-3p/KIT axis. Our research may give a potential therapeutic target for AML.