
Abstract
PURPOSE:
Acute myeloid leukemia (AML) is a type of hematologic malignancy. This study was attempt to explore the effect of long noncoding RNA GAS6 antisense RNA1 (GAS6-AS1) on pediatric AML and the regulation mechanisms.
METHODS:
GAS6-AS1, microRNA-370-3p (miR-370-3p), and Tetraspanin3 (TSPAN3) expression in bone marrow (BM) tissues and cells was determined by qRT-PCR. The correlation between GAS6-AS1 and clinicopathological features of pediatric patients with AML was assessed. In vitro, viability and migration and invasion of AML cells were evaluated via MTT and transwell assays, respectively. Interactions among GAS6-AS1, miR-370-3p, and TSPAN3 were revealed by dual-luciferase reporter assays. Western blot was applied to confirm the protein expression of TSPAN3.
RESULTS:
GAS6-AS1 and TSPAN3 expression was elevated in BM tissues of pediatric patients with AML and AML cells, but miR-370-3p expression was reduced. GAS6-AS1 expression was positively related to French-American-British (FAB) classification in pediatric patients with AML. In vitro, GAS6-AS1 deficiency restrained the viability, migration, and invasion of AML cells. Additionally, GAS6-AS1 mediated miR-370-3p expression indeed and TSPAN3 was identified as a target of miR-370-3p. Furthermore, miR-370-3p overexpression repressed the protein expression of TSPAN3. The feedback experiments demonstrated that miR-370-3p inhibition or TSPAN3 overexpression mitigated the suppressive effect of sh-GAS6-AS1 on the tumorigenesis of AML cells.
CONCLUSION:
GAS6-AS1 silencing restrained AML cell viability, migration, and invasion by targeting miR-370-3p/TSPAN3 axis, affording a novel therapeutic target for pediatric AML.
Introduction
Acute myeloid leukemia (AML) is a common malignant and aggressive hematologic tumors [1]. Owing to extremely diminished healthy blood cells, AML patients generally suffer from severe infection, easy bleeding, and anemia [2]. Pediatric AML accounts for approximately 30% of pediatric leukemia in China [3]. For decades, marrow transplant and intensive chemotherapy are considered to be the main treatment options for AML patients [4]. Although the overall survival and complete remission rates of pediatric patients with AML exceeds 60 and 90 %, the cure rates for many subtypes of pediatric AML are still very low [5]. Thus, it is essential to explore the pediatric AML pathogenesis and identify novel therapeutic targets.
Long noncoding RNAs (lncRNAs) are involved in various hematological malignancies, including multiple myeloma [6], acute lymphoblastic leukemia [7], and AML [8]. LncRNA GAS6 antisense RNA1 (GAS6-AS1) has been shown to participate in diverse cancers. For instances, GAS6-AS1 accelerates the tumorigenesis of gastric cancer through attenuating GAS6 expression [9]. GAS6-AS1 regulates microRNA-585 (miR-585) to elevate EIF5A2 expression, enhancing viability and preventing apoptosis of hepatocellular carcinoma cells [10]. However, the underlying mechanism of GAS6-AS1 in the AML progression remains unclear.
Some miRNAs have anti-cancer function in AML. MiR-34a inhibits autophagy and promotes apoptosis via regulating HMGB1 in AML cells [11]. MiR-23a-3p inhibits the tumorigenesis of AML cells via modulating SMC1A [12]. MiR-650 exerts anti-AML activity via attenuating cell proliferation by Gfi1 targeting [13]. Notably, lncRNA TUG1 deficiency decreases AML cell viability and metastasis through elevating miR-370-3p [14]. However, whether GAS6-AS1 can mediate AML through interacting with miR-370-3p remains undefined.
Tetraspanin3 (TSPAN3), a member of tetraspanin superfamily, takes part in the development of solid tumors and hematologic malignancies [15]. TSPAN3 acts as a therapeutic target for tumor. Inhibition of TSPAN3 markedly reduces proliferation of colon cancer cell [16]. Interestingly, miR-139-5p constrains the leukemogenesis via suppressing TSPAN3 expression in AML cells [17]. However, the potential regulatory mechanism of GAS6-AS1 in AML, regulated by miR-370-3p/TSPAN3 axis, remains unknown.
Herein, we assessed the expression and function of GAS6-AS1 in pediatric AML. The relationship between GAS6-AS1 and miR-370-3p was confirmed. Meanwhile, we verified whether TSPAN3 is a downstream target of miR-370-3p. A GAS6-AS1/miR-370-3p/TSPAN3 regulatory axis in pediatric AML was discovered. Our study may reveal a new therapeutic target for pediatric AML.
Materials and methods
Sample collection
Fifty-nine pediatric patients with AML at our hospital from October 2016 to January 2019 were recruited in our study. Simultaneously, thirty non-hematologic malignancy patients who underwent bone marrow (BM) biopsy acted as Control. BM tissues were gained during the biopsy from Control and AML patients. We separated the AML patients cohort based on the median GAS6-AS1 exp-ression into low GAS6-AS1 (n = 29) and high GAS6-AS1 (n = 30) groups. This study was permitted by our hospital ethics committee, and informed consents were obtained from each participants and their guardian.
Cell culture
HS-5 human BM stromal cell line and four human AML cell lines (HL-60, U937, KG-1, and NB4) were obtained from the American Type Culture Collection (Manassas, VA, USA). The cells were cultured in Dulbecco’s Modified Eagle Medium (DMEM, Invitrogen, Carlsbad, CA, USA) with 10% fetal bovine serum (FBS, Invitrogen) at 37°C containing 5% CO2.
Cell transfection
The short hairpin (sh)-GAS6-AS1, sh-GAS6-AS1#2, sh-negative control (NC), miR-370-3p mimics, miR-370-3p inhibitor, miR-NC, inhibitor NC, pcDNA3.1 (pcDNA)-TSPAN3, and pcDNA-NC were gained from Genepharma (Shanghai, China). HL-60 and KG-1 cells grown to 75% confluence were transfected or co-transfected with the aforementioned agents using Lipofectamine 3000 (Invitrogen). Cells without transfection acted as Blank.
Quantitative real time polymerase chain reaction (qRT-PCR) and western blot
We implemented qRT-PCR and western blot as previously described [18]. The primers were illustrated in Table 1. GAPDH, U6, and β-actin were used to normalize the expression of GAS6-AS1, miR-370-3p, and TSPAN3, respectively. The antibodies (Sigma, St. Louis, MO, USA) of western blot included anti-TSPAN3 primary antibody (1:500, SAB2700939), anti-α-tubulin antibody (1:4000, T5168), and HRP-conjugate secondary antibody (1:5000, 12–348). The immunoblots were measured via enhanced chemiluminescence, and quantified through ImageLab software (Bio-Rad, Hercules, CA, USA).
Primers sequences
Primers sequences
HL-60 and KG-1 cells were seeded into 96-well plates at a density of 2×103 cells/well and cultured with 5% CO2 for 0, 24, 48, 72, and 96 h at 37°C. Cell viability was assessed using the MTT cell proli-feration assay kit (Sigma).
Transwell assay
Migration assay was performed with transwell chamber (8 nm, Corning Inc., Corning, NY, USA). The transfected or untransfected cells were seeded into 6-well plate. Next, 1×105 cells of each sample were seeded into the upper chamber with serum-free DMEM. DMEM containing 10% FBS was added into the lower chamber. After incubation for 24 hours at 37°C, cells were fixed with 4% paraformaldehyde (Sigma) and stained with 0.1% crystal violet for 30 min followed by washing with PBS 3 times. For invasion assay, matrigel (Sigma) was coated in the transwell.
Dual-luciferase reporter assay
The potential binding sites of GAS6-AS1 and miR-370-3p or miR-370-3p and TSPAN3 were predicted by Starbase or TargetScan, respectively. GAS6-AS1 and TSPAN3 with WT or MUT miR-370-3p-binding sites were generated and fused to the psiCHECK-2 vectors (Ke Lei Biological Technology, Shanghai, China). HL-60 and KG-1 cells were co-transfected with above luciferase vectors and miR-NC or miR-370-3p mimics using Lipofectamine 3000 (Invitrogen).
Statistical analysis
Statistical data was implemented on GraphPad Prism 7.0 (GraphPad Software) and were displayed as mean±standard deviation. The differences between two groups or among multiple groups were assessed by Student’s t-test or one-way ANOVA followed by Tukey’s post-hoc test. The correlation significance was confirmed by Pearson correlation analysis. A P-value <0.05 was considered statistically significant.
Results
GAS6-AS1 expression was enhanced in AML patients
To explore whether GAS6-AS1 is differently expressed between AML patients and Control, qRT-PCR was performed. As depicted in Fig. 1A, GAS6-AS1 expression in BM tissues was clearly higher in AML patients compared to Control. As illustrated in Table 2, GAS6-AS1 expression had obviously correlation with French-American-British (FAB) classification in AML patients (P < 0.01). Additionally, the GAS6-AS1 expression was dramatically up-regulated in HL-60, U937, KG-1, and NB4 cells compared to the HS-5 cells (P < 0.01, Fig. 1B). HL-60 and KG-1 cells were chosen for the function experiments of GAS6-AS1 because of high GAS6-AS1 expression.
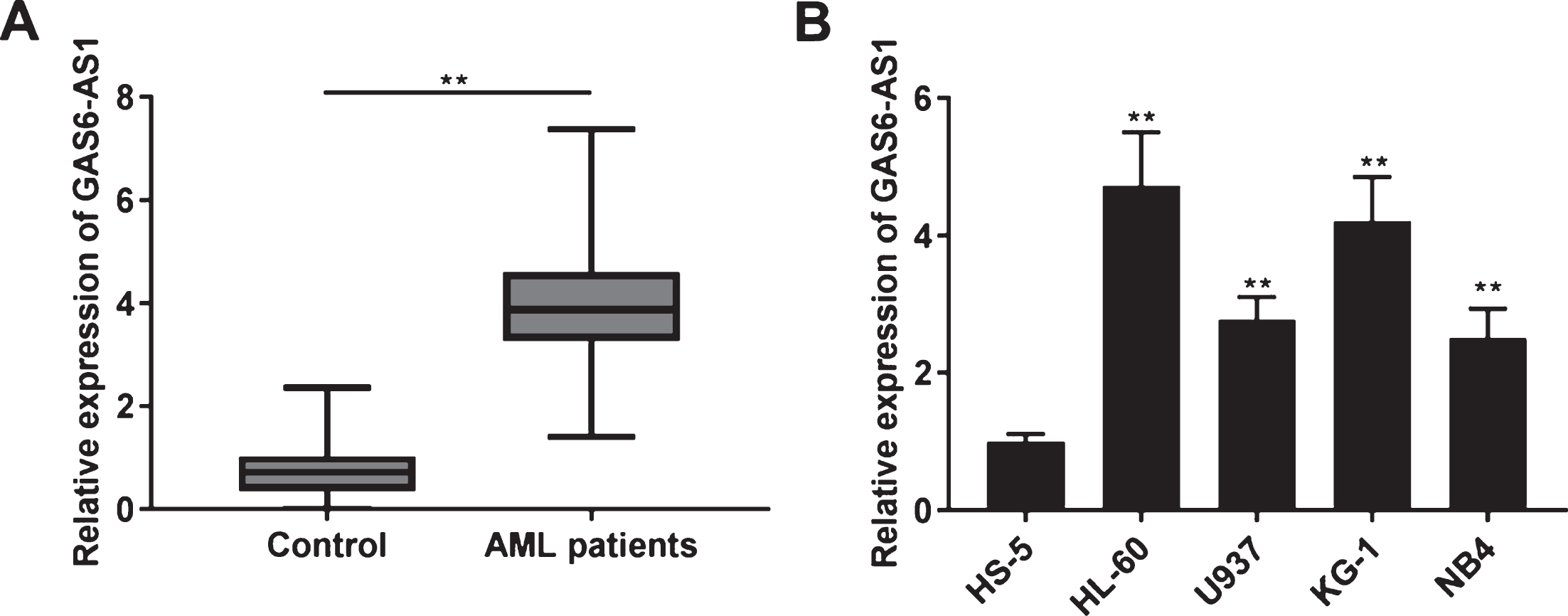
GAS6-AS1 expression was enhanced in bone marrow (BM) tissues of acute myeloid leukemia (AML) patients. (A) The expression of GAS6-AS1 in BM tissues of AML patients and Control was measured by qRT-PCR. **P < 0.01 vs. Control; (B) QRT-PCR was implemented to measure the expression of GAS6-AS1 in HS-5, HL-60, U937, KG-1, and NB4 cells. **P < 0.01 vs. HS-5.
Correlation between GAS6-AS1 expression and clinicopathological features in pediatric patients with acute myeloid leukemia
Note: **P < 0.01. FAB classification, French-American-British classification.
GAS6-AS1 expression was visibly silenced in HL-60 and KG-1 cells by transfection of sh-GAS6-AS1 and sh-GAS6-AS1#2 (P < 0.01, Fig. 2A). Sh-GAS6-AS1 was chosen for following experiments because of its high silence efficiency. MTT assay revealed that sh-GAS6-AS1 markedly declined the HL-60 and KG-1 cell viability after 96 h of culture (P < 0.01, Fig. 2B). Furthermore, migration and invasion of HL-60 and KG-1 cells were considerably suppressed following GAS6-AS1 silencing (P < 0.01, Fig. 2C and D).
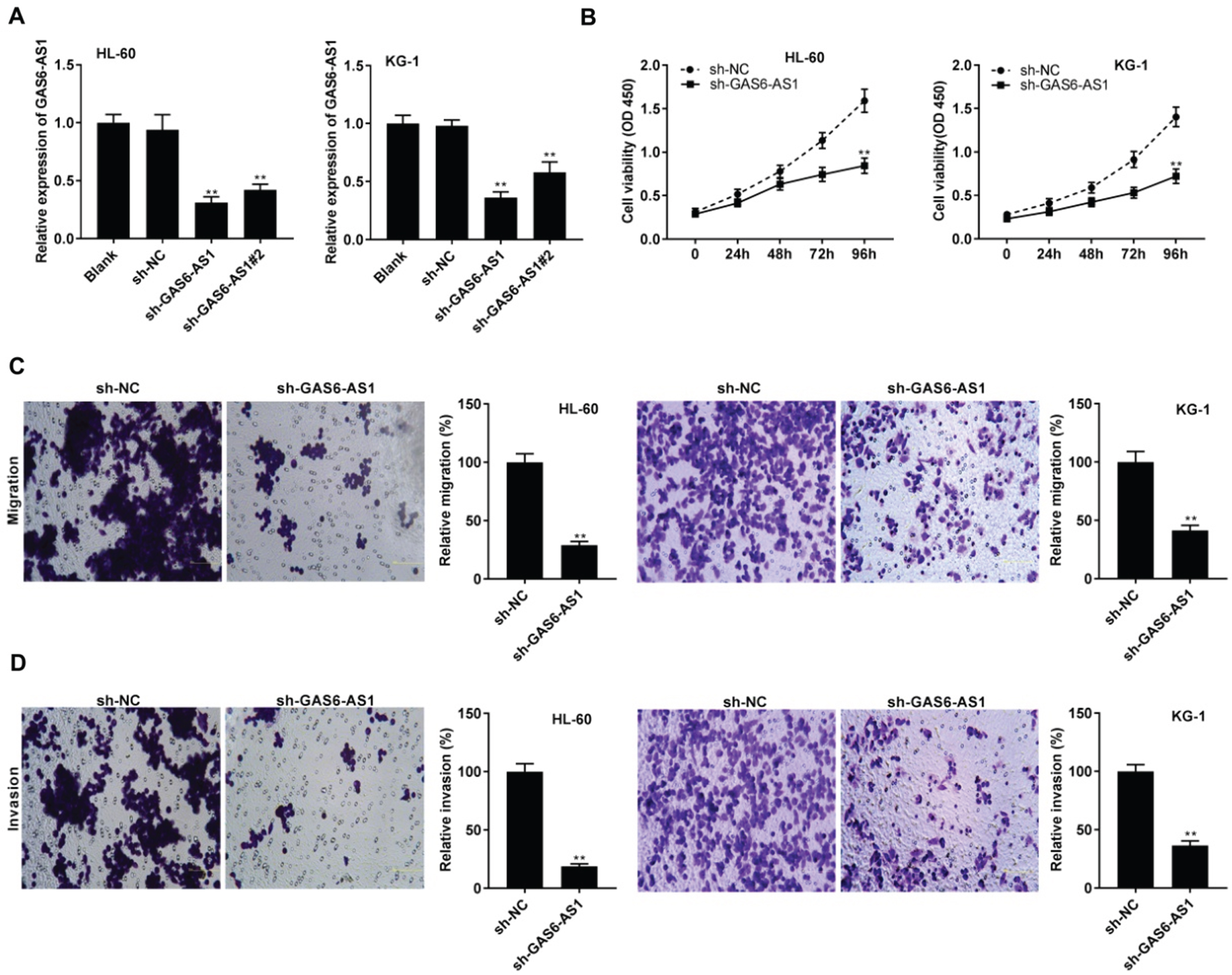
GAS6-AS1 silencing inhibited tumorigenesis of acute myeloid leukemia (AML) cells. (A) The transfection efficiency of sh-NC, sh-GAS6-AS1, and sh-GAS6-AS1#2 in HL-60 and KG-1 cells was assessed by qRT-PCR. **P < 0.01 vs. sh-NC; (B) The viability of HL-60 and KG-1 cells was detected by MTT assay. **P < 0.01 vs. sh-NC; (C and D) The migration and invasion of HL-60 and KG-1 cells were measured by transwell assay. **P < 0.01 vs. sh-NC.
Starbase was used to predict the binding site for GAS6-AS1 on the 3’ UTR of miR-370-3p (Fig. 3A). GAS6-AS1 silencing visibly enhanced the miR-370-3p expression in HL-60 and KG-1 cells (P < 0.01, Fig. 3B). MiR-370-3p mimics dramatically repressed the luciferase activity of WT GAS6-AS1 reporter vector in HL-60 and KG-1 cells (P < 0.01, Fig. 3C). The miR-370-3p expression in BM tissues was clearly inhibited in AML patients compared to Control (P < 0.01, Fig. 3D). The expression of GAS6-AS1 and miR-370-3p was conversely correlated in AML patients (r =–0.3826, p = 0.0028, Fig. 3E). Moreover, miR-370-3p expression was considerably attenuated in HL-60, U937, KG-1, and NB4 cells compared to HS-5 cells (P < 0.01, Fig. 3F). HL-60 and KG-1 cells were chosen for the function experiments of miR-370-3p because of low miR-370-3p expression.

MiR-370-3p was targeted by GAS6-AS1. (A) Starbase exhibited the predicted binding site between GAS6-AS1 and miR-370-3p; (B) The expression of miR-370-3p was up-regulated by the transfection of sh-GAS6-AS1 in HL-60 and KG-1 cells. **P < 0.01 vs. sh-NC; (C) Dual-luciferase reporter assay was performed to confirm the relative luciferase activity in HL-60 and KG-1 cells. **P < 0.01 vs. miR-NC; (D) QRT-PCR was performed to obtain the expression of miR-370-3p in bone marrow (BM) tissues of acute myeloid leukemia (AML) patients and Control. **P < 0.01 vs. Control; (E) The expression of GAS6-AS1 was negatively correlated with miR-370-3p; (F) The expression of miR-370-3p in HS-5, HL-60, U937, KG-1, and NB4 cells was detected by qRT-PCR. **P < 0.01 vs. HS-5.
MiR-370-3p was overexpressed by transfecting with miR-370-3p mimics in HL-60 and KG-1 cells (P < 0.01, Fig. 4A). MiR-370-3p overexpression could obviously decrease HL-60 and KG-1 cell viability after 72 and 96 h of culture (P < 0.01, Fig. 4B). Furthermore, the migration and invasion of HL-60 and KG-1 cells were significantly reduced following miR-370-3p up-regulation (P < 0.01, Figs. 4C and D).
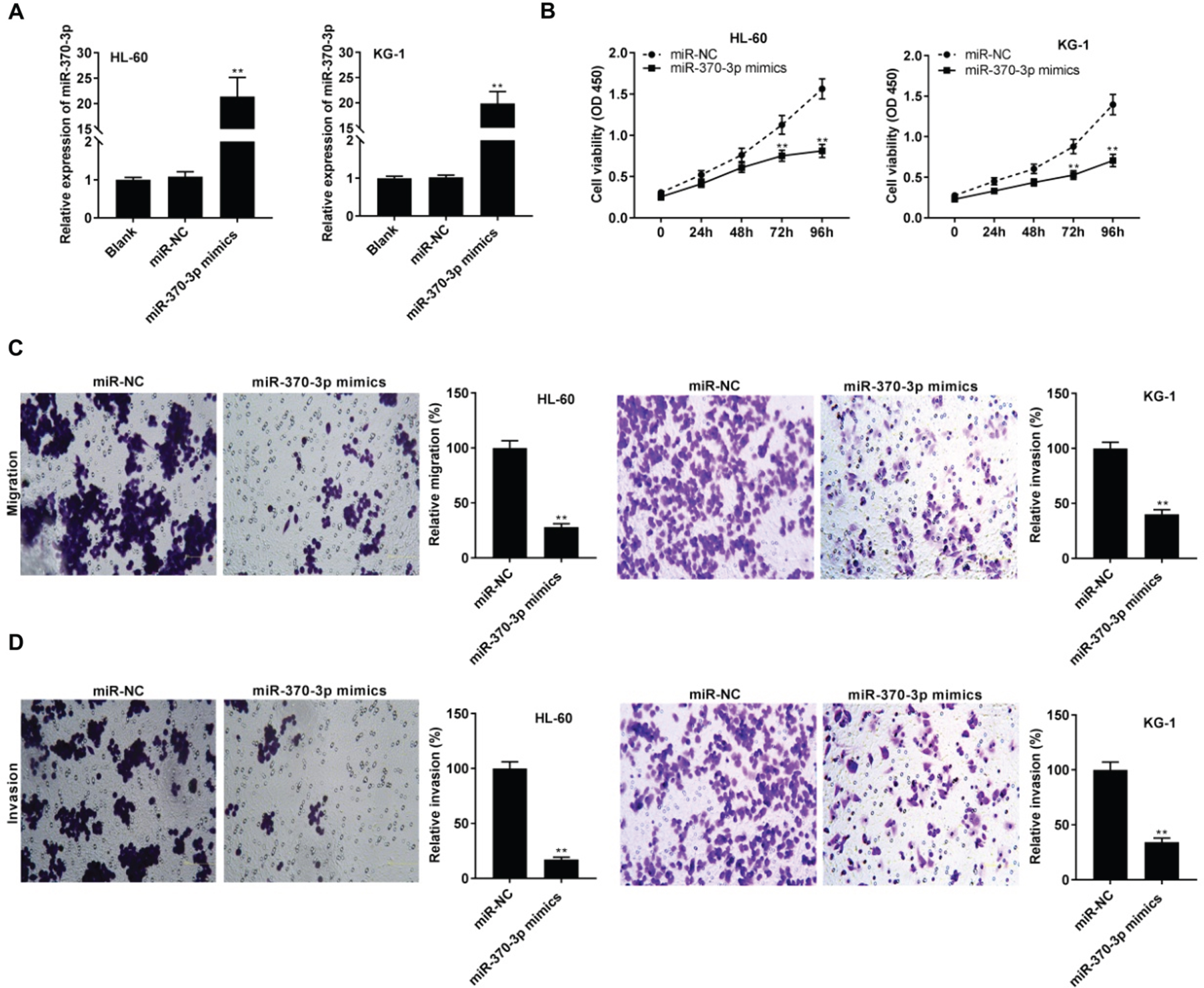
MiR-370-3p inhibited the tumorigenesis of acute myeloid leukemia (AML) cells. (A) The transfection efficiency of miR-NC and miR-370-3p mimics was evaluated using qRT-PCR in HL-60 and KG-1 cells. **P < 0.01 vs. miR-NC; (B) The viability of HL-60 and KG-1 cells was measured by MTT assay. **P < 0.01 vs. miR-NC; (C and D) The migration and invasion of HL-60 and KG-1 cells were detected by transwell assay. **P < 0.01 vs. miR-NC.
TargetScan was used to predict the binding site for miR-370-3p on the 3’ UTR of TSPAN3 (Fig. 5A). MiR-370-3p overexpression clearly attenuated the relative luciferase activity of the reporter with TSPAN3 WT in HL-60 and KG-1 cells (P < 0.01, Fig. 5B). MiR-370-3p overexpression visibly supp-ressed the TSPAN3 protein expression in HL-60 and KG-1 cells (P < 0.01, Fig. 5C). Moreover, TSPAN3 expression in BM tissues was higher in AML patients than that in Control (P < 0. 01, Fig. 5D). The expression of miR-370-3p and TSPAN3 was conversely correlated in AML patients (r =–0.3208, P = 0.0132, Fig. 5E). Interestingly, a positive correlation between GAS6-AS1 and TSPAN3 expression in AML patients was appeared (r = 0.3932, P = 0.0021, Fig. 5F). Compared with the HS-5 cells, TSPAN3 expression was considerably up-regulated in HL-60, U937, KG-1, and NB4 cells (P < 0.01, Fig. 5G).
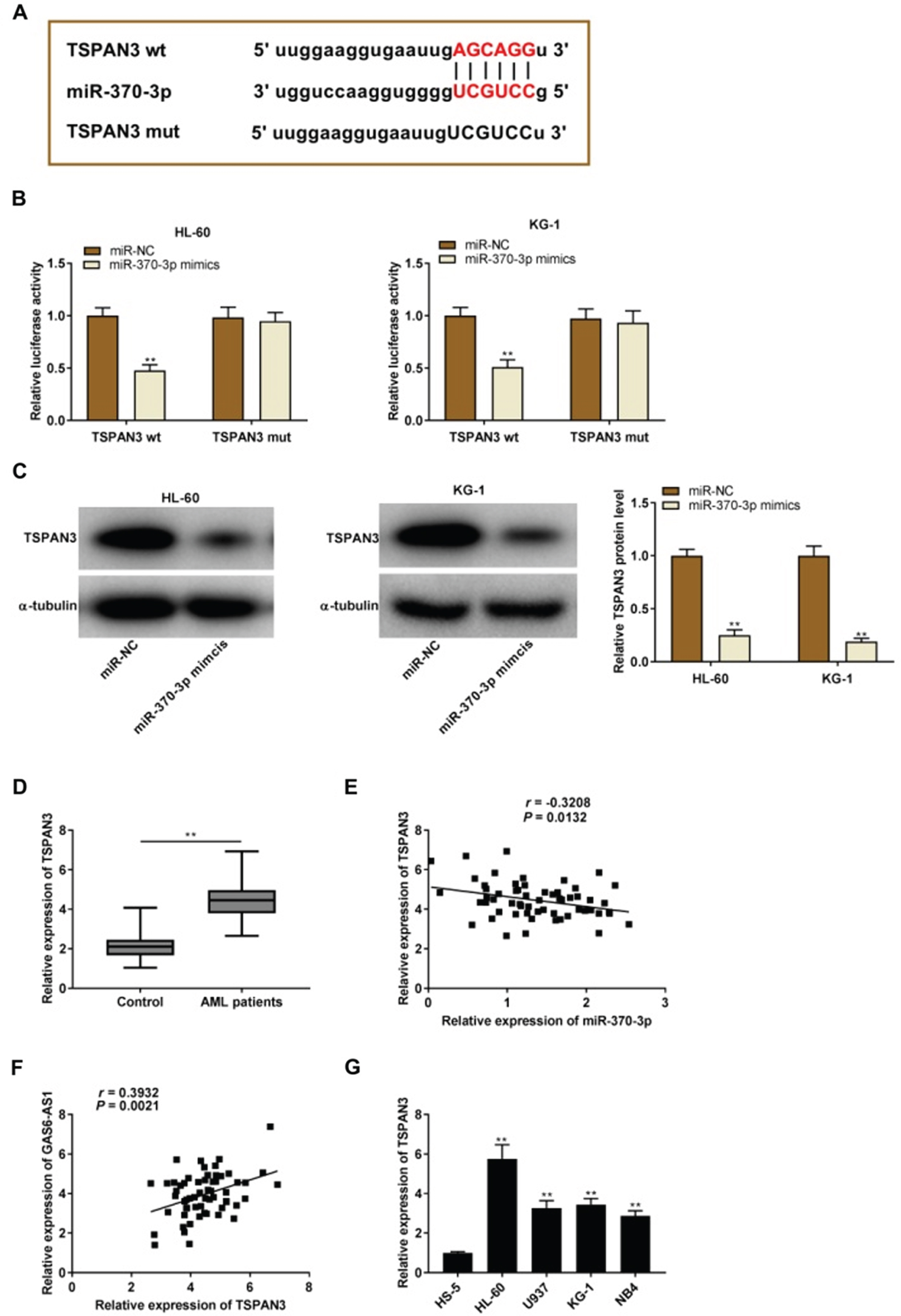
TSPAN3 was directly targeted by miR-370-3p. (A) TargetScan displayed the predicted binding site between miR-370-3p and TSPAN3; (B) Relative luciferase activity in HL-60 and KG-1 cells was detected by dual-luciferase reporter assay. **P < 0.01 vs. miR-NC; (C) The protein expression of TSPAN3 in HL-60 and KG-1 cells was tested by western blot. **P < 0.01 vs. miR-NC. (D) QRT-PCR was used to measure the expression of TSPAN3 in bone marrow (BM) tissues of acute myeloid leukemia (AML) patients and Control. (E) The expression of miR-370-3p was negatively correlated with TSPAN3 in BM tissues of AML patients; (F) The expression of GAS6-AS1 was positively correlated with TSPAN3 in BM tissues of AML patients; (G) QRT-PCR was performed to detect the expression of TSPAN3 in HS-5, HL-60, U937, KG-1, and NB4 cells. **P < 0.01 vs. HS-5.
The TSPAN3 expression was markedly enhanced following transfection with pcDNA-TSPAN3 in HL-60 cells (P < 0.01, Fig. 6A). Transfection of miR-370-3p inhibitor dramatically repressed miR-370-3p expression in HL-60 cells (P < 0.01, Fig. 6B). To verify whether GAS6-AS1 regulated the miR-370-3p/TSPAN3 axis in AML, the feedback experiments were performed in HL-60 cells. As illustrated in Fig. 6C-E, GAS6-AS1 silencing visibly reduced the viability, migration, and invasion of HL-60 cells (P < 0.01). MiR-370-3p deficiency or TSPAN3 up-regulation mitigated the reduction effect of GAS6-AS1 silencing on the tumorigenesis of HL-60 cells (P < 0.01).
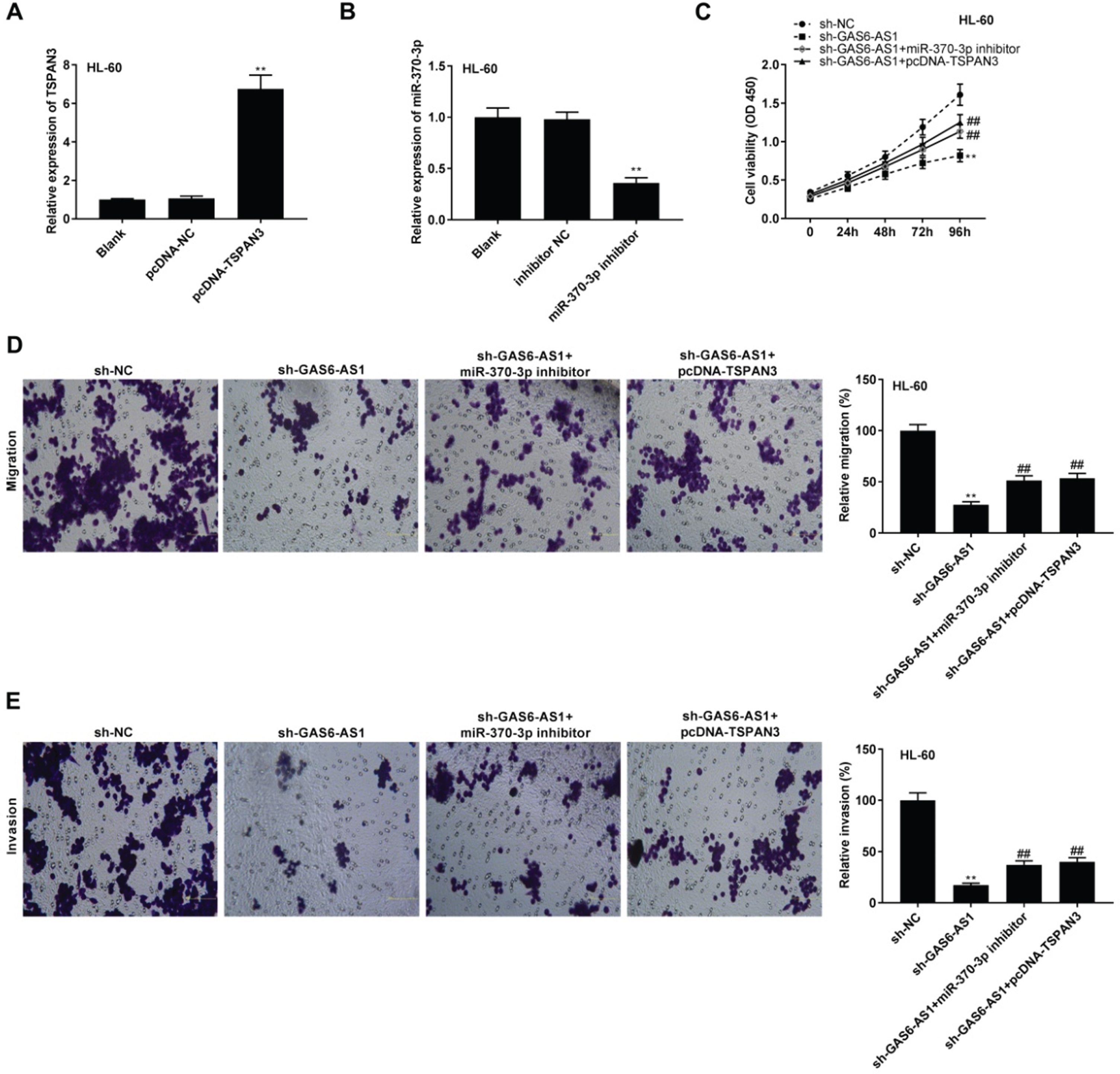
GAS6-AS1 silencing inhibited tumorigenesis of acute myeloid leukemia (AML) cells by regulating the miR-370-3p/TSPAN3 axis. (A) The transfection efficiency of pcDNA-NC and pcDNA-TSPAN3 in HL-60 cells was evaluated by qRT-PCR. **P < 0.01 vs. pcDNA-NC; (B) The transfection efficiency of inhibitor NC and miR-370-3p inhibitor in HL-60 cells was assessed by qRT-PCR. **P < 0.01 vs. inhibitor NC; (C-E) MiR-370-3p inhibition or TSPAN3 overexpression reversed the inhibitory effects of GAS6-AS1 silencing on viability, migration, and invasion of HL-60 cells. **P < 0.01 vs. sh-NC; # #P < 0.01 vs. sh-GAS6-AS1.
Up-regulation of lncRNAs, such as HOXB-AS3 [19], LINP1 [20], and SNHG5 [21] have been proved to display a pivotal influence on AML pathogenesis. Here, GAS6-AS1 expression was clearly enhanced in AML patients and AML cells. Additionally, GAS6-AS1 was found to be related to FAB classification in AML patients, which was similar to previous studies. High lncRNA-CRNDE expression is positively associated with classification in AML patients [22]. LncRNA SNHG5 overexpression usually occurs in AML patients with advanced FAB classification [23]. High expression of lncRNA CCDC26 in AML patients is considerably related to risk stratification [24]. Above all, we conjecture that GAS6-AS1 may be correlated with AML development. Inhibition of lncRNAs suppresses the AML pathogenesis. LncRNA H19 knockdown exerts anti-growth and pro-apoptotic functions in AML cells [25]. LncRNA HOXA-AS2 silencing attenuates tumorigenesis of AML through hindering chemoresistance [26]. GAS6-AS1 exerts carcinogenic function in certain tumors. GAS6-AS1 interacts with GAS6, promoting gastric cancer cell growth and reducing cell cycle arrest [9]. GAS6-AS1 acts as an oncogenic lncRNA that promoted hepatocellular carcinoma tumorigenesis via mediated the EIF5A2 expression by sponging miR-585 [10]. In this study, GAS6-AS1 silencing markedly hampered the viability, migration, and invasion of AML cells. In summary, we suggest that GAS6-AS1 deficiency may restrain the tumorigenesis of AML.
Prior studies have revealed that lncRNAs regulate miRNAs, exerting its oncogenic effects in AML. For instances, lncRNA H19 enhances the AML cell proliferation via repressing miR-29a-3p expression [27]. LncRNA ANRIL potentiates AML cell invasion and migration by decreasing miR-34a expression [28]. LncRNA KCNQ1OT1 deficiency up-regulates miR-326 to repress AML cell growth and induce cell death [29]. In this study, sh-GAS6-AS1 enhanced miR-370-3p in AML, indicating that GAS6-AS1 may be involved in AML by similar mechanisms above. MiR-370-3p exerts tumor suppressor effect on diverse cancers. For instances, miR-370-3p attenuates glioma cell proliferation via regulating β-catenin [30]. MiR-370-3p increases growth and decreases apoptosis of chronic myelogenous leukaemia cell by inhibiting PDLIM1 [31]. Importantly, lncRNA TUG1 interacts with miR-370-3p to induce the malignant behaviors of AML cells [14]. In this study, miR-370-3p expression was inhibited and conversely associated with GAS6-AS1 expression in BM tissues of AML patients, indicating that GAS6-AS1 may exert cancer-promoting function by decreasing miR-370-3p in AML cells. Moreover, we revealed that miR-370-3p repressed the viability, migration, and invasion of AML cells, and the miR-370-3p deficiency weakened the anti-tumor effect of GAS6-AS1 deficiency in AML cells. To sum up, we indicate that GAS6-AS1 silencing may retard tumorigenesis by enhancing miR-370-3p in AML cells.
TSPAN3 can serve as an oncogenic gene, and TSPAN3 expression is up-regulated in diverse tumors, such as desmoid tumors [32], colon cancer [16], and esophageal cancer [33]. Here, we discovered that TSPAN3 expression was obviously up-regulated in BM tissues of AML patients. It has been demonstrated that TSPAN3 partakes in leukemia progression. For instances, TSPAN3 knockdown attenuates leukemia stem cell self-renewal and elevates survival in mouse models of acute myelogenous leukemia [34]. MiR-139-5p overexpression results in suppression in cell growth of AML by inhibiting TSPAN3 expression [17]. Importantly, lncRNA KCNQ1OT1 promotes the progression and chemoresistance in AML via modulating the miR-193a-3p/TSPAN3 axis [35]. In this study, TSPAN3 was a target of miR-370-3p, the expression of miR-370-3p and TSPAN3 was conversely correlated. We speculate that miR-370-3p modulates AML pathogenesis via regulating TSPAN3. Furthermore, the TSPAN3 expression was positively related to GAS6-AS1. Given that the targeted relationship between miR-370-3p and GAS6-AS1, we hypothesize that GAS6-AS1 may mediate TSPAN3 through sponging miR-370-3p. Encouragingly, the feedback data discovered that TSPAN3 up-regulation mitigated the anti-tumor effect of sh-GAS6-AS1 on AML cells. Taken together, we suggest that GAS6-AS1 silencing may exert its anti-tumor function via targeting miR-370-3p/TSPAN3 axis in AML.
In conclusion, GAS6-AS1 expression was elevated in BM tissues of pediatric patients with AML and AML cells. GAS6-AS1 silencing attenuated the AML tumorigenesis by targeting miR-370-3p/TSPAN3 axis in vitro. Our research may provide a potential therapeutic target for AML.