
Abstract
In the present study, the progressive alteration of cognition and the mechanisms of reduction in long-term potentiation (LTP) in spontaneous obese KK-Ay type 2 diabetic mice were investigated. In the study, 3-, 5-, and 7-month-old KK-Ay mice were used. The results indicated that KK-Ay mice showed cognitive deficits in the Morris water maze test beginning at the age of 3 months. LTP was significantly impaired in KK-Ay mice during whole study period (3 to 7 months). The above deficits were reversible at an early stage (3 to 5 months old) by diet intervention. Moreover, we found the underlying mechanisms of LTP impairment in KK-Ay mice might be attributed to abnormal phosphorylation or expression of postsynaptic glutamate receptor subunits instead of alteration of basal synaptic transmission. The expression levels of NR1, NR2A, and NR2B subunits of N-methyl-d-aspartate receptors (NMDARs) were unchanged while the Tyr-dependent phosphorylation of both NR2A and NR2B subunits were significantly reduced in KK-Ay mice. The level of p-Src expression mediating this process was decreased, and the level of αCaMKII autophosphorylation was also reduced. Meanwhile, the GluR1 of α-amino-3-hydroxy-5-methyl-4-isoxazole propionic acid receptors (AMPARs) was decreased, and GluR2 was significantly increased. These data suggest that deficits in synaptic plasticity in KK-Ay mice may arise from the abnormal phosphorylation of the NR2 subunits and the alteration of subunit composition of AMPARs. Diet intervention at an early stage of diabetes might alleviate the cognitive deficits and LTP reduction in KK-Ay mice.
Keywords
INTRODUCTION
Type 2 diabetes mellitus (T2DM) is not only associated with the cardiovascular and peripheral nervous systems but is also a major risk factor of the central nervous system (CNS), so-called “diabetic encephalopathy (DE)” [1]. One of the most prominent clinical manifestations of DE is moderate cognitive dysfunction in humans. A growing body of evidence indicated an association between T2DM and dementia as well as Alzheimer’s disease (AD) [2–5]. Epidemiological data reveals that T2DM increases the long-term risk of dementia by nearly 2-fold, and one in ten cases of dementia in the world may be attributable to the effects of T2DM [6]. In view of the worldwide increase in T2DM incidence, DE has become a serious medical and social issue and has gained great public concern in recent years. Hence it is important to reveal the pathological mechanisms in DE and to identify the targets that can reduce or abrogate this process.
A growing body of evidence points to a deficit in hippocampal synapses in diabetes [1, 8]. Impairment of long-term potentiation (LTP) might be linked to the spatial learning and memory impairment in diabetic rodents. Previous studies showed that hippocampal LTP was severely impaired in diabetic rodent models [9]. It is well known that activity-dependent synaptic plasticity is mainly mediated by ionotropic glutamate receptors: α-amino-3-hydroxy- 5-methyl-4-isoxazole propionic acid receptors (AMPARs) and N-methyl-d-aspartate receptors (NMDARs). Studies underlying the LTP deficits in streptozotocin-induced diabetic (STZ-DM) rats have addressed abnormal reorganization of the glutamate receptor complexes [8]. However, previous studies have led to controversial results depending on the age of DM onset [10, 11]. Furthermore, in STZ-DM rats, a model induced by diabetogenic agents, it is difficult to mimic T2DM seen in humans, which is a heterogeneous and multifactorial disease [12]. To further explore the link between the duration of hyperglycemia and cognitive deficits, the obese T2DM model of KK-Ay mice with different ages was used to elucidate the possible roles played by glutamate receptorsin DE.
The KK-Ay mouse, a typical model of obese insulin-resistant T2DM based on a polygenic background, was established by transferring the yellow obese gene (Ay allele) into the Kuo Kondo (KK) mouse. The KK mouse comes from the Japanese KK mouse, a strain inbred for large body size [13]. The mechanism for obesity caused by the Ay allele is thought to inhibit the action of α-melanocyte-stimulating hormone signal [14, 15]. KK mice with Ay mutation can spontaneously develop severe obesity, hyperglycemia, hypertriglyceridemia, and hyperinsulinemia [16, 17]. It is a useful model for investigating the pathology of obesity- associated diabetes mimicking human obesity and T2DM [17, 18]. This animal model has been recently applied in pharmacological studies.
In the present study, 3-, 5-, and 7-month-old KK-Ay mice were used. We observed the effects of obese T2DM on hippocampus-dependent spatial learning, memory, and LTP reduction. The underlying mechanisms of LTP impairment during the development of the course of diabetes were also investigated, including pre- and post-synaptic transmission and the expressions of NMDARs and AMPARs. In addition, we observed whether the cognitive deficits in KK-Ay mice could be reversed by dietary intervention.
MATERIALS AND METHODS
Animals and treatment
Male KK/Upj-Ay/J mice (Beijing HFK Bioscience Co. Ltd.) were fed with high fat diet until the experiments were carried out at 3, 5, and 7 months. Age-matched male C57BL/6J mice (WT mice) were generally employed as non-diabetic control for the KK-Ay mice. Dietary intervention group: the male KK-Ay mice were given a normal diet (low fat) for 2 month beginning at 3 months of age. Mice were group-housed in an animal room at a constant temperature of 23±1°C, and maintained at a 12-h light/dark cycle per day. All animals were given food and water ad libitum. All experiments were approved and performed in accordance with the institutional guidelines of the Experimental Animal Center of the Chinese Academy of Medical Science, Beijing, China.
Morris water maze
The Morris water maze task was used to evaluate the changes in learning and memory in KK-Ay mice [19]. The apparatus consisted of a circular metal pool (120 cm in diameter and 50 cm high), filled with water made opaque by the addition of milk powder. The water temperature was kept at 23±1°C. A translucent acrylic platform (10 cm in diameter), located in the center of the Northwest quadrant during training, was placed 1.5 cm under the surface of the water. There were prominent visible cues around the room. The mouse was gently released with its nose against the wall into the water from one of the four preplanned starting positions (north, south, east, or west). The starting position for each trial was pseudorandomly chosen and counterbalanced across all the experimental groups.
Spatial training of the hidden platform in the water maze was performed for 5 consecutive days. On each day, the mice were allowed two consecutive training trials. Each mouse was allowed to swim for a maximum duration of 120 s in each trial to find the hidden platform. If the mice found the platform within the 120 s, it was allowed to stay on the platform for 30 s. If a mouse failed to find the platform within 120 s, the training was terminated, a maximum score of 120 s was assigned, and the mouse was manually guided to the hidden platform staying for 30 s. The latency to locate the platform were recorded.
On the sixth day, to assess long-term memory consolidation respectively, the platform was removed and the mice, placed into the pool from the quadrant opposite to the training quadrant, were allowed to search for the platform for 120 s (probe test). The time in the platform quadrant and latency time to cross platform location were recorded to measure the spatial memory maintenance without the influence of chance encounters with the platform.
Hippocampal electrophysiology
Mice were killed by decapitation after a short period of inhalation anesthesia with Diethyl ether. Brains were rapidly removed and immersed in ice-cold medium (in mM): Sucrose 213, KCl 3, NaH2PO4 1, MgCl2 5, CaCl2 0.5, NaHCO3 26, and glucose 10. Transverse slices (400 μm-thick slices) for electrophysiological study were prepared from male KK-Ay mouse and their respective controls by using a vibratome (Campden Instruments, Loughsborough, UK) as described [20], and incubated in an artificial cerebrospinal fluid (ACSF) containing (in mM): NaCl 125, KCl 5, NaH2PO4 1.2, MgCl2 1.3, CaCl2 2.6, NaHCO3 26, and glucose 10, saturated with 95% O2/5% CO2 at room temperature for 1 h.
Then, the slices were transferred to the recording chamber and perfused with ACSF at a rate of 2–4 ml/min. Hippocampal CA1 pyramidal neurons were visualized with a 40× water-immersion objective lens under a microscope (Eclipse FN1, Nikon, Japan) equipped with infrared differential interference contrast optics. Extracellular recording were obtained with an EPC10 USB Patch Clamp amplifier (HEKA Elektronik, German). The recording electrode was placed in the stratum radiatum recording field excitatory postsynaptic potentials (fEPSPs). A concentric bipolar stimulation electrode (FHC, USA) was placed the afferent fibers of the stratum radiatum. The signals were filtered at 2 kHz (–3 dB). I–V curve ranging from threshold level to the maximum response level was made prior to each experiment. Next, the stimulus intensity was adjusted to evoke fEPSPs of half maximal amplitude, and was used throughout the measurements. Stimulation frequency was 0.033 Hz. After baseline recordings for 15 min, a high-frequency train of stimuli (100 Hz for 1 s) was applied, and fEPSPs were recorded for another 60 min. The average slope of the fEPSP at baseline was set at 100%, and changes in slope were expressed as percent change from baseline.
Paired-pulse facilitation (PPF) in the CA1 field of the hippocampus was obtained by giving a paired pulse with an interstimulus interval from 20–1000 ms. In the present study, PPF were recorded by giving various interpulse intervals (20, 40, 60, 80, 100, 300, 500, 700, and 900 ms). The facilitation ratio was obtained by computing the ratio of the slope of the second pulse to the slope of the firstslope.
Western blotting analysis
Six to eight mice from each group were decapitated after a short period of inhalation anesthesia with Diethyl ether. Cortex and hippocampus were dissected on ice. The left hemibrain was used for electrophysiological study, and then the right hemibrain was rapidly frozen and stored at –80°C for biochemical analysis. Samples was weighed and homogenized in 10 vol of a lysis buffer (150 mM NaCl, 50 mM Tris, 1 mM EDTA, 1% NP40, 0.5% sodium deoxycholate, 0.1% SDS, 10 mM sodium fluoride, 1 mM sodium orthocanadate, and 1x protease inhibitor cocktail (pH 7.4)) by ultrasonic irradiation. The samples were centrifuged at 12000 r/min for 30 min. Protein concentrations were measured with a BCA Protein Assay kit (Applygen Technologies Inc., Beijing, China). Homogenates (60 μg per lane) were separated on 10% sodium dodecylsulfate (SDS)-polyacrylamide gels, electrophoretically transferred to PVDF membranes, blocked with 5% fat-free milk (fat-free dry milk diluted in TBST) for 1 h, and subsequently the blots were incubated with primary antibodies overnight at 4°C. After being washed with TBST for 5 times, the protein bands were visualized by the application of secondary antibodies (horseradish peroxidase-linked anti-mouse, anti-rabbit or IgG) at room temperature for 1 h. The signals were detected using an enhanced chemiluminescence (ECL) kit, scanned using an LAS3000 Fujifilm imaging system (Fujifilm, Tokyo, Japan) and analyzed by densitometric evaluation using Quantity-One software (Bio-Rad, Hercules, CA, USA). The values were normalized to β-actin intensitylevels.
Statistical analysis
All data were expressed as mean±SEM. LTP recording tests were analyzed by two-way ANOVA with repeated measures to detect differences between groups. Other statistical significance was evaluated via Student’s t-test or one-way ANOVA. A value of p < 0.05 was considered statistically significant.
RESULTS
Changes of the biochemical indexes in KK-Ay mice
All KK-Ay mice, at 3, 5, and 7 months of age, presented with typical metabolic syndrome such as severe obesity, hyperglycemia, hypertriglyceridemia, hypercholesterolemia, and hyperinsulinemia. Body weight, random blood glucose, triglyceride, cholesterol, and insulin in KK-Ay mice were significantly higher than that in WT mice (Fig. 1A-E). These results suggested that KK-Ay mouse used in our study is a reliable diabetic animal model. Furthermore, the expression levels of classical hallmarks of AD, including Aβ, p-Tau (S199), p-Tau (T205), and p-Tau (S404) were significantly increased in 5-month-old KK-Ay mice compared with aged-matched WT mice (Fig. 1F-I). Because, at this age KK-Ay mice show higher incidence and more stable pathologicalcondition.
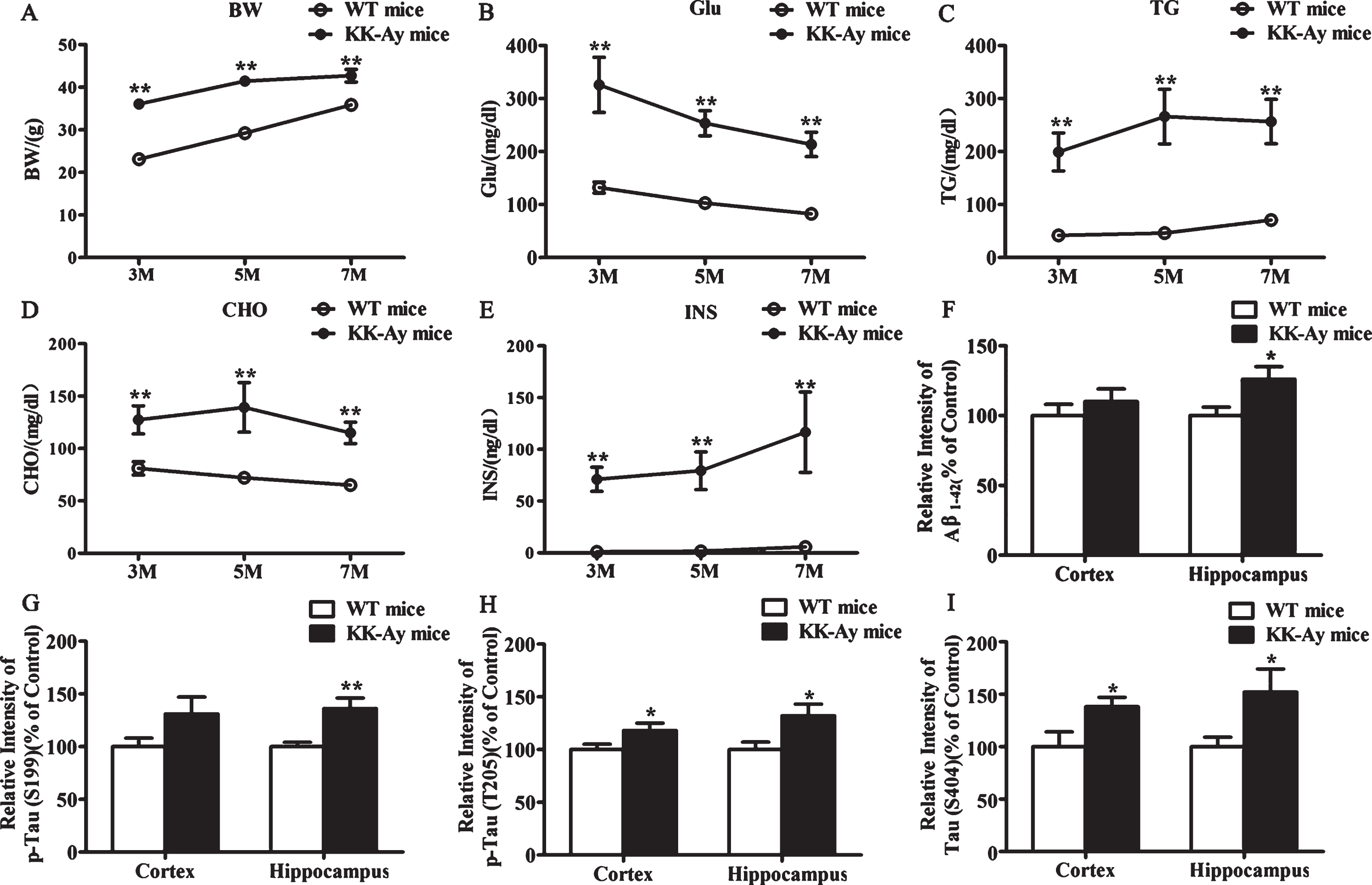
The changes of biochemical indexes in KK-Ay mice. A-E) KK-Ay mice presented typical metabolic syndrome. Body weight (BW), random blood glucose (Glu), cholesterol (CHO), triglycerides (TG), and insulin (INS) were significantly enhanced in 3-, 5-, and 7-month-old KK-Ay mice (n = 11–16 for each group). F-I) Quantitative analysis of the protein expression of Aβ and phosphorylated tau in western blotting. The level of Aβ and phosphorylated tau at Ser199, Thr205, and Ser404 were significantly increased in the hippocampus of 5-month-old KK-Ay mice compared with age-matched WT mice. (n = 8 for each group). *p < 0.05, **p < 0.01 versus WT mice group.
Course of development of spatial learningand memory deficits in KK-Ay mice
The effects of T2DM on the spatial learning and memory were evaluated. The escape latency to find the hidden platform in all mice during the water maze acquisition training is shown in Fig. 2A. All mice effectively improved their spatial learning across the 5-day training period. Compared to the age-matched WT mice, the KK-Ay mice of different diabetic duration all showed longer escape latencies, implying a significant learning deficit in diabetic group (Fig. 2A). The results also demonstrated that the KK-Ay mice had been subjected to cognitive deficits since the age of 3 months. However, the swimming speed of KK-Ay mice was not slower than the WT mice (Fig. 2B), excluding the possibility that the effect of T2DM on spatial learning and memory in KK-Ay mice was attributable to sensorimotor abnormalities.
In the probe trial, to assess the memory deficits of the diabetic mice, the time that an animal crossed the position of the hidden platform at the first time was recorded. The first crossing time of KK-Ay mice showed an increase compared with the age-matched WT mice, and showed a significant difference in the 7-month-old group, indicating the memory deficits in the 7-month-old group was more significant than that in the 3- and 5-month-old group (Fig. 2C). These data suggested the spatial learning and memory deficits in KK-Ay mice were diabetic duration-related.
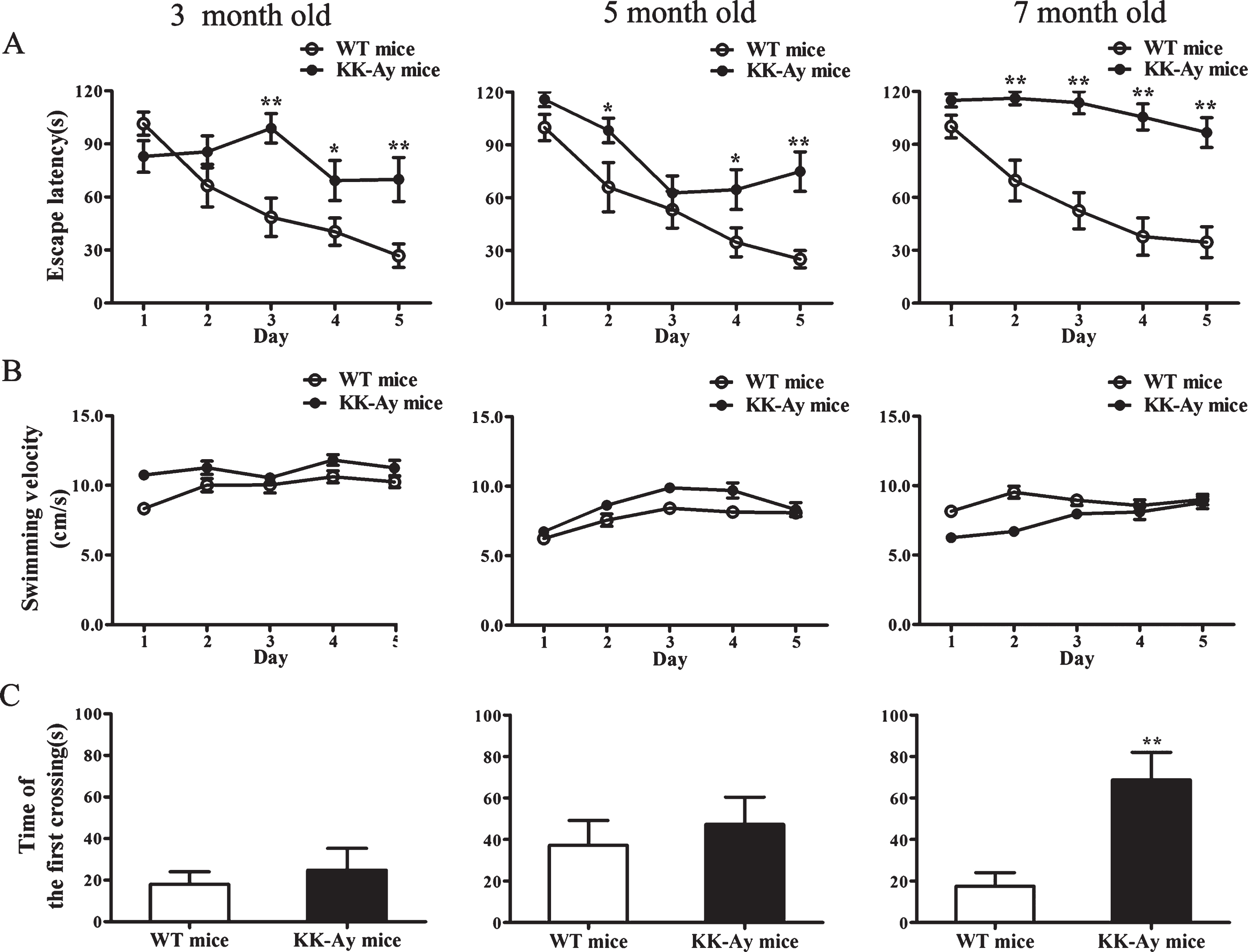
Impaired cognitive behaviors of KK-Ay mice in the Morris water maze. The KK-Ay mice showed significant cognitive impairment at the three ages. A) Acquisition of spatial learning was assessed in the Morris water maze hidden platform task. The escape latency of KK-Ay mice was higher than WT mice, indicating learning deficits. B) No motor function abnormalities of KK-Ay mice were observed as reflected by the swimming speed. C) The first crossing time of KK-Ay mice was increased compared with the age-matched WT mice, and reached a significant difference in the 7-month-old group. *p < 0.05, **p < 0.01 versus WT mice, n = 12–15 for WT mice and n = 11–15 for KK-Ay mice.
Course of development of LTP impairment in KK-Ay mice
Hippocampal synaptic functional plasticity impairment in KK-Ay mice was investigated via LTP recording after the behavior test. We compared LTP between KK-Ay mice and age-matched WT mice at three ages. The effect of T2DM duration on expression of LTP in CA1-field is depicted in Fig. 3. After 15 min baseline recording, LTP was induced successfully by giving high frequency simulation (HFS) in both WT mice and KK-Ay mice. Nevertheless, LTP was significantly impaired in both inducing and maintaining stage in KK-Ay mice since the age of 3 months, showing a tendency of deterioration as the course prolonging, compared with age-matched WT mice (3-, 5-, and 7-month-old KK-Ay mice: 139.1±6.9, 129.1±6.3, 126.9±5.9, respectively; aged-matched WT mice: 156.9±7.0%, 159.2±6.9%, 153.4±7.1%, respectively. p < 0.05). These data indicated that LTP was impaired during the development ofT2DM.
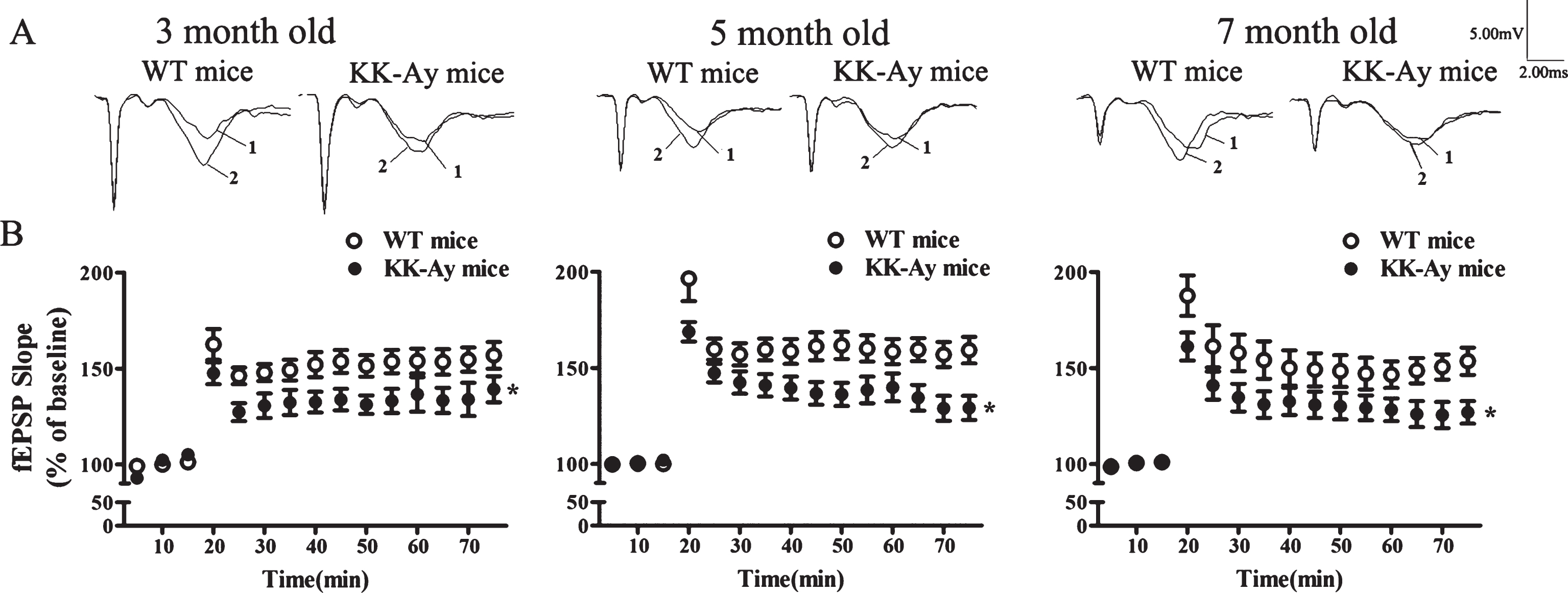
Course of development of LTP impairment in KK-Ay mice. The KK-Ay mice displayed impaired LTP in the hippocampal CA1 area during the course of diabetes. A) Representative traces before (1) and after (2) LTP induction. B) HFS-induced LTP in CA1 area of the hippocampus in KK-Ay mice (filled circles) compared with their controls (open circles). The increase in slope after HFS was significantly impaired in KK-Ay mice compared with WT mice. *p < 0.05 versus WT group, n = 16∼21/10∼13 for WT mice, n = 15∼24/10∼14 for KK-Ay mice.
Cognitive deficits could be relieved in KK-Ay mice by diet intervention
To investigate whether cognitive deficits in KK-Ay mice were irreversible damage, the effects of diet intervention on cognitive deficits were then observed. It is well known that healthy diet is crucial to T2DM care. KK-Ay mice were given a normal diet as a dietary intervention group beginning at 3 months of age when significant cognitive deficits had already been shown (Figs. 2, 3). Meanwhile, the model group was given a high-fat diet for 2 months. In the dietary intervention group, the random blood glucose, triglycerides, and cholesterol were significantly lower than the model group (Fig. 4A). Additionally, in the behavioral test and LTP recording, the model group showed significant impairment in cognitive deficits compared to the control group. Moreover, the behavioral performance was improved but was not statistically significant (p = 0.058) (Fig. 4B), while LTP impairment were significantly relieved in the dietary intervention group, compared with the model group (p < 0.01) (Fig. 4C). These results optimistically suggested that the cognitive deficits in T2DM might be reversible and could be prevented and treated at an early stage.
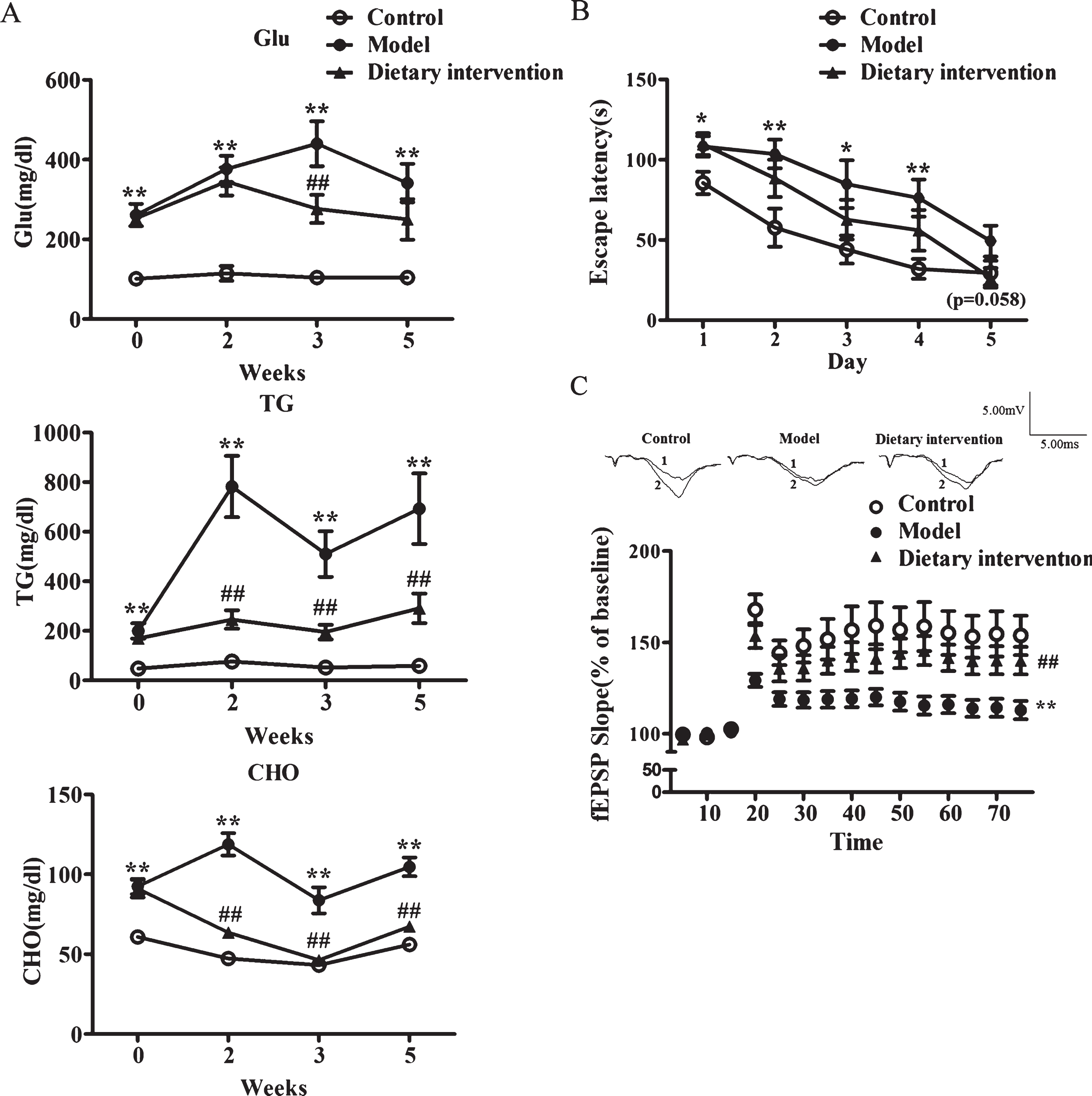
The effects of dietary intervention on the cognitive deficits in KK-Ay mice. A) The level of random blood glucose (Glu), triglycerides (TG), and cholesterol (CHO) were monitored. B) Acquisition of spatial learning was assessed in the Morris water maze hidden platform task. C) HFS-induced LTP in CA1 area of the hippocampus. The upper were representative traces before (1) and after (2) LTP induction are shown. In biochemical indexes test, n = 15 in control group, n = 11 in model group, and n = 13 in dietary intervention group. In Morris water maze test, n = 15 in control group, n = 11 in model group and n = 12 in dietary intervention group. In LTP recording test, n = 13/10 in control group, n = 19/10 in model group, and n = 19/9 in dietary intervention group. *p < 0.05, **p < 0.01 versus control group and # #p < 0.01 versus model group.
Basal synaptic transmission at the SC-CA1 pathway in KK-Ay mice
The impaired LTP in hippocampal CA1 area is normally related to the alteration at the presynaptic and/or postsynaptic level. We thus investigated whether presynaptic activity was impaired during LTP induction in KK-Ay mice. PPF is a short-term plasticity of synapses, a phenomenon of the synaptic enhancement of the second response during two short-interval consecutive pulses due to presynaptic enhancement of transmitter release [21, 22]. In the study, we examined PPF by giving a short interpulse interval (20∼900 ms). The strength of PPF was not modulated by chronic T2DM in KK-Ay mice at different stages of the course, compared with the WT mice, indicating no alteration in the probability of transmitter release (Fig. 5). Moreover, KK-Ay mice showed no change in presynaptic fiber volley amplitude at different three ages (Fig. 6A, B). These results demonstrated that T2DM mice of KK-Ay did not interfere with presynaptic properties. The postsynaptic changes were further examined. The input-output curves showed a decrease tendency in 7-month-old KK-Ay mice with no statistical significance (Fig. 6A, C). Taken together, basal synaptic transmission does not change in KK-Ay mice during the course of diabetes.
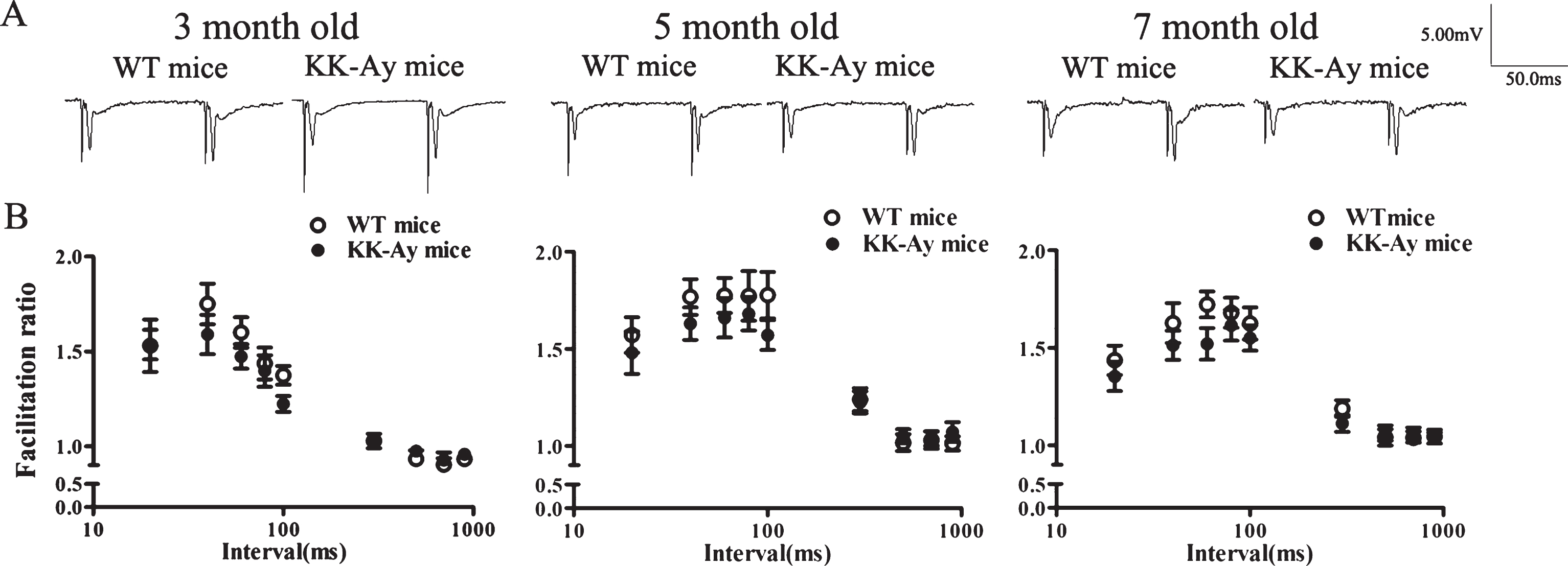
Paired-pulse facilitation (PPF) in the hippocampal CA1 area of KK-Ay mice during the course of diabetes. A) Representative traces by giving an interpulse interval of 80 ms. B) Facilitation ratios were plotted against interpulse intervals. PPF did not change significantly in KK-Ay mice (n = 15∼24/10∼14), compared with the WT mice (n = 16∼;21/10∼;13).

Synaptic excitability in the hippocampal CA1 area of KK-Ay mice during the course of diabetes. A) Representative traces by giving a series of stimulus from 0.02 mA to 0.2 mA. FV, presynaptic fiber volley; fEPSP, field postsynaptic excitatory potential. B) These presynaptic fiber volley amplitudes did not change significantly in KK-Ay mice at three ages. C) The input-output curve was obtained by plotting the fEPSP slope against the stimulus intensity, which showed a decreased tendency in 7-month-old KK-Ay mice, compared with WT mice. n = 16∼21/10∼13 for WT mice and n = 14∼24/10∼14 for KK-Ay mice.
Abnormal expression of proteins involved in LTP induction
To explore the possible mechanisms of LTP impairment induced by T2DM, the expression of both NMDARs and AMPARs subunits were detected by western blotting. In KK-Ay mice, the expression of NR1, NR2A, and NR2B subunits of NMDARs were unchanged, while the phosphorylation of NR2A and NR2B subunits were significantly reduced both in the cortex and hippocampus at three ages (Fig. 7). Furthermore, the p-Src expression mediating the phosphorylation of these subunits in the cortex and hippocampus in 5-month-old KK-Ay mice was significantly reduced with no alteration of the total Src expression. The basal CaMKII autophosphorylation, crucial for the LTP initiation, was downregulated in the hippocampus of KK-Ay mice, while the total αCaMKII did not change significantly(Fig. 8).
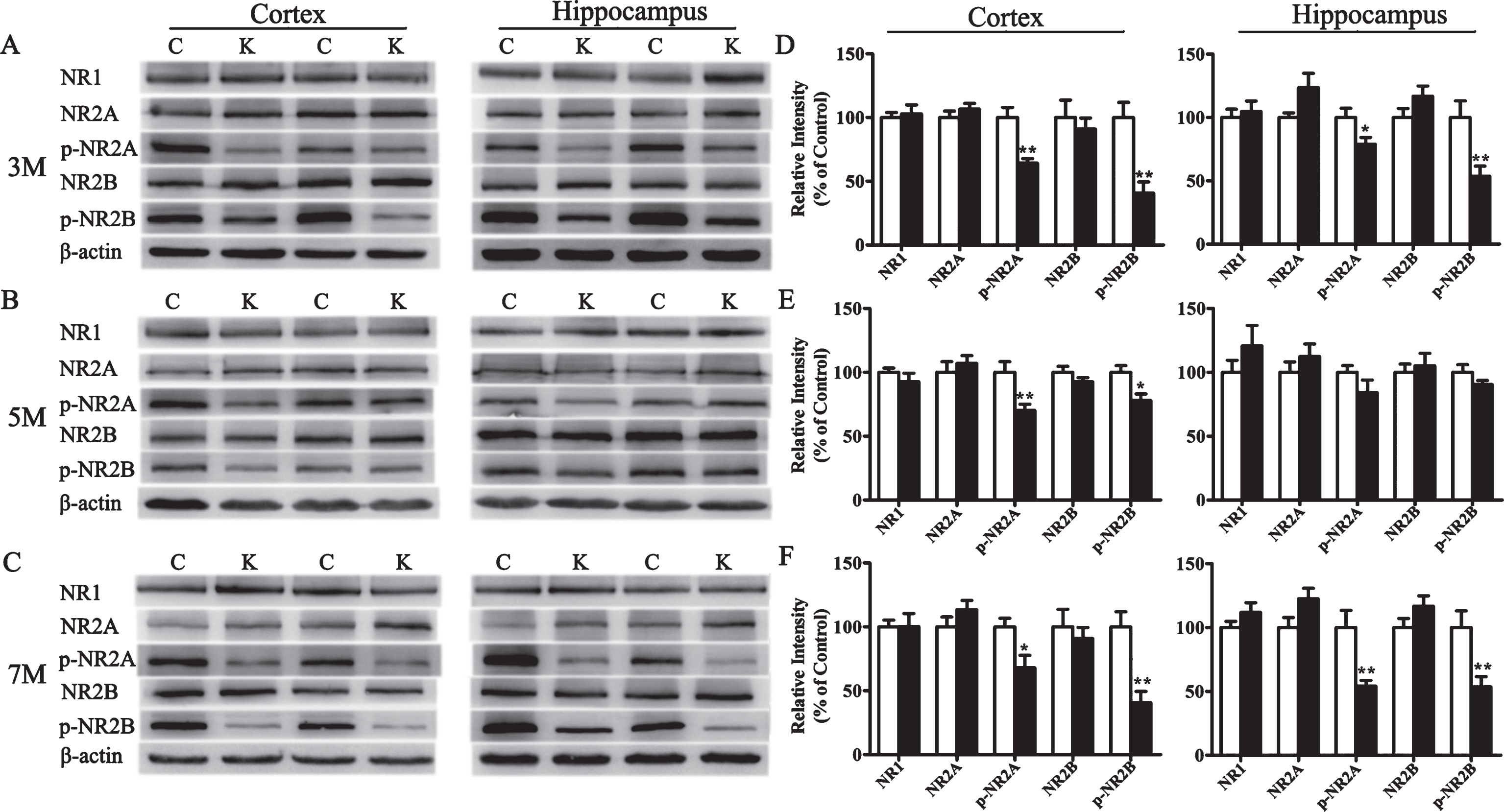
Changes of NMDARs subunits expression in KK-Ay mice during the course of diabetes. A-C) Representative western blots of NR1, p-NR2B, NR2B, p-NR2A, and NR2A in the cortex and hippocampus homogenates of KK-Ay mice at different ages respectively are shown. D-F) Quantified results were normalized to β-actin expression. Values were expressed as percentages compared with WT mice (set to 100%). C, WT mice; K, KK-Ay mice; 3M, 3-month-old mice; 5M, 5-month-old mice; 7M, 7-month-old mice. *p < 0.05, **p < 0.01 versus age-matched WT group, n = 7∼8 mice per group.
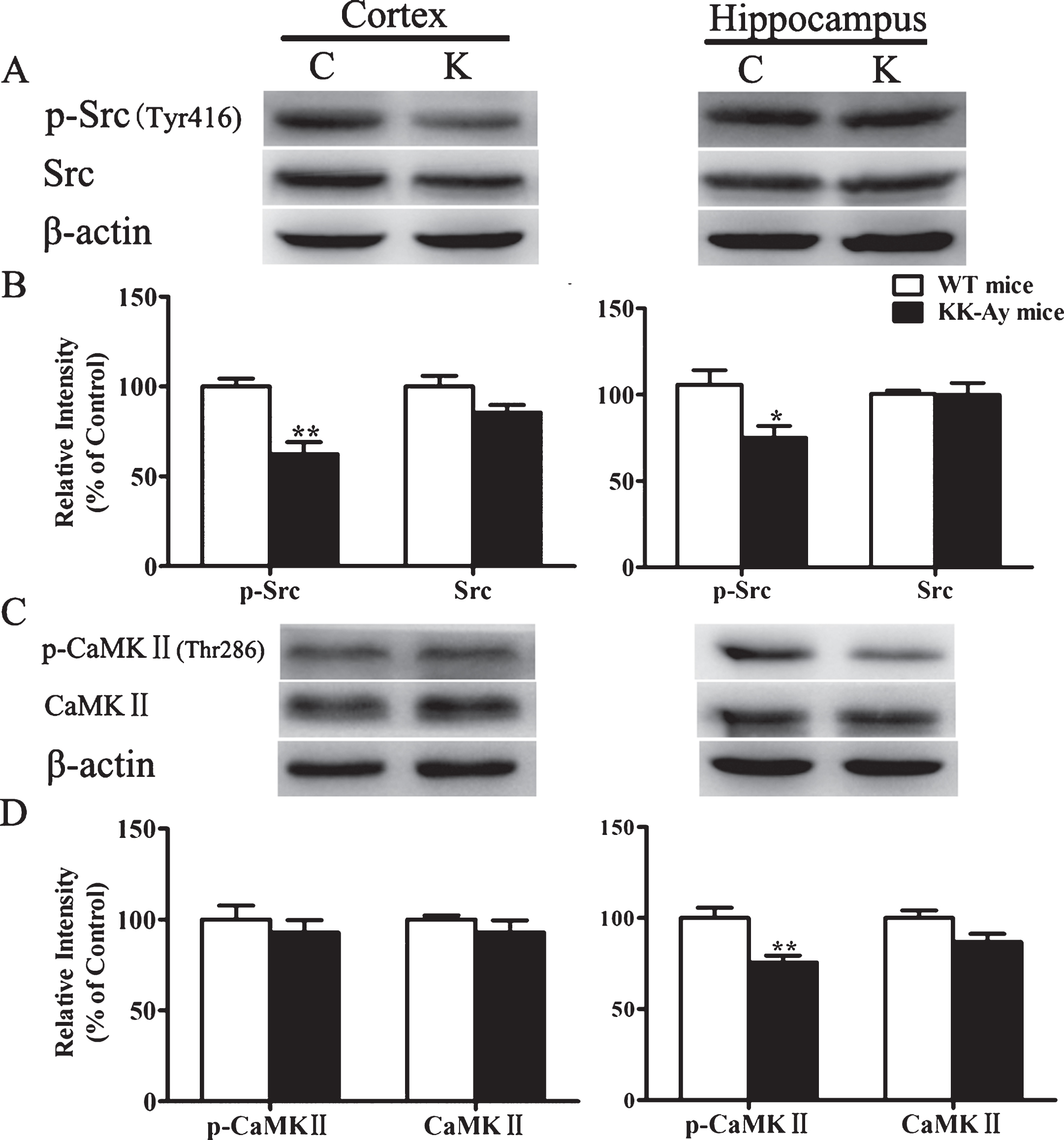
Changes of kinases expression involved in LTP induction in 5-month-old KK-Ay mice. A, C) Representative western blots of Src, p-Src, CaMKII, and p-CaMKII in the cortex and hippocampus homogenates of KK-Ay mice respectively are shown. B, D) Quantified results were normalized to β-actin expression. Values were expressed as percentages compared with WT mice (set to 100%). C, WT mice; K, KK-Ay mice. *p < 0.05, **p < 0.01 versus age-matched WT group, n = 5∼7 mice per group.
Moreover, the expression of AMPARs subunit GluR1 was significantly decreased in the hippocampus and GluR2 was significantly elevated in the cortex and hippocampus at different stages of diabetes in KK-Ay mice (Fig. 9). These results demonstrated that the changes of LTP induction in KK-Ay mice might be involved in reorganization of postsynaptic glutamate receptors.
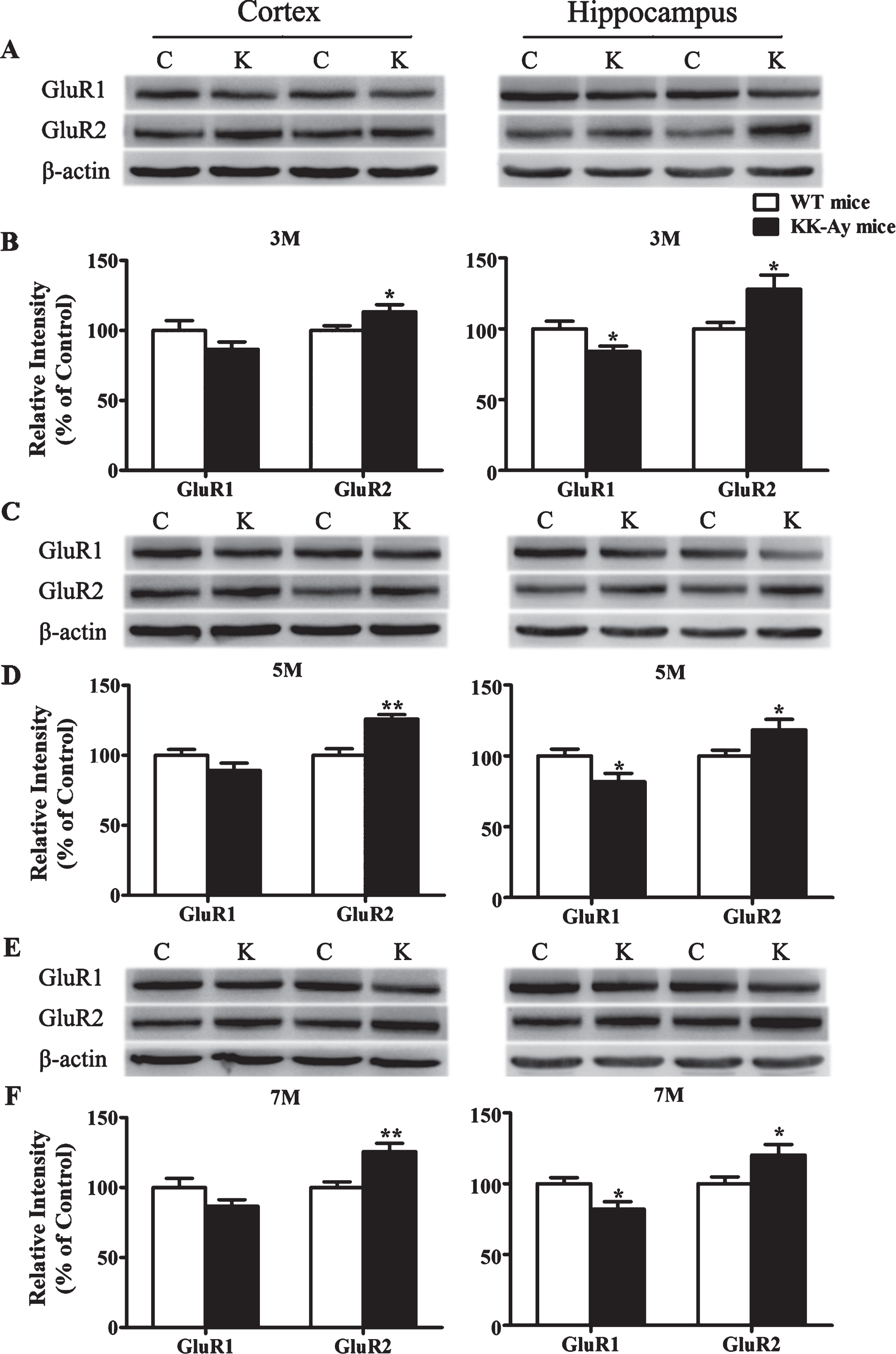
Changes of AMPARs subunits expression in KK-Ay mice during the course of diabetes. A, C, E) Representative western blots of GluR1 and GluR2 in the cortex and hippocampus homogenates of KK-Ay mice at different ages respectively are shown. B, D, F) Quantified results were normalized to β-actin expression. Values were expressed as percentages compared with WT mice (set to 100%). C, WT mice; K, KK-Ay mice; 3M, 3-month-old mice; 5M 5-month-old mice; 7M, 7-month-old mice. *p < 0.05, **p < 0.01 versus age-matched WT group. n = 7∼8 mice per group.
DISCUSSION
T2DM is associated with a high risk of the CNS, characterized by moderate learning and memory deficits [1, 24]. In recent years, rodent models of diabetes have pushed the studies for neuronal mechanisms of learning and memory impairment [9], such as STZ-induced diabetes models and obese monogenic rodent models of spontaneous T2DM like ob/ob (db/db) mice and Zucker rats, etc. [9]. However, as well-known, non-insulin-dependent T2DM in human is a heterogeneous and multifactorial disease [12]. KK-Ay mice, as the major heralds of the category of obese T2DM models with polygenic background, are regarded as suitable models for exploring the mechanisms of obese T2DM and its complications [13]. However, the study of obese diabetes inducing cognitive deficits was scarcely reported in KK-Ay mice.
Up to now, the most studies about diabetic dementia were carried out on STZ-DM mice or rats and few of them were carried out on the models of metabolic disorder. The diverse reports about onset of the disease and the mechanisms of synaptic dysfunction were obtained. In the present study, KK-Ay mice showed spatial learning and cognitive impairment at 3 months of age. Meantime, LTP was significantly impaired and postsynaptic NMDA/AMPA receptor subtypes were specificly changed in KK-Ay mice. These results indicated that spatial learning and cognitive impairment might link to the lesion of functional synaptic plasticity in KK-Ay mice.
LTP impairment attributes to the alteration of presynaptic and/or postsynaptic structure and function. It is generally admitted that PPF refers to the change of the presynaptic processes involved in neurotransmitter release [25]. Our present results showed no alteration of the strength of PPF in KK-Ay mice, which was consistent with studies carried out in STZ-DM rats in similar experimental conditions[10, 26]. In addition, presynaptic fiber volley amplitude was also not changed in KK-Ay mice at different stages of the course, which agreed with the result of STZ-DM rats [11]. These data further strengthen the notion that rather than presynaptic modifications, deficits in LTP in diabetic animals are mainly due to postsynaptic abnormalities. Previous studies carried out in STZ-DM rats have led to controversial results in basal excitatory synaptic transmission on the age of DM onset [11, 26–28]. We thus examined the input-output curve indicating the postsynaptic excitability at different ages, and it just showed a decrease tendency in 7-month-old KK-Ay mice. Our data suggested that the LTP deficits in KK-Ay mice might not be correlated with the basal synaptic transmission.
In addition to membrane excitability, the postsynaptic mechanisms underlying the LTP induction involve in receptors located on synaptic surface [7, 29]. Previous studies have demonstrated that the synaptic functional deficits in the hippocampus of diabetic rodents involved in glutamate receptors, including NMDARs and AMPARs [1, 7]. NMDAR, mediating the induction of long-term synaptic plasticity, is a heteromeric complexes composed of three classes of subunits, NR1, NR2(A∼D), and NR3 [30]. The direction of NMDAR-dependent plasticity is influenced by many factors including subunitcomposition and phosphorylation state [31, 32]. Abnormal expression of NMDAR subunits has been proved in STZ-DM rats [1]. Therein, NR1, the core subunit to form the receptor channel required for brain development and survival, was not altered in STZ-DM rats [11]. NR2A and NR2B, the major regulatory subunits of NMDARs crucial for LTP induction, have been found to be either unaltered [11] or decreased in STZ-DM rats [33, 34]. In the present study, we found that the expression levels of NR1, NR2A, or NR2B subunits of NMDARs were not changed in the KK-Ay mice at different ages. However, the phosphorylation level of NR2A (Y1325) and NR2B (Y1427) subunits were significantly reduced since 3 months of age. Previous studies have indicated that tyrosine phosphorylation of NR2A and NR2B subunits could regulate the activity of NMDARs, and it might involve LTP induction, synaptic plasticity, and memory formation [29]. The phosphorylated NR2A potentiates NMDAR function while phosphorylated NR2B (Y1472) inhibits NR2B-mediated endocytosis and promotes surface expression of NMDARs [35, 36]. Therefore, both functions were weakened in KK-Ay mice.
The Src family of kinases (SFKs), major kinases to phosphorylate the NR2 subunits, is reported to be a crucial point of convergence for signaling pathways in the regulation of NMDAR-dependent synaptic transmission and plasticity [35, 37]. The present study showed that the expression of p-Src state,mediating the phosphorylation of NR2s subunits, was significantly reduced in the cortex and hippocampus in 5-month-old KK-Ay mice. It indicated that the impairment of LTP induction in KK-Ay mice might be associated with the reductions in Tyr-dependent phosphorylation of NR2s subunits. Further, the basal CaMKII autophosphorylation, displaying constitutive activity in enhancing NMDARs function by binding to NR2s and promoting AMPARs trafficking/function by phosphorylation process crucial for the LTP initiation, was downregulated consistent with previous studies in STZ-DM rats [38–40]. The other mechanisms involved in the disturbances of Tyr-dependent phosphorylation of NR2s subunits in the KK-Ay mice remain to be determined.
It is known that the variation in the number and subunit composition of AMPARs would also affect the formation and stability of LTP [41]. The most predominant AMPAR subtype in CA1 pyramidal cells is a heteromer assembled as two identical heterodimers with GluR1/2 in controlling AMPAR surface expression and synaptic plasticity [41]. The GluR1 subunit is the foremost in the AMPARs providing the driving force for activity-dependent trafficking of AMPARs into synapses [42]. Constitutive deletion of the GluR1 subunit prevented LTP expression in the hippocampus [43]. It has been proven that GluR1 subunit expression and phosphorylation level was significantly downregulated in the AD transgenic mice, resulting in the dysfunction of AMPARs trafficking, a critical event for the establishment of LTP [44, 45]. Furthermore, GluR1 protein levels were decreased [34] or unaltered in STZ-DM rats [11]. In the present study, GluR1 was significantly decreased in the hippocampus of KK-Ay mice, which would affect the level of subsequent phosphorylation to translocate AMPARs into synaptic sites. The GluR2 subunit renders them Ca2 +-impermeable, while GluR2-lacking AMPARs (Ca2 +-permeable) offer a route for Ca2 + entry and mediates excitotoxicity under pathological conditions [46]. Some studies demonstrated that overexpression of GluR2 delayed signs of neurotoxicity and lengthened survival. Moreover, the GluR2 subunit is mobile, and the stable expression of LTP is based on insertion of conventional GluR2-containing AMPARs into membrane [42, 48]. We found that the expression of GluR2 subunit in KK-Ay mice was increased significantly, in contrast to that in an AD model. A downregulation of GluR2 subunit expression was shown in AD transgenic mice [45]. The upregulation of GluR2 subunit in KK-Ay mice might result from a compensatory effect leading to a relative attenuation of cognitive impairment as that in subcortical ischemic vascular dementia [49]. These changes may be more similar to that of vascular dementia. The dementia of KK-Ay mice might result from pathological changes in small cerebral blood vessels, and also from AD-like alterations. Further mechanisms of T2DM-induced dementia remain to be studied.
Finally, we demonstrated that the cognitive deficits in KK-Ay mice could be relieved by dietary intervention. Behavior performance and LTP activity were improved. Meanwhile, blood glucose, triglycerides, and cholesterol were reduced significantly. It indicates that the cognitive deficits of T2DM could be recovered at an early stage of the disease.
In conclusion, KK-Ay mice manifested cognitive deficits during the development of the disease, closely related with the impairment of synaptic function. It might be recovered by dietary intervention. Impaired LTP might be related to alteration of phosphorylation levels of NR2A and NR2B as well as the pathological expression of AMPARs.