
Abstract
Background:
Alzheimer’s disease (AD) is the leading cause of degenerative dementia in the aging population. Patients with AD have alterations in cerebral hemodynamic function including reduced cerebral blood flow (CBF) and cerebral metabolic rate. Therefore, improved cerebrovascular function may be an attractive goal for pharmaceutical intervention in AD.
Objective:
We wished to observe the acute effects of sildenafil on cerebrovascular function and brain metabolism in patients with AD.
Methods:
We used several novel non-invasive MRI techniques to investigate the alterations of CBF, cerebral metabolic rate of oxygen (CMRO2), and cerebrovascular reactivity (CVR) after a single dose of sildenafil administration in order to assess its physiological effects in patients with AD. CBF, CMRO2, and CVR measurements using MRI were performed before and one hour after the oral administration of 50 mg sildenafil. Baseline Montreal Cognitive Assessment score was also obtained.
Results:
Complete CBF and CMRO2 data were obtained in twelve patients. Complete CVR data were obtained in eight patients. Global CBF and CMRO2 significantly increased (p = 0.03, p = 0.05, respectively) following sildenafil administration. Voxel-wise analyses of CBF maps showed that increased CBF was most pronounced in the bilateral medial temporal lobes. CVR significantly decreased after administration of sildenafil.
Conclusion:
Our data suggest that a single dose of sildenafil improves cerebral hemodynamic function and increases cerebral oxygen metabolism in patients with AD.
Keywords
INTRODUCTION
Patients with Alzheimer’s disease (AD) have alterations in cerebral hemodynamic function including reduced cerebral blood flow (CBF), increased cerebrovascular resistance, and reduced cerebral metabolic rate as compared to healthy controls [1 –4]. Epidemiologic studies have shown that AD and vascular disease share many risk factors including hypertension, diabetes, hyperhomocysteinemia, obesity, and hyperlipidemia [5 –7]. One study [1] revealed that compared to healthy controls, CBF and the variability of cerebral arterial velocity in AD patients is reduced while cerebrovascular resistance is increased. Patients with AD also have reduced vascular response to CO2 in frontal regions, distinct from the well-established reductions in CBF seen in the temporoparietal and posterior cingulate regions [2, 4]. Cerebrovascular disease may simply be an independent process that interacts with AD pathology in an additive manner to produce cognitive dysfunction [8]; however, others have suggested more direct links with AD pathology through cerebral amyloid angiopathy, dysregulation of the neurovascular unit, hypoxia, interruption of the blood-brain barrier, and other failures of endothelial function including impaired clearance of amyloid-β across the blood-brain barrier [9, 10]. Therefore, improved cerebrovascular function is an attractive goal for a pharmaceutical intervention in AD.
Sildenafil is known as a highly selective inhibitor of cyclic guanosine monophosphate (cGMP) specific phosphodiesterase type 5 (PDE-5) that enhances nitric oxide (NO)-mediated vasodilatation. cGMP is the second messenger of NO and, a principal mediator of smooth muscle relaxation. Sildenafil is FDA approved for the treatment of erectile dysfunction in men as well as pulmonary arterial hypertension in both men and women [11, 12]. Recently, some investigators have focused on the effect of sildenafil intervention on ischemic brain. In preclinical studies, PDE5 inhibitors have shown promise in animal models of cerebral ischemia. Several studies have shown improved functional outcomes in rat models of stroke [13 –16], even when treatment with a PDE5 inhibitor was delayed by as much as 7 days [15]. Human studies of the CNS effects of PDE5 inhibitors have directly addressed effects on CBF. In normal healthy subjects, i.e., those without a baseline disturbance in cerebral hemodynamics, sildenafil does not affect basal CBF [17 –19]; but has been shown to increase cerebrovascular reactivity to breath holding [20]. In patients with pulmonary hypertension who had reduced CBF and attenuated vascular reactivity, sildenafil had a normalizing effect on cerebrovascular reactivity indicating improved neurovascular coupling [21].
Of additional relevance to the current work, several animal studies [22, 23] suggested that upregulation of the NO pathway may be protective in AD. One effective way to upregulate the NO pathway is by increasing cGMP levels through inhibitors of PDE5. Animal studies have shown that the selective PDE5 inhibitors raise hippocampal cGMP levels and improve memory in aged rats [24]. Sildenafil has been tested in multiple transgenic mouse models of AD (APP/PS1 double transgenic, Tg2576, and J20 mice) as well as a senescence-accelerated mouse model of age dependent cognitive impairment [25 –28]. In all of these studies, daily treatment with sildenafil restored cognitive function. In one study, after treatment for 3 weeks, sildenafil rescued the memory deficits of these mice and this improvement persisted even 9–12 weeks after the treatment had been stopped. In contrast to the human studies of CBF, these studies suggest that the therapeutic impact of PDE5 inhibition in these animal models may include non-vascular mechanisms of action as well. It is possible, therefore, that sildenafil could have a multi-pronged impact on AD pathophysiology on both sides of the blood-brain barrier.
Sildenafil has not been investigated in patients with AD. However, before an expensive large-scale clinical trial is conducted to evaluate the therapeutic effect of sildenafil, preliminary evidence that sildenafil can improve brain function and physiology needs to be obtained. In particular, biomarkers are useful in detecting early changes in disease and treatment, and may also provide insights on potential mechanisms by which sildenafil may improve cognitive function in AD.
The goal of this pilot study, therefore, is to observe the impact of a single dose of sildenafil on known vascular and metabolic abnormalities in patients with AD in order to obtain evidence of any potential benefit. We measured CBF using both global and regional magnetic resonance imaging (MRI) methods. The global method is relatively straightforward in CBF quantification, thus can ensure that the observed CBF change is due to blood flow, rather than other factors such as transit time. The regional method can identify brain regions that manifest the most pronounced effect. Recognizing that increased blood flow does not mean that the brain is metabolically more active, we also measured the brain’s metabolic rate of oxygen (CMRO2), using a recently developed MRI technique. Finally, other evidence that would suggest a vasodilatory effect of sildenafil to the brain would be to measure the blood vessel’s vascular reserve. An acute vasodilatory effect of sildenafil would cause a reduction in vascular reserve. We therefore assessed vascular reserve before and after sildenafil administration using an index, cerebrovascular reactivity (CVR), which was measured by a brief CO2 inhalation inside the MRI scanner. We hypothesized that a single dose of sildenafil would increase CBF at baseline, increase cerebrovascular reactivity, and increase metabolic activity as measured by O2 consumption. If supported by these data, then such markers could then be incorporated as additional outcome measures in a future clinical trial of chronic daily treatment of AD with sildenafil.
METHODS
Participants
This study was conducted under approval by the Institutional Review Board (IRB) of UT Southwestern Medical Center and was in accord with the Helsinki Declaration of 1975. A total of 14 subjects aged 62–87 years (mean±SD 71.6±7.5) were recruited from the early AD cohort of the UT Southwestern Alzheimer’s Disease Center (ADC). All subjects had a Clinical Dementia Rating of either 0.5 or 1 and a Mini-Mental Status Exam score of ≥20 at their most recent ADC research visit, within the past year. The Montreal Cognitive Assessment (MoCA) was obtained on the day of the experiment prior to the first imaging session. All subjects were able to undergo MRI scanning and gave informed written consent before participating in the study. They did not have any safety contraindications for MRI such as metal implants, pacemaker, neurostimulator, body piercings, or claustrophobia. Patients were excluded if they had a contraindication for receiving sildenafil or if they were on a daily PDE5 inhibitor such as low dose tadalafil (Cialis). Patients who used “on demand” sildenafil or other PDE5 inhibitors for erectile dysfunction were required to refrain from taking it for at least 7 days prior to testing.
Procedures
This study consisted of one visit that included two sessions. The first session started in the morning. Blood pressure was measured before they entered into the MRI scanner room. Three brain physiological markers were measured by advanced MRI techniques, including CBF, cerebral metabolic rate of oxygen (CMRO2) and cerebrovascular reactivity (CVR) to CO2. After the first session, participants were taken out of the magnet and given a single 50 mg dose of sildenafil. One hour later, the second session started with repeated measurement of CBF, CMRO2, and CVR. Blood pressure was measured one more time before subjects entered into the scanner room.
All experiments were conducted on a 3T MR system (Philips Healthcare, Best, The Netherlands). The body coil was used for radiofrequency transmission and a 32-channel sensitivity encoding (SENSE) head coil for receiving. Foam padding was used to stabilize the head to minimize motion. A localizer scan was performed for slice positioning and a coil sensitivity scan was conducted for SENSE reconstruction. A 3D T1-weighted Magnetization-prepared-rapid-acquisition-of-gradient-echo (MPRAGE) scan was performed for anatomical reference and the estimation of brain volume. The MPRAGE sequence used the following imaging parameters: repetition time (TR) of 8.1 ms, an echo time (TE) of 3.7 ms, a flip angle of 12°, a shot interval of 2100 ms, an inversion time (TI) of 1100 ms, with a voxel size of 1×1×1 mm3, 160 slices with a sagittal slice orientation.
Measurement of CBF
CBF of the participant was assessed with two different techniques. Global CBF was measured with a phase-contrast MRI technique [29]. Phase-contrast MRI measures bulk blood flow at the level of internal carotid and vertebral arteries, and reflects blood supply of the brain as a whole. We therefore use the term “global CBF” to refer to this measure. Regional CBF was measured with a Pseudo-Continuous Arterial Spin Labeling (pCASL) technique [30, 31]. pCASL MRI measures regional distributions of labeled arterial water in the brain and reflects local perfusion. Thus we use the term “regional CBF” to refer to the pCASL measure.
In this study, we performed four PC-MRI scans, with each targeting one of the following four vessels, left/right internal carotid artery (ICA) and left/right vertebral artery (VA) (Fig. 1A). An automatic algorithm was used for slice positioning [29]. For data analysis, a region-of-interest (ROI) was then drawn on each of the 4 arteries based on the magnitude image [32]. The ROI mask was applied to the velocity map and the integration of the velocity within the ROI (i.e., velocity×area) yielded CBF in units of milliliters per minute. To obtain unit volume CBF value, it is necessary to adjust for brain volume, which we obtained by employing FSL (FMRIB Software Library, Oxford University, UK) to segment the high-resolution T1 image into gray matter, white matter, and cerebrospinal fluid. The brain’s parenchymal volume was given by the sum of gray and white matter volumes, and converted to the weight of the brain by assuming a parenchyma density of 1.06 g/ml [33]. Normalized unit volume CBF (in ml/100 g/min) which has accounted for brain volume differences across subjects and groups were used for further CMRO2 calculation.
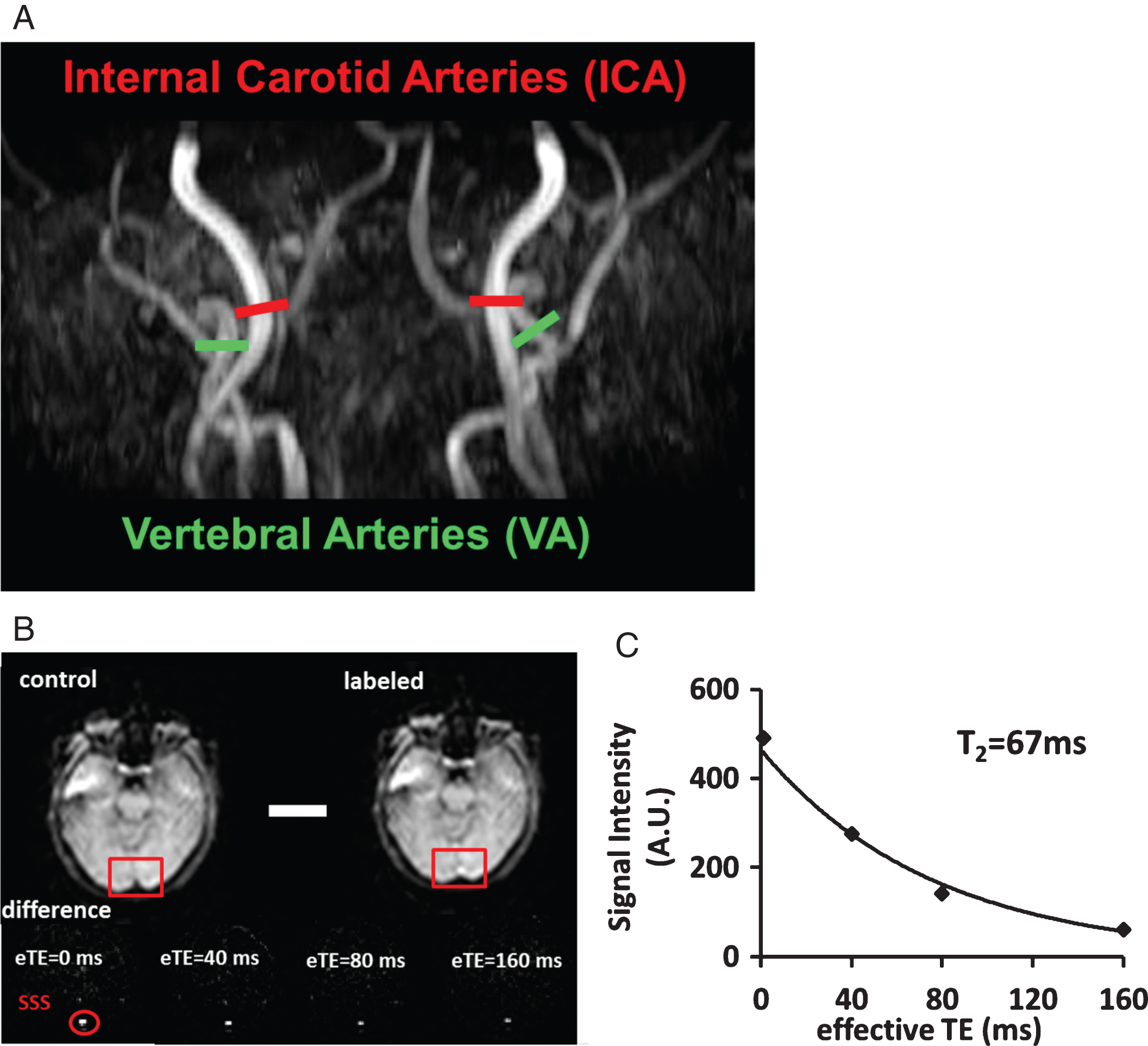
Representative MR images in one participant. A) Procedures to measurement of global CBF. Four PC MRI scans, red bars for Internal Carotid Arteries (ICA) and green bars for Vertebral Arteries (VA) are positioned perpendicular to the respective feeding arteries on an angiogram image (shown in black-and-white). B) Measurement of venous T2 value using T2 relaxation under spin tagging (TRUST) MRI. Upper panel shows raw images of control and labeled scans. The red boxes illustrate the manually drawn ROI of superior sagittal sinus (SSS). Lower panel shows difference images, i.e., control-labeled. eTE = effective echo time. Red circle highlighted the location of the SSS. C) Monoexponential fitting of the signal intensity in SSS as a function of eTE yields blood T2 value.
In the pCASL scan, forty pairs of control and labeled images were acquired using a multi-slice echo-planar imaging (EPI) acquisition. Imaging parameters for pCASL experiments were: single-shot gradient-echo EPI, field of view (FOV) = 240× 240 mm2, matrix = 80×80, voxel size = 3×3 mm2, 29 slices acquired in ascending order, thickness = 5 mm, labeling duration 1650 ms, post-labeling delay 1525 ms, TR/TE = 4205/13.81 ms, flip angle (FA) = 90°, and scan duration = 5 min 49 s. This image acquisition protocol generally follows the recommendations of the recently published ASL consensus paper [34]. pCASL data analysis used a standard kinetic model and voxel-by-voxel CBF in ml/100 g/min was quantified [34].
Measurement of CMRO2
CMRO2 reflects the amount of oxygen consumed by the brain. CMRO2 measurement traditionally requires the use of multiple PET radiotracers as well as arterial blood sampling, which makes it difficult to be used for the assessment of medication effects. The present study used a novel MRI technique that allows tracer-free measurement of CMRO2 and can be repeated as many times as needed. CMRO2 of each subject was measured following techniques originally developed by Xu et al. [35] and recently improved by Liu et al. [36]. Global CMRO2 (in units of μmol O2/min/100 g brain tissue) was quantified based on Fick principle of arteriovenous difference in oxygen content [37]: CMRO2 = CBF×(Ya-Yv)×Ch, where CBF is total cerebral blood flow and is already measured with phase-contrast as described above; Ya is the arterial blood oxygenation, and was measured using a pulse oximeter (In Vivo, Gainesville, FL), and Ch is a constant representing the capacity of blood to carry O2 and is well established in physiology literature [38]. Here we used Ch values of 8.155μmol O2/ml blood for females and 8.562 for males based on assumed hematocrit 0.40 and 0.42 respectively [38].
Yv is the global oxygen saturation percentage in venous blood and was noninvasively measured by a T2-relaxation-under-spin-tagging (TRUST) MRI technique that was recently developed and validated in our laboratory [39, 40]. The TRUST technique is similar to ASL but the labeling slab is placed above the imaging slice to label venous instead of arterial blood. Pure venous blood signal can then be assessed in the superior sagittal sinus (SSS) by subtracting the labeled image from the control image (Fig. 1B). The venous blood signals were fitted to a monoexponential function to obtain T2 (Fig. 1C), which was in turn converted to Yv via the well-known relationship between blood T2 and oxygenation [40, 41]. The imaging parameters were: voxel size 3.44×3.44×5 mm3, TR = 3000 ms, TI = 1022 ms, four effective TEs: 0, 40, 80, 160 ms, labeling thickness 100 mm, gap 22.5 mm, and scan duration 1.2 min.
Measurement of CVR
CVR response to CO2 was measured using hypercapnia (inhalation of a gas mixture containing 5% CO2, 21% O2, and 74% N2) challenge. The details of the CVR measurement were described previously [42, 43]. Briefly, the hypercapnic gas was contained in a Douglas bag and the valve on the bag helps to achieve gas switching between room air and hypercapnia, while the subject is inside the MRI scanner. The challenge gas was delivered to subjects through a two-way valve and mouthpiece combination (Hans Rudolph, 2600 series, Shawnee, KS, USA). Subjects breathed hypercapnic gas and room air via a mouthpiece in an interleaved manner (60 s CO2, 60 s room air, repeated four times). Blood-Oxygen-Level-Dependent (BOLD) images were acquired continuously during the entire experimental period. The imaging parameters were: TR/TE/FA = 1500 ms/30 ms/60°, voxel size = 3.44×3.44×5 mm3, FOV = 220×220 mm2, 29 slices, and thickness = 5 mm. End-tidal CO2 (EtCO2) is an indicator of the vasodilatory input to the brain which was measured and recorded using a capnography monitor (Capnograd, model 1265, Novametrix Medical Systems, Wallingford, CT) during the experiment. Other physiologic parameters including respiratory rate, heart rate and arterial oxygenation were also monitored and recorded by a physiology monitor (MEDRAD Inc., Pittsburgh, PA). In data analysis, linear regression was performed between the voxel-wise BOLD time course and the EtCO2 time course, yielding a voxel-by-voxel map of CVR in the units of % BOLD/mmHg CO2.
Statistical analyses
The null hypotheses for all analyses were that there were no differences comparing variables before and after sildenafil administration. Global changes in CBF and CMRO2 between the two time-points (before and after administration of sildenafil) were assessed by paired t tests. Two-tailed p-values of less than 0.05 were considered significant. Voxel-wise analyses of CBF maps were conducted using a paired t-test in SPM. Results were considered statistically significant at a voxel wise threshold of p < 0.005 and a cluster size >100 voxels, corresponding to family wise error (FWE) corrected p-value of 0.001. If significant clusters were observed, these clusters were then defined as the functional mask for further ROI analysis.
For comparison of CVR maps between two time-points, ROI analysis was performed using Montreal Neurological Institute (MNI) template space ROIs defined by Automated Anatomical Labeling (AAL) software [44]. ROIs consisted of two hemispheres of frontal, temporal, parietal and occipital lobes, subcortical gray matter, and insula. Because of the relatively small sample size for the CVR data and our a priori hypothesis that CVR will decrease due to diminished vascular reserve by the acute vasodilatory effect of sildenafil, a one-tail threshold was used in the CVR analysis.
Correlations between the Montreal Cognitive Assessment (MoCA) scores, obtained on the same day as imaging, as well as the Clinical Dementia Rating (CDR) and CDR sum of boxes from the ADC visit closest in time to the day of imaging and the alterations of three brain MRI markers were assessed.
RESULTS
Two participants refused to continue the scans after the first session, and four participants did not undergo the CVR part because they could not follow the instructions for holding the mouthpiece correctly. Blood pressure between two time-points did not show any changes (see Table 1 for values). Seventy-five percent of participants carried at least one APOE ɛ4 allele. Participants’ demographic information in all experiments is summarized in Table 1.
Participant demographic information by MR measurement categories
MoCA, Montreal Cognitive Assessment; MAP, mean arterial pressure.
We first examined whether the administration of sildenafil altered blood supply to the brain. Twelve participants completed CBF and CMRO2 procedure. The global CBF as measured by PC-MRI was 55.3±2.8 ml/100 g/min (mean±SEM) at the baseline and significantly increased (p = 0.03) to 59.0±2.3 ml/100 g/min (mean±SEM) after administration of sildenafil (Fig. 2A), an increase of 7.9% ±3.7%. We then analyzed regional CBF maps measured with ASL MRI to examine which brain region(s) manifests the most pronounced CBF enhancement. Voxel-wise analyses suggested that increases in CBF were most significant in bilateral medial temporal lobes (MTL) (Fig. 2B). The center coordinators of the clusters were [16 , 36] and [20 , 28], respectively. When we lowered the threshold, other regions in the frontal, temporal, and parietal lobes could also be detected (see Supplementary Figure 1). However, it should be emphasized that the MTL cluster was clearly the most significant region, which showed a CBF increase of 22.8% (from 47.5±4.2 ml/100 g/min to 58.3±5.6 ml/100 g/min, mean±SEM, p < 0.001) after taking the drug.
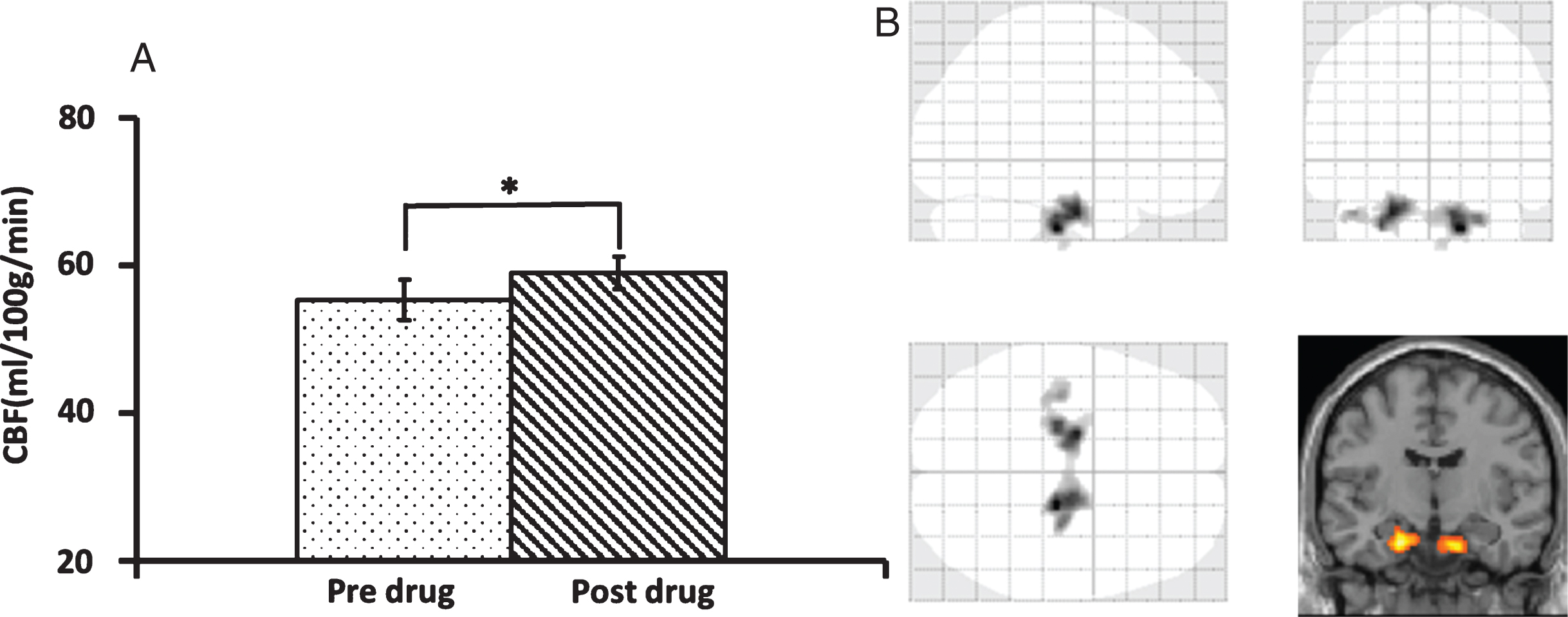
Comparison of cerebral blood flow (CBF) between two time-points. A) Global CBF comparison using Phase Contrast-MRI technique. The asterisk indicates p < 0.05. B) Voxel-wise analyses demonstrate a higher CBF in the bilateral medial temporal lobes after sildenafil.
Next, we examined whether the additional oxygen delivered due to increased blood supply was actually taken up by the brain for its oxidative metabolism. Group averaged Ya and Yv was 96.6% ±0.4% and 60.3% ±1.2% respectively before sildenafil administration. No significant statistical differences were detected after administration of sildenafil (post-Ya = 96.6% ±0.4%, post-Yv = 61.4% ±1.0%). CMRO2 significantly increased by 5.1% ±2.3% (p = 0.05) (Fig. 3A). We found a significant correlation between baseline MoCA scores and CMRO2 alterations (Fig. 3B); however, the correlations between global CDR and CDR sum of boxes with CMRO2 were not significant.
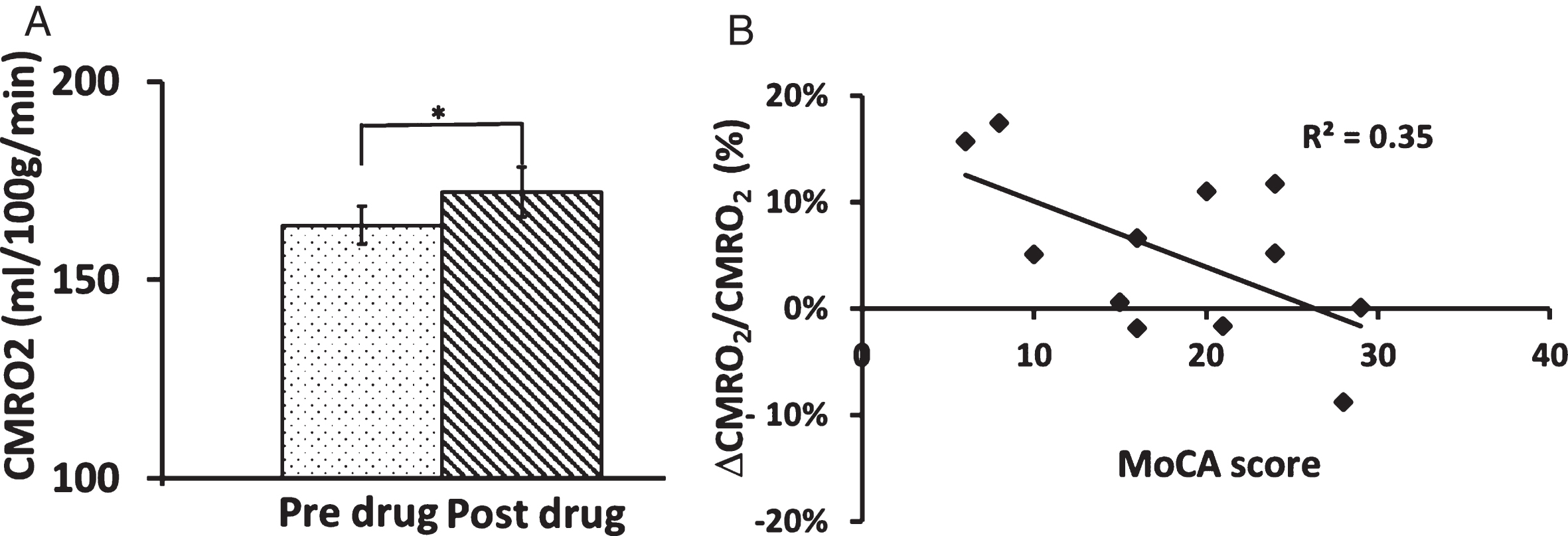
Effect of sildenafil administration on brain metabolic rate. A) Comparison of Cerebral metabolic rate of oxygen (CMRO2) between two time-points. CMRO2 increased after administration of sildenafil. B) The degree of CMRO2 change was significantly correlated with the individual’s baseline Montreal Cognitive Assessment (MoCA) scores (p = 0.04). The error bar is standard error of the mean.
Vasodilation associated with sildenafil administration would reduce vascular reserve, which was assessed with CVR. Baseline and gas-inhalation end-tidal CO2 (etCO2) values before sildenafil administration were 38.7±1.3 mmHg (mean±SEM) and 48.4±1.1 mmHg, respectively. These values were not different from those after sildenafil administration (baseline 38.8±1.1 mmHg; gas-inhalation 48.1±1.0 mmHg). Whole-brain averaged CVR values showed a trend of lower CVR after taking drugs (paired t test, one tail p = 0.05). When examining regional CVR values by major brain lobes, all lobes manifested a lower CVR value after sildenafil administration, although not all are significant (one tail p = 0.02 in temporal lobe, p = 0.05 in insula, p = 0.06 for occipital lobe, p = 0.07 for parietal lobe, p = 0.08 for frontal lobe, and p = 0.13 for limbic system). Voxel-wise comparison revealed that CVR was significantly lower after administration of sildenafil in several brain regions that were located in precuneus, limbic lobe, temporal lobe, and parietal lobe (Fig. 4). No brain clusters showed a CVR increase after sildenafil administration. Since CBF comparison showed MTL-prominent enhancement, we also applied the CBF functional ROI to the CVR maps to see how CVR changed in the areas with the most pronounced increase in CBF. It showed that there was a trend of decreased CVR (paired t test, one tail p = 0.03) in the functional CBF ROI (from 0.16±0.02% /mmHg CO2 to 0.13±0.02% /mmHg CO2).
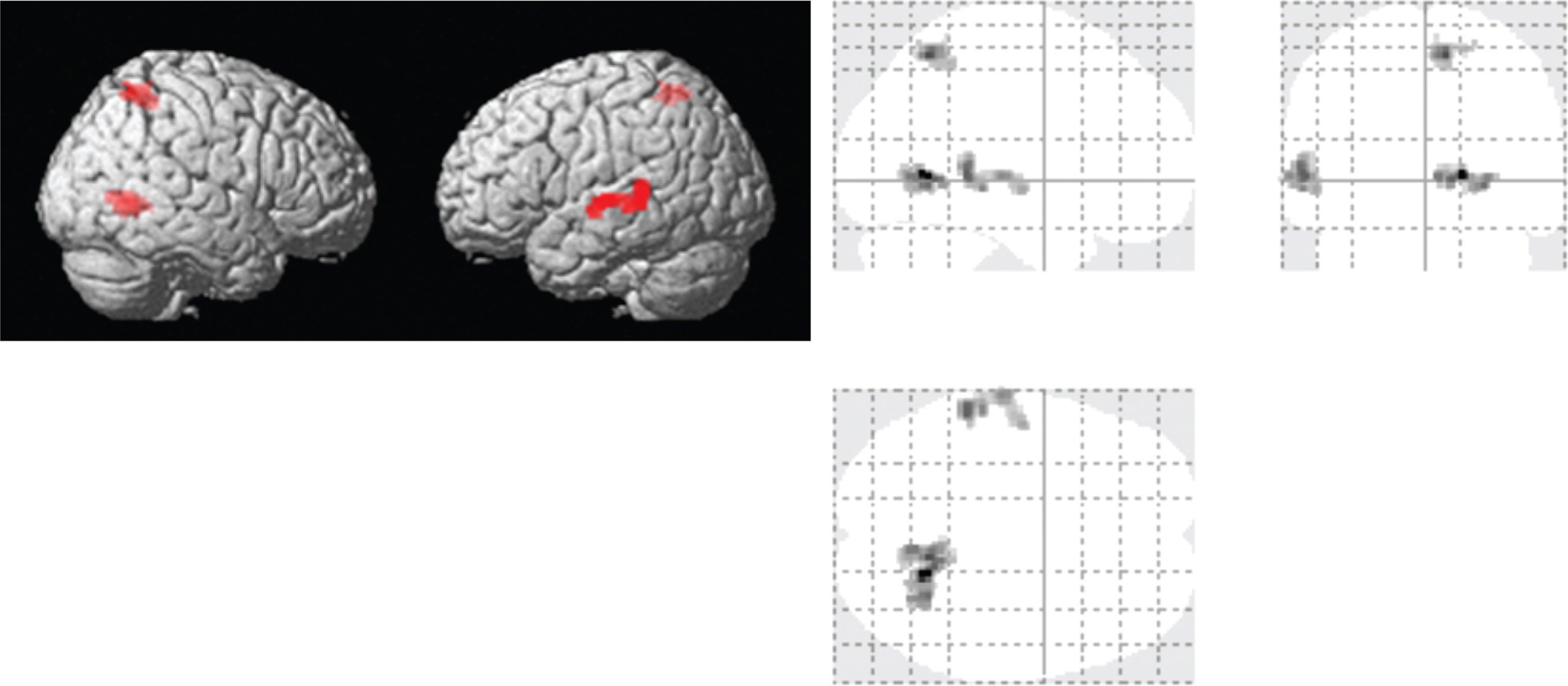
Voxel based paired t-tests of CVR maps (n = 8) between the pre- and post-sessions. Both rendered review and glass-brain overlay are shown. Red color indicates clusters where CVR significantly decreased after administration of sildenafil.
DISCUSSION
The aim of this study was to evaluate the effect of a single, 50 mg dose of sildenafil on cerebral hemodynamic and metabolic function in patients with AD. We applied several advanced techniques to examine imaging biomarkers including CBF, CMRO2, and CVR. The results showed that CBF was augmented after administration of sildenafil in AD patients. This enhancement in oxygen supply resulted in more oxygen utilization by the brain as manifested by an increase in CMRO2, the effect of which was greater in more severely affected patients. The CBF increase was most pronounced in the bilateral medial temporal lobes, although several other regions could also be identified when we lowered the statistical threshold. CVR decreased after sildenafil administration throughout the brain, suggesting that increased CBF was achieved at a cost of vascular reserve.
Central nervous system (CNS) effects of PDE5 inhibitors have been studied both in animal models and in humans. Several studies reported similar CBF results as ours. For example, Li et al. [45] investigated the long-term response of sildenafil at a dose of 10 mg/kg on rats with ischemic brain lesions. They reported that after 1 week of treatment, sildenafil enhances angiogenesis and increases CBF in the ischemic boundary region. Ding et al. [46] also reported that after 7 days of sildenafil treatment, elevated CBF was observed in rats with stroke. The MRI results were subsequently confirmed by immunohistochemical measurement of endothelial antigen-staining. In a human study, Nagdyman et al. [47] investigated CNS effect of sildenafil on 15 infants. They measured their oxygenated hemoglobin (HbO2), deoxygenated hemoglobin (HHb), and total hemoglobin (tHb) concentration after cardiac surgery. Significant increase of cerebral HbO2 and tHb were observed at the beginning of i.v. sildenafil administration as well as a concomitant decrease in HHb which suggested that sildenafil may increase CBF. However, there are few other reports that contradict our findings. Kruuse et al. [19] and Arnavaz et al. [48] reported no effects of sildenafil on CBF and blood velocity after oral administration in normal healthy subjects. In those studies, blood flow velocity was recorded in the middle cerebral artery (MCA) by transcranial Doppler (TCD), and regional CBF in the perfusion area of the MCA was measured using single photon emission computed tomography (SPECT). In the present study, CBF was measured with MRI using two methods, i.e., phase-contrast MRI and ASL MRI. Different subjects’ conditions and measurement techniques could be the reason for this discrepancy. A multi-model study conducted on the same participants should be performed in the future to investigate these possibilities.
Moreover, our findings are well in line with previous observations that the initial blood flow response to cortical activity is mainly governed by an improvement of the NO system [49]. In the brain, NO is one of the primary signals in regulation of CBF in the brain. It is derived from endothelial cells, neurons and astrocytes. NO binds to guanylyl cyclase and activates it to produce the second messenger cGMP, which ultimately results in relaxation of vascular smooth muscle, resulting in vasodilatation [50]. PDE5 catalyzes the selective hydrolysis of cGMP and thereby terminates the vasodilatory response [50]. PDE5 is expressed in several brain regions that are associated with cognitive function, such as the hippocampus, cortex, and cerebellum [51].
Potential therapeutic and protective effects of PDE5 inhibitors in AD follow from multiple potential mechanisms. In line with our findings, a PDE5 inhibitor could reduce chronic hypoperfusion and resultant hypoxia through a vasodilatory mechanism. This raises the question, however, of whether this treatment directly impacts AD pathophysiology or may it just treat an atherosclerotic comorbidity. There is evidence directly linking hypoxia to amyloidogenic processing of the amyloid-β protein precursor (AβPP) [52]. Hypoxia-inducible factor-1α (HIF-1α) leads to increased β-secretase 1 (BACE-1) transcription, which occurs through HIF-1α binding to the hypoxia-responsive element in the promotor region of the BACE-1 gene [53]. While BACE-1 cleavage of AβPP is the rate limiting step in amyloid-β production, hypoxia and ischemia also increases expression and activation of γ-secretase through multiple proposed mechanisms [52]. In addition to hypoxia enhancing the amyloidogenic pathway of AβPP processing, there is also evidence that it inhibits the non-amyloidogenic pathway. The α-secretase activity is primarily due to two members of the disintegrin and metalloproteinase domain-containing protein (ADAM), ADAM10, and ADAM17. These proteins initiate the non-amyloidogenic pathway by cleaving AβPP within the amyloid-β domain resulting in the release of the soluble N-terminal ectodomain (sAβPPα) [52, 54]. In turn, sAβPPα has a number of effects including inhibition of BACE-1 [55]. Hypoxia decreases the expression of both the ADAM10 and ADAM17 proteins resulting in downregulation of the non-amyloidogenic pathway while simultaneously allowing the activation of the amyloidogenic pathway [56 –58]. Furthermore, there is evidence from transgenic mouse models of AD that amyloid-β interferes with cerebrovascular responses to vasodilatory stimuli [59, 60]. Taken together, these observations imply a potential feed forward loop in which hypoperfusion and resultant hypoxia increase amyloidogenesis which in turn further inhibits vasodilatory function. If a PDE5 inhibitor can interrupt this cascade it would potentially have a direct, positive effect on AD pathophysiology.
While our primary intent with this study was to look at vascular mechanisms of benefit, there are other potential mechanisms by which a PDE5 inhibitor, such as sildenafil, could have a therapeutic effect involving NO signaling from non-epithelial sources. There are three nitric oxide synthases (NOS) in the brain: endothelial NOS, neuronal NOS and inducible NOS (iNOS). Typically, iNOS in the brain is localized to microglia, but in AD is expressed in neurons and astrocytes [22 , 61–63]. Increased expression of iNOS and increased NO was generally assumed to have a negative, inflammatory impact on AD. Surprisingly, when knock out mice missing the gene that codes for iNOS, NOS2, were crossed with the Swedish familial AD double mutation (APPsw) mice (Tg2576), to produce mice with the Swedish mutation on a NOS2 null background, there was an increase in total brain amyloid-β compared to the APPsw mice as well as abundant tau pathology including hyperphosphorylated tau and aggregated tau which is not seen in APPsw mice [22]. The lack of NO signaling, therefore, results in increased AD pathology including tau pathology, which is characteristic of AD but not characteristic of the APPsw mouse model that typically lacks tau pathology. This makes it plausible that enhancing NO signaling may suppress AD pathology. Several other plausible molecular mechanisms have been reported. A NO dependent mechanism involving cGMP activation of protein kinase G (PKG) has been shown to inhibit BACE-1 transcription directly, thereby linking NO signaling to reduced amyloid-β production [64]. Tau pathology may be mechanistically linked to NO signaling through a chemical modification of the tau protein. In addition to activating guanylate cyclase to produce cGMP, NO also reacts with cGMP to form 8-Nitroguanosine 3’,5’-cyclic monophosphate (8-nitro-cGMP). This compound is capable of producing post translational modifications of some proteins by reacting with the sulfhydryl of a cysteine residue forming a protein cysteine-cGMP adduct, a process termed s-guanylation [65]. Recent work has demonstrated that tau can undergo s-guanylation and that the resultant s-guanylated tau will not form granules or fibrils and hence cannot aggregate to form β sheet structures, but can nonetheless participate in microtubule formation at near normal levels [66]. These protective mechanisms could potentially be enhanced by a PDE5 inhibitor in a way that is independent of its effects on cerebrovascular function.
In addition to the potentially disease modifying mechanisms already discussed, sildenafil or other PDE5 inhibitors could also produce a symptomatic benefit through enhancement of the molecular mechanisms underlying memory such as long term potentiation (LTP). Although not without considerable controversy, NO signaling has been proposed as the primary mechanism for presynaptic enhancement of synapse strength during LTP [67]. While a number of studies strongly support NO’s role in LTP, others have not and assert the view that LTP is solely a postsynaptic phenomenon [68 –71]. Some researchers suggest that these discrepancies are due to differing methods and differing types of LTP being studied with some forms utilizing NO signaling and others not [69 , 72]. Because of the possible role of NO signaling through cGMP in LTP, a number of studies addressed the effects of PDE5 inhibitors on learning and memory in animal models and found that these agents did indeed improve memory [73 –78]. While this mechanism would not be disease modifying in AD, the possibility of improving memory would still be a worthy symptomatic goal.
It is widely recognized that the MTL is a key brain region in the pathogenesis of AD and consequent memory loss [79]. In our study, increased CBF was observed to be most pronounced in the bilateral MTL. Interestingly, in a study of CBF in a mouse model of AD, hypertension, previously induced by chronic administration of angiotensin II, resulted in significantly reduced blood flow in the hippocampus but not the cortex of AD mice. This effect only occurred in the hypertensive AD mice (not in non-hypertensive AD or wild type mice) and was unrelated to the presence or absence of concurrent antihypertensive treatment [80]. This suggests an interaction between AD pathology and hypertension that predominantly affects hippocampal blood flow and may further suggest that an enhanced vasodilatory response in this region may be expected in AD patients. The targeting of increased CBF to this area of pathology may explain why some studies of normal human subjects did not detect an impact on CBF by sildenafil.
Increased CBF may have resulted from 1) vasodilatation irrespective of neuronal demand (independent of neurovascular coupling), 2) vasodilatation in response to unmet neuronal demand (normalization of neurovascular coupling), 3) vasodilatation in response to increased neuronal demand secondary to non-vascular cGMP mediated effects such as those associated with LTP previously discussed (intact neurovascular coupling with increased neuronal demand) or a combination of 2 or more of these. While our methods cannot fully distinguish between these possibilities, looking at CMRO2 in relationship to CBF can distinguish whether or not the first option, vasodilatation independent of neurovascular coupling, is a likely explanation, as vasodilatation per se should not affect CMRO2. The results revealed that global CMRO2 significantly increased along with CBF in response to a single dose of sildenafil implying that there were either metabolic demands that were not being supported by available blood flow at baseline which was corrected by sildenafil or that sildenafil increased metabolic demand over baseline and blood flow increased to meet the additional demand. The contention that sildenafil directly dilated the cerebral vasculature irrespective of neurovascular coupling is not supported by our CMRO2 findings and would not be expected based on the known mechanism of action. Sildenafil does not promote NO production, as would amyl nitrite for example [81]. Rather, it amplifies the effect of NO by prolonging the life of its second messenger. Without NO being elaborated, presumably via mechanisms of neurovascular coupling, it is unlikely that sildenafil would have much of an effect. Our CMRO2 data, therefore, provides physiological relevance to our observations of increased CBF.
Cerebrovascular reactivity (CVR) is another important marker of cerebral vascular function indexing reserve and can be measured as the CBF response to either hypo- or hypercapnia. It reflects the capability of intracerebral blood vessels to dilate in response to hypercapnia or to constrict in response to hypocapnia and usually provides more useful information in addition to traditional baseline blood flow measurement when assessing vascular factors in brain disorders [2]. In humans with AD, vasoconstriction in response to hypocapnia was previously shown to be similar to controls, while vasodilatation in response to hypercarbia was reduced, indicating selective impairment of vasodilatation [82]. In the present study, we applied a novel CO2-inhalation MRI technique to measure CVR non-invasively and our data showed that it significantly decreased after administration of sildenafil. This finding suggests that the administration of sildenafil in AD patients dilated their arterial resistance vessels and consequently resulted in an increase in resting CBF. However, in achieving that, the vessel is sacrificing its reserve capacity such that, when vasoactive stimulus is applied on top of that, the vessel’s ability to dilate further is diminished. Alternatively, just as AD patients have differential responses to hypo- and hypercarbia compared to normal controls, they may have differential responses to pharmaceutical vasoconstrictors and vasodilators as well. Our observations were made after a single dose and it is quite possible that chronic administration may allow for other physiological adaptations to occur that could restore reserve capacity.
A limitation of the present study is that not all parameters necessary for the estimation of CMRO2 were experimentally determined. Specifically, we did not measure hematocrit in the participants. Instead, we assumed that female and male participants had hematocrit of 0.40 and 0.42, respectively. Major deviations from these assumed values could cause bias in the estimated CMRO2. Another limitation is that our CVR measurement required the delivery of CO2-enriched air to AD participants inside the MRI scanner. This resulted in some subject dropouts and reduced sample size. In this study, we used a mouthpiece to deliver the gas. An alternative approach would be to use a face mask. Our experience with the face mask method was that the small size of the head coil sometimes limits the placement of the face mask on the subject. Our group is actively exploring non-gas methods to measure cerebrovascular reactivity [83]. A third limitation of this study is that the metabolic parameters, i.e., OEF and CMRO2, are global only, without spatial resolution. It should be pointed out that technically it is challenging to measure OEF and CMRO2 without using any exogenous tracers. Thus, our CMRO2 technique represents the state-of-the-art in terms of non-invasive physiological imaging technologies and the results do provide a confirmation that enhanced oxygen delivery (via CBF) resulted in more oxygen consumption by the brain tissue. Nonetheless, a lack of spatial information makes it difficult to determine which brain region(s) contributes to the observed increase in global metabolic rate and, therefore, this is a limitation.
In conclusion, for the specific case of PDE5 inhibitors there may be both vascular and non-vascular mechanisms of action that may be relevant for potential therapeutic effects in AD. In our study, we chose to focus on the measurement of vascular effects. The present study assessed CBF and global CMRO2 before and after a single 50 mg dose of sildenafil with several non-invasive, novel MRI techniques and revealed that sildenafil could improve CBF in AD patients, especially in the bilateral medial temporal lobes. Moreover, global CMRO2 also significantly increased after taking drug which indicates that, for the brain as a whole, sildenafil improves oxygen consumption in AD patients. Additionally, our CVR results suggest that the enhancement in CBF mentioned above was achieved at a cost of cerebral vascular reserve, as CVR is reduced following sildenafil administration. Taken together, the findings from this study suggested that perfusion-enhancing medications such as sildenafil present a potential opportunity for therapeutic interventions in AD. MRI-based vascular and metabolic measures may be used as target engagement or treatment monitoring markers in such interventional trials. Among the MRI measures investigated, pCASL MRI appears to provide the most promising index given its regional sensitivity and the convenience of the measurement.
Footnotes
ACKNOWLEDGMENTS
The authors are grateful to Yamei Cheng for helping with experiments, to Kristin Martin-Cook for her efforts in the initiation of the study, and to Dr. Samarpita Sengupta for editorial assistance.
Funding for this research was provided by NIH P30 AG012300, NIH R01 MH084021, NIH R01 NS067015, NIH R01 AG042753, and NIH R21 NS078656.