
Abstract
Alzheimer’s disease (AD) represents the most frequent neurodegenerative disease of the human brain worldwide. Currently practiced treatment strategies for AD only include some less effective symptomatic therapeutic interventions, which unable to counteract the disease course of AD. New therapeutic attempts aimed to prevent, reduce, or remove the extracellular depositions of the amyloid-β protein did not elicit beneficial effects on cognitive deficits or functional decline of AD. In view of the failure of these amyloid-β-based therapeutic trials and the close correlation between the brain pathology of the cytoskeletal tau protein and clinical AD symptoms, therapeutic attention has since shifted to the tau cytoskeletal protein as a novel drug target. The abnormal hyperphosphorylation and intraneuronal aggregation of this protein are early events in the evolution of the AD-related neurofibrillary pathology, and the brain spread of the AD-related tau aggregation pathology may possibly follow a corruptive protein templating and seeding-like mechanism according to the prion hypothesis. Accordingly, immunotherapeutic targeting of the tau aggregation pathology during the very early pre-tangle phase is currently considered to represent an effective and promising therapeutic approach for AD. Recent studies have shown that the initial immunoreactive tau aggregation pathology already prevails in several subcortical regions in the absence of any cytoskeletal changes in the cerebral cortex. Thus, it may be hypothesized that the subcortical brain regions represent the “port of entry” for the pathogenetic agent from which the disease ascends anterogradely as an “interconnectivity pathology”.
Keywords
ALZHEIMER’S DISEASE
The clinical and major neuropathological features of Alzheimer’s disease (AD) were first described in a seminal report by the German psychiatrist and neuroscientist Alois Alzheimer [i.e., amyloiddepositions and neurofibrillary tangles (NFT)] in 1907 and by Oskar Fisher (i.e., senile plaques) in the same year [1–8]. AD accounts for more than 65% of patients suffering from late-life dementia. It commonly begins insidiously in late adulthood and subsequently takes a progressive, relentless, and disabling disease course, resulting in a serious and irreversible decline of many essential mnestic and cognitive functions. Thus, AD constitutes a tremendous burden for the patients, their families and caregivers, involved health systems, and social economies [2, 7–16].
AD is a heterogeneous disorder that includes an early-onset form and a much more common late-onset form. Afflicting more than 95% of AD patients, the exact molecular biological causes of the late-onset AD form are still enigmatic. It is therefore regarded to occur sporadically, whereby advanced age and the presence of the APOE ɛ4 allele represent the strongest risk factors. The early-onset AD form concerns less than 5% of AD patients and is caused by specific point mutations in the transmembrane amyloid precursor protein, presenilin-1, or presenilin-2 [2, 18].
The symptoms emerging during the clinical phase of AD comprise severe disturbances of the normal wake/sleep patterns and sleep architectonics, extrapyramidal motor and oculomotor dysfunctions, dysphagia, severe autonomic symptoms and weight loss, a large variety of neuropsychiatric symptoms (e.g., depression, apathy, agitation, anxiety, delusions, hallucinations), and eventually the progressive deterioration of vital cognitive and memory functions. These symptoms of AD sooner or later lead to progressive limitations of the autonomy, judgement, ability to reason, reduced accountability, loss of orientation, disintegration of the personality, encroachments of activities of daily living, and dependency on supervision and care [2, 7–10]. This clinical phase of AD commonly becomes obvious only in late adulthood. It is preceded by a long, asymptomatic, pre-clinical, and a prodromal phase of approximately 30–40 years with beginning mild symptoms (i.e., little neuropsychiatric, neurological, or somatic symptoms). The late symptomatic clinical phase of AD lasts approximately 10–15 years before the death of the multimorbid patients. A frequent cause of their death is dysphagia-related aspiration pneumonia [3, 19].
At the moment, clinical treatment strategies commonly are initiated only during the late symptomatic clinical phase of AD and include rather limitedsymptomatic therapeutic interventions to control select AD symptoms or to serve neuroprotection (e.g., acetylcholinesterase inhibitors to increase the concentration and duration of acetylcholine at sites of neurotransmission, NMDA receptor antagonists for treatment of moderate-to severe AD, which regulates excitatory glutamatergic function). However, all of these symptomatic treatment approaches are associated with side effects and are initiated too late for the meanwhile severely affected patients and have at best minimal if any effect on the course of AD[2, 19].
THE MAIN NEUROPATHOLOGICAL FEATURES OF ALZHEIMER’S DISEASE
The major neuropathological features of AD are: the early onset of selective and ongoing neuronal loss at strategically important cortical and subcortical brain sites (i.e., transentorhinal and entorhinal regions, hippocampus, amygdala; cholinergic nuclei of the basal forebrain; dopaminergic substantia nigra and ventral tegmental area of the midbrain; noradrenergic locus coeruleus of the pons; serotonergic midbrain and pontine raphe nuclei; cholinergic pedunculopontine nucleus), the intraneuronal accumulations of the aggregations of the hyperphosphorylated tau cytoskeletal protein and the extraneuronal depositions of the fibrillary and insoluble amyloid-β protein [17, 19–31].
TAU CYTOSKELETAL PROTEIN AND AD-RELATED NEUROFIBRILLARY PATHOLOGY
The tau cytoskeletal protein is abundantly expressed in axons of central nervous system neurons and is also present in the somatodendritic compartment of neurons, oligodendrocytes, and non-neural tissues. Tau binds to axonal microtubules and is therefore an important microtubule associated protein (MAP). As a MAP, the normal tau protein is an integral constituent of the axonal cytoskeleton of nerve cells and guarantees the accurate polymerization and maintenance of the structural stability of the microtubule rails of axonal transport mechanisms. One of the most important post-translational modifications of the tau protein is its phosphorylation at distinct sites of the molecule, which involves the cooperation of multiple protein kinases and phosphatases and regulates the biological activity of tau (Fig. 1). This interplay between kinases and phosphatases inexplicably loses its balance in AD and ultimately leads to the hyperphosphorylation of the tau cytoskeletal protein, conformational changes of tau, its insolubility, the tendency of tau to self-aggregation, and the deposition of its aggregated accumulations in diseased nerve cells. The pathological hyperphosphorylation of tau impairs its interaction with microtubules, and results in the deteriorations of intra-axonal transport mechanisms[3, 32–34].
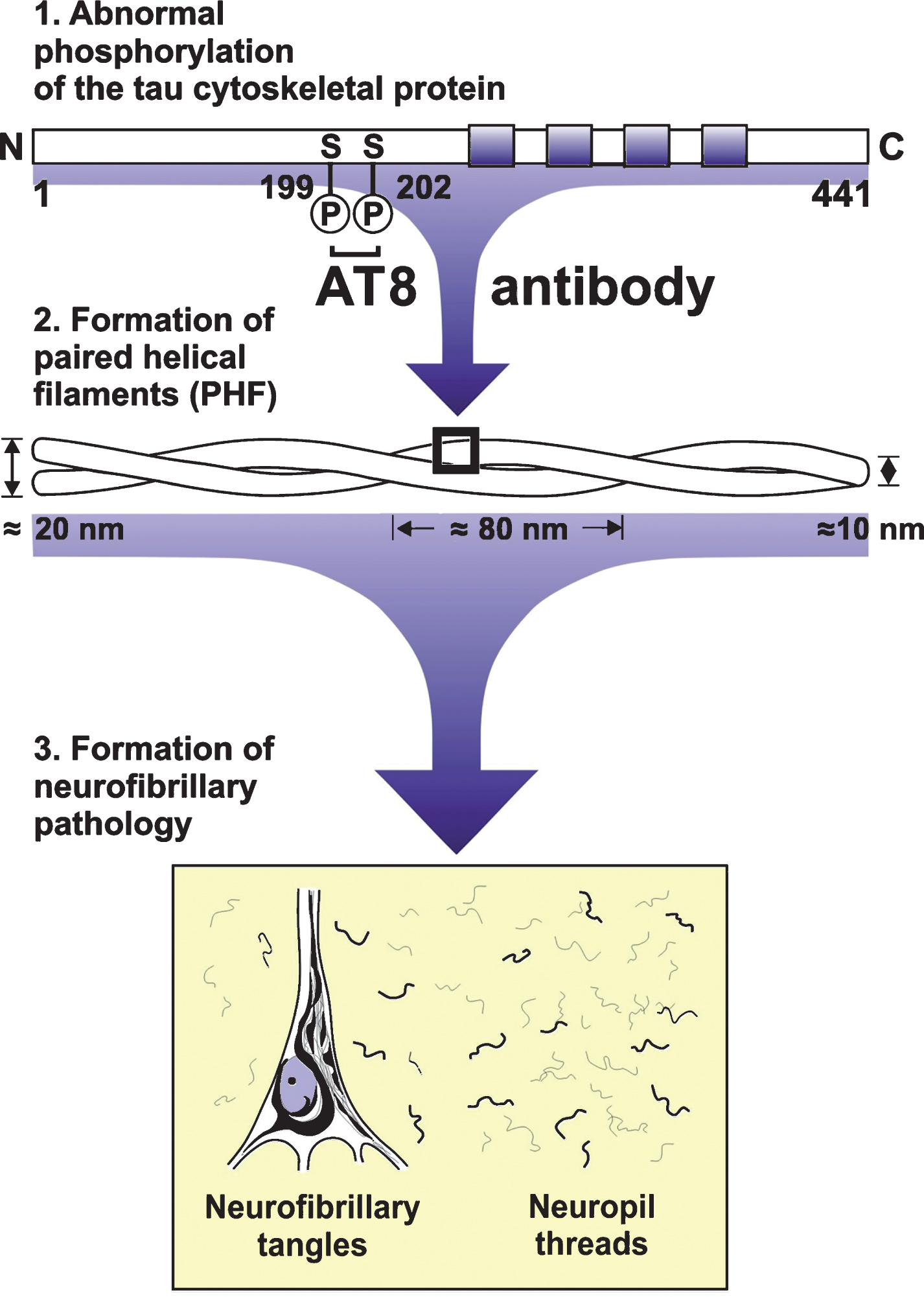
Sequence of the intraneuronal formation of Alzheimer’s disease (AD)-related neurofibrillary pathology. The tau cytoskeletal protein is categorized as a microtubule associated protein and an integral constituent of the axonal cytoskeleton of nerve cells. One of the most important post-translational modifications of the tau protein is its phosphorylation at distinct sites of the molecule. Owing to an imbalance of involved protein kinases and phosphatases, the tau cytoskeletal protein becomes hyperphosphorylated in AD. This hyperphosphorylation results in conformational changes of tau, its insolubility, self-aggregation, and deposition of its aggregated accumulations in diseased nerve cells. The hyperphosphorylated tau cytoskeletal protein contributes to the structural core of paired helical filaments. These abnormally-wound helical filaments constitute the major protein incorporated into the insoluble, non-degradabl,e and argyrophilic neurofibrillary tangles in neuronal perikarya and dendritic neuropil threads, which are formed by the intraneuronal cross-linkage of these paired helical filaments. AT8, AT8 antibody; C, C-terminus; N, N-terminus; S, Serine.
Electron-microscopically, neurofibrillary pathology consists of abnormally wound helically filaments (i.e., paired helical filaments, PHF), whose intraneuronal cross-linkage forms the insoluble, non-degradable and argyrophilic NFT in neuronal perikarya and dendritic neuropil threads (NT) (Figs. 1 and 2). [3, 35]. “Ghost” or “tombstone” tangles are most frequently present in early and severely affected cortical brain regions (e.g., entorhinal region, hippocampus) and are regarded as evidence that the occurrence of these NFT is associated with the demise of diseased nerve cells. These extraneuronal, argyrophilic tangles represent non-degradable, filamentous residues of previously intraneuronal NFT and remain in the brain neuropil at the position of nerve cells that died earlier. The mature intraneuronal NFT and NT and the ghost or tombstone tangles can be highlighted with a high sensitivity by traditional and advanced silver techniques (e.g., Bielschowsky staining, Gallyas staining) (Fig. 2) [3, 35].
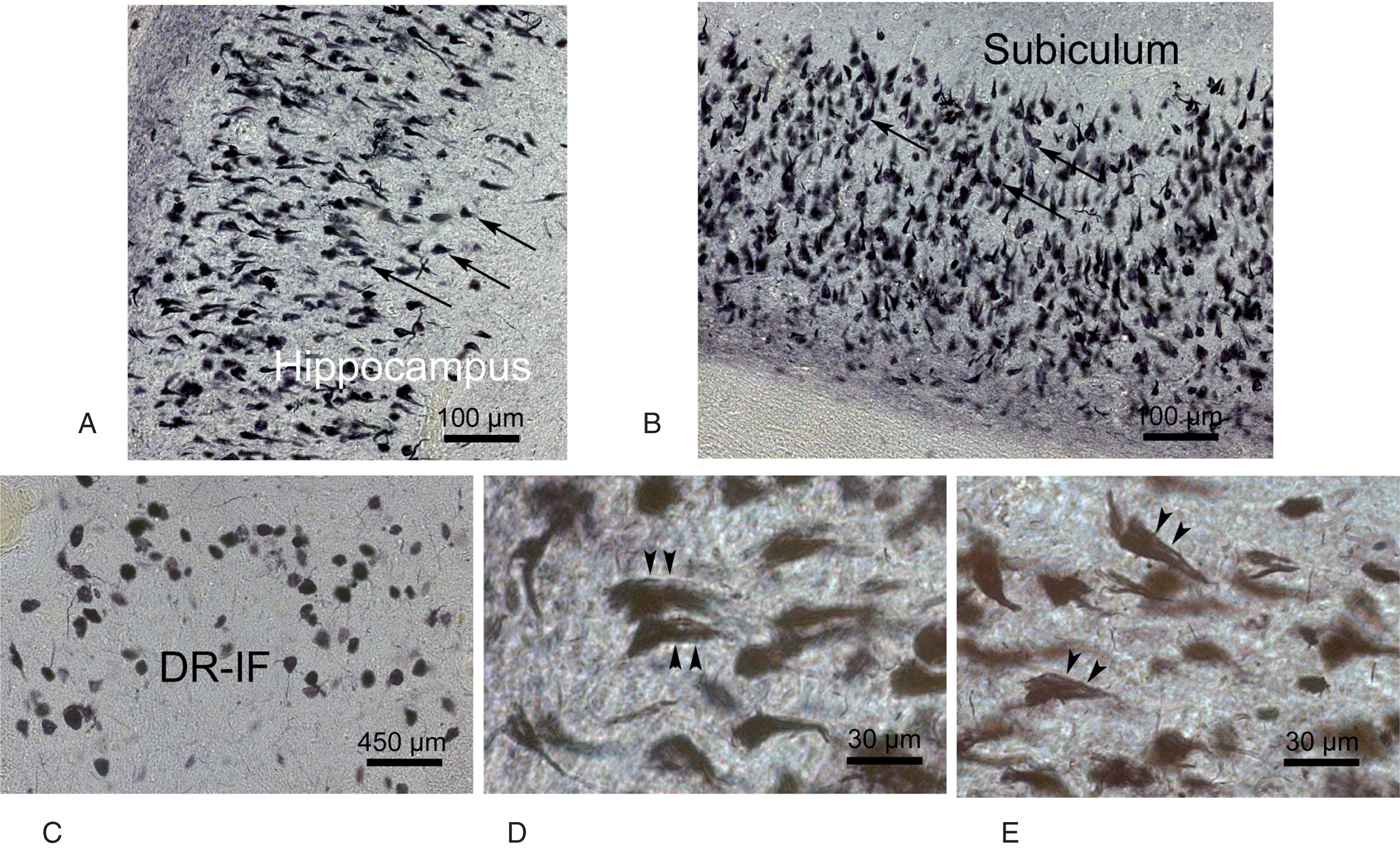
The insoluble, non-degradable and argyrophilic neurofibrillary pathology in a demented Alzheimer’s disease (AD) patient in the clinical Braak stage V. A) Dense network of predominant flame-shaped argyrophilic intraneuronal neurofibrillary tangles (NFT) (arrows) in the hippocampus of a demented AD patient in the advanced Braak stage V. B) An abundance of argyrophilic, flame-shaped intraneuronal NFT (arrows) in the adjacent subiculum of the same demented AD patient. C) Numerous argyrophilic, globose intraneuronal NFT (arrows) in the interfascicular part of the midbrain dorsal raphe nucleus (DR-IF). Arrowheads point to argyrophilic, dendritic neuropil threads. C, D) Extraneuronal, argyrophilic non-degradable, filamentous residues of previously intraneuronal NFT (i.e., ghost or tombstone tangles) in the severely affected hippocampus of this demented AD patient (A-D: 100 μm PEG sections, Gallyas silver impregnation).
Hyperphosphorylated tau protein contributes to the structural core of PHF and constitutes the major protein of NFT and NT. Antibodies raised against hyperphosphorylated tau protein (e.g., AT8 antibody) decorate mature AD-related NFT with nearly the same sensitivity as the traditional and/or advanced silver techniques (Fig. 1). They also allow for the demonstration of even traces of non-argyrophilic, initial accumulations of the cytoskeletal tau protein (i.e., pretangle material) in the somata, dendrites, and axons of affected nerve cells. Therefore, anti-tau antibodies reveal the very early events in the pathological cascades of the evolution of AD-related neurofibrillary or cytoskeletal pathology (Fig. 3)[3, 35].

The immunoreactive tau cytoskeletal pathology in the allocortical predilection sites of asymptomatic individuals in the preclinical Braak stage I. Nerve cells of the superficial pre-alpha layers of the allocortical transentorhinal and entorhinal regions affected by the immunoreactive tau cytoskeletal pathology in healthy individuals in the preclinical Braak stage I. A) Entorhinal region. B) Transentorhinal region (arrows). C) Transentorhinal region (A-C: 100 μm PEG sections, anti tau immunocytochemistry with the AT8 antibody).
As with the AD-related β-amyloidosis, the evolution and brain spread of AD-related tau cytoskeletal pathology is by no means a random event. The underlying disease process is highly systematic and follows a predictable path throughout the brain, apparently along strictly defined anatomical pathways. It affects only select subcortical nuclei and areas and neuronal layers of the cerebral cortex in highly stereotypical temporal and spatial sequences. This predetermined, domino-like and inter-individually consistent brain spread ultimately results in a pathognomonic topographic distribution pattern of the tau cytoskeletal pathology in the brains of AD patients (Fig. 4), which correlates significantly with the clinical symptoms of AD and contributes substantially to the severe cognitive and mnestic impairments of AD patients[2, 38].

The immunoreactive tau cytoskeletal pathology in subcortical nuclei of demented patients in Braak stage V. The tau cytoskeletal pathology immunoreactive for the monoclonal AT8 antibody in select subcortical nuclei of the limbic system of demented individuals in Braak stage V. A) Nucleus of the horizontal band of Broca (DBH). B) Nucleus of the vertical band of Broca (DBV). C) Hypothalamic tuberomammillary (TM) and lateral tuberal nuclei (LT). D) Anterodorsal nucleus of the thalamus (AT) (A-D: 100 μm PEG sections, anti tau immunocytochemistry with the AT8 antibody).
THE BRAAK AND BRAAK AD STAGING SYSTEM
The Braak and Braak staging system describes the consistent targeted spatial spread and increasing severity of the AD-related cytoskeletal pathology in the cerebral cortex. In the cerebral cortex, the allocortical transentorhinal region is regarded as the first (Braak stage I) and the allocortical entorhinal region (Braak stage II) as the second cortical region, which exhibit nerve cells immunoreactive for the tau cytoskeletal pathology. From the allocortical transentorhinal and entorhinal regions, the AD-related cortical cytoskeletal pathology extends into the hippocampal formation (Braak stage III) and ultimately reaches the association (Braak stages IV and V) and primary fields of the neocortex (Braak stage VI). Although argyrophilic NFT also become evident in brainstem nuclei in the very early AD phases, the Braak and Braak staging system did not include all of these argyrophilic subcortical NFT [8, 30]. As cross-sectional studies repeatedly demonstrated, the Braak and Braak staging system roughly correlates with the clinical course AD: Braak stages I and II correspond to the asymptomatic, preclinical phase of AD, Braak stages III and IV to the prodromal phase of AD or incipient AD, and Braak stages V and VI to the clinical phase of AD (Fig. 5) [2, 39].
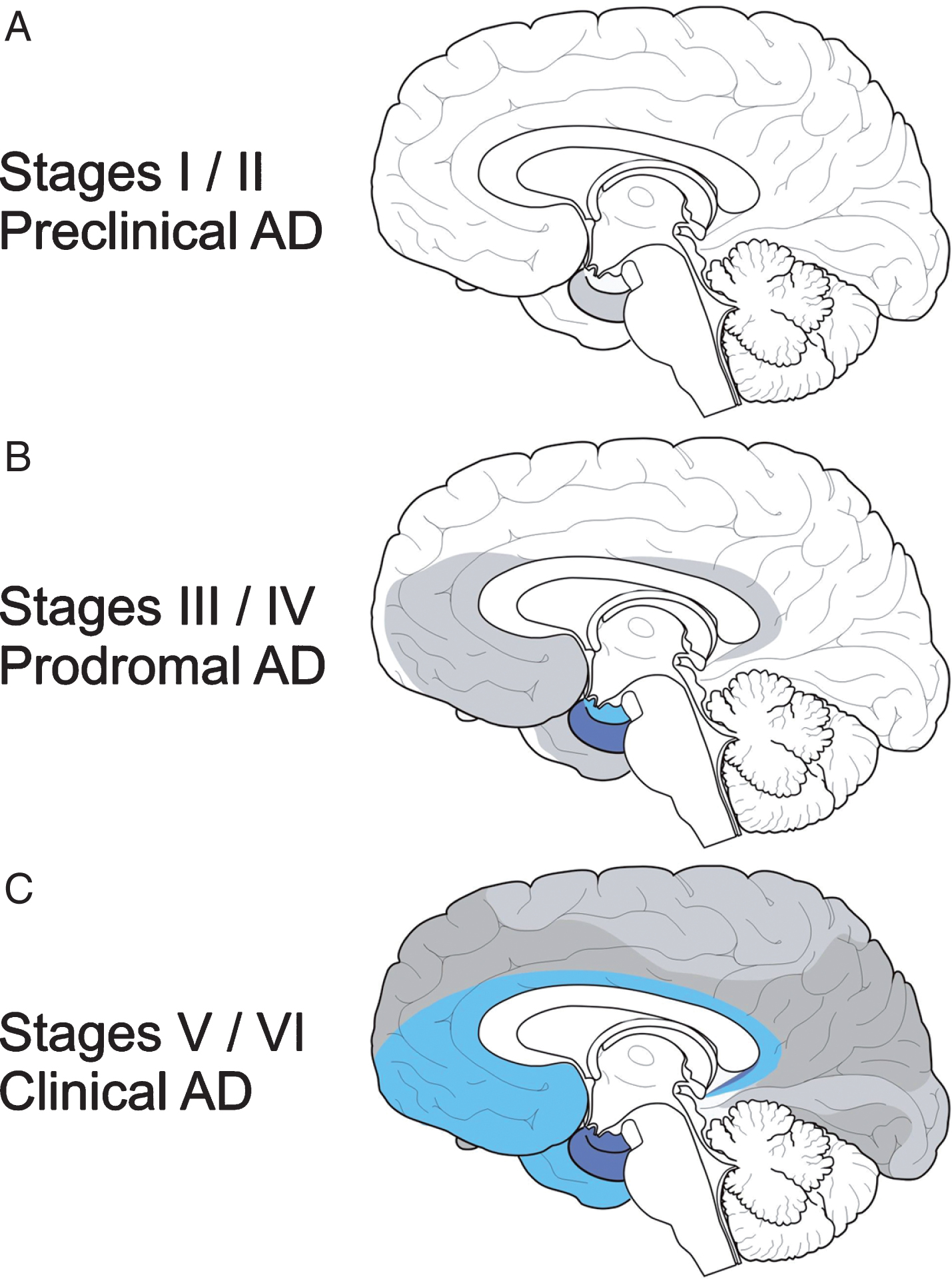
The evolution of the cytoskeletal pathology in the cerebral cortex and the Braak and Braak staging system. In the cerebral cortex, the allocortical transentorhinal region is the first (Braak stage I) and the allocortical entorhinal region (Braak stage II) the second cortical region affected by the tau cytoskeletal pathology. In Braak stage III, the hippocampal formation also becomes involved. In Braak stages IV and V, the Alzheimer’s disease (AD)-related tau cytoskeletal pathology reaches the association cortex and in Braak stage VI, the primary neocortical fields. Braak stages I and II correspond to the asymptomatic, preclinical phase of AD, Braak stages III and IV to the prodromal phase of AD or incipient AD, and Braak stages V and VI to the clinical phase of AD.
Although Braak and colleagues proposed a modification to the original Braak staging system and introduced the brainstem locus coeruleus as the possible begin of the AD-related cytoskeletal pathology [3], they did not recognize the real extent of Braak stage 0 cytoskeletal pathology as recently revealed by Stratmann et al. [9].
Dementia is regarded as the core symptom of AD and occurs comparatively late during AD (i.e., in the final clinical phase of AD) and is commonly associated with Braak stage AD stage V or VI cortical tau cytoskeletal pathologies (Fig. 5). Therefore, AD is still considered as an age-associated disease. However, the Braak and Braak staging system and further postmortem cross-sectional studies of the evolution of AD-related tau cytoskeletal pathology show that the underlying disease process begins earlier and is by no means restricted to the clinical phase or the period of advanced age (i.e., Braak stages V and VI). In contrast, this pathological process is already operating in the protracted asymptomatic pre-clinical (i.e., Braak stages I and II) and in the prodromal phase of AD with the first mild symptoms (i.e., Braak stages III and IV). It thus precedes the onset of the dementing syndrome by approximately three to four decades and often already starts in early adulthood (Figs. 5, 6) [2, 40].
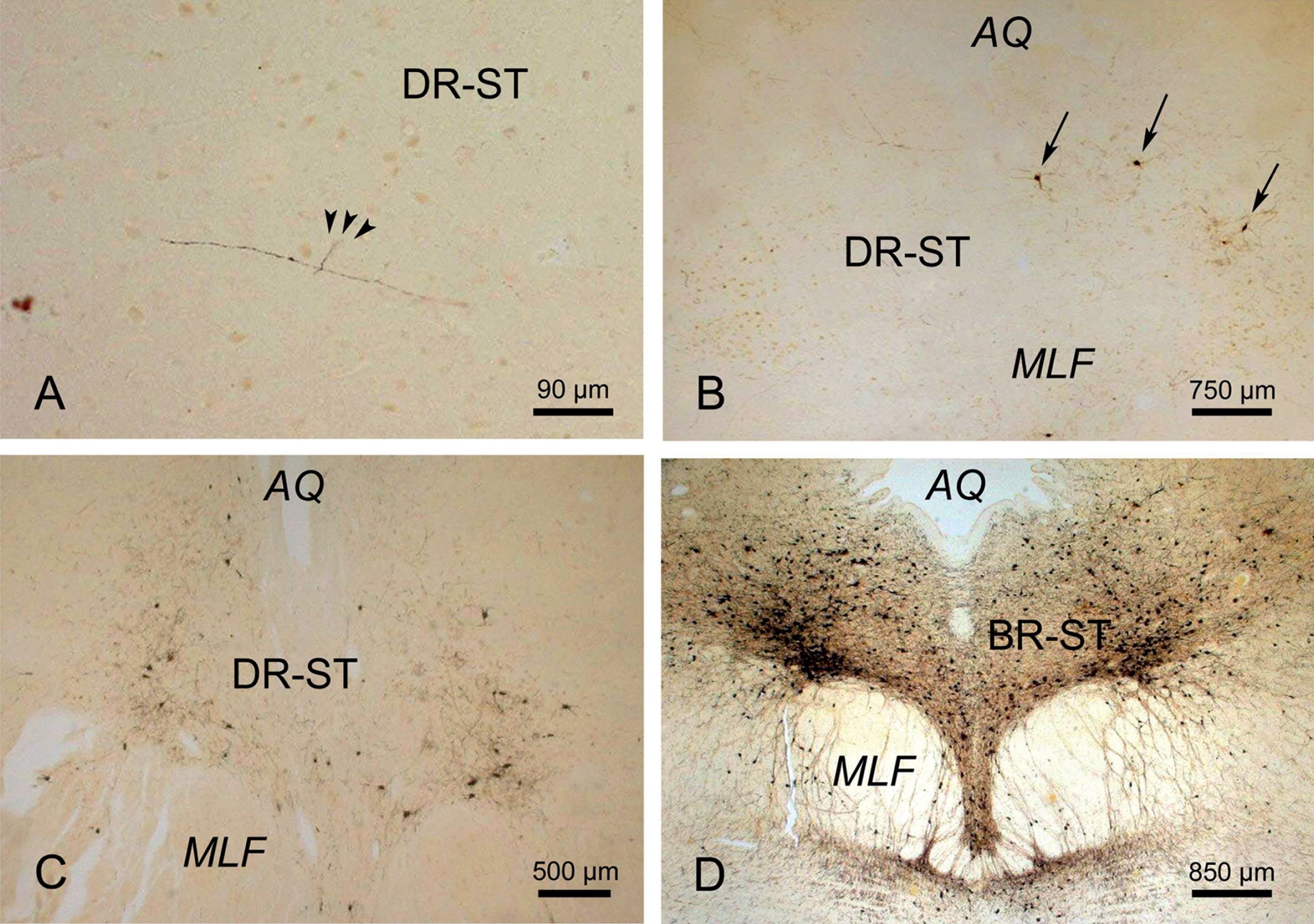
The evolution of the immunoreactive Alzheimer’s disease (AD)-related tau cytoskeletal pathology in the midbrain supratrochlear part of the dorsal raphe nuclei. The progression of the tau cytoskeletal pathology immunoreactive for the monoclonal AT8 antibody from the early precortical into the late clinical phases of AD. A) Initial discrete traces of tau immunoreactive cytoskeletal changes (arrowheads) in the supratrochlear part of the dorsal raphe nucleus (DR-ST) of an individual in the asymptomatic, precortical Braak stage 0. B) AT8 immunopositive nerve cells (arrows) in the DR-ST of an individual in the preclinical Braak stage I. C) Numerous tau immunopositive nerve cells in the DR-ST of an individual in prodromal or incipient AD (i.e., Braak stage III). D) Well-developed immunoreactive tau cytoskeletal pathology in the DR-ST of an individual in the advanced clinical Braak stage V. (A-D: 100 μm PEG sections, anti tau immunocytochemistry with the AT8 antibody). AQ, aqueduct; MLF, medial longitudinal fascicle.
The typical clinical symptoms of AD develop insidiously in the prodromal phase of the disease. The rates at which people decline from the prodromal to the clinical disease phase, however, differ inter-individually substantially and are still not fully understood. However, known risk factors that are associated with a faster decline despite the same degree of brain pathology include decreased reserve capacity of the brain (i.e., is number of neurons and their synaptic and dendritic arborization together with lifestyle-related cognitive strategies), reduced brain size, low educational and occupational attainment, low mental ability in early life, and reduced mental and physical activity during late life [10, 13].
AMYLOID-β PROTEIN, THE AMYLOID CASCADE THEORY, AND THE THAL AD PHASES
The significance of the tau cytoskeletal pathology in AD for a long time remained in the shadow of the amyloid cascade theory. Amyloid-β is an extracellular, proteinaceous deposit, which is posttranslationally cleaved from the amyloid-β protein precursor by the proteolytic enzymes β- and γ-secretases. The amyloid-β protein precursor is an integral transmembrane protein and has been implicated as a regulator of synaptic formation and repair and apparently mediates the interaction between cargoes and the motor protein kinesin of anterograde axonal transport mechanisms [2, 41–43].
Insoluble, extraneuronal amyloid-β plaques can be present in the brains of symptomatic AD patients as well as in individuals in the pre-clinical and prodromal phases of AD and can be visualized by classical histological techniques (i.e., Congo red) or immunocytochemical techniques (Fig. 7). The most popular theory for the pathomechanism of AD, the amyloid cascade hypothesis, deals with a pathogenetic link between the evolution of the intraneuronal tau pathology and the extracellular neuropil deposits of the insoluble amyloid-β protein. This simplified, causal theory has for a long time served as the platform for many AD therapeutic trials and suggests that the accumulation of amyloid-β peptides is the primary pathogenic driver that induces and triggers a cascade of fatal downstream changes. These downstream changes eventually culminate in the development of the AD-related tau cytoskeletal pathology, synaptic and neuronal loss [2, 41–44].
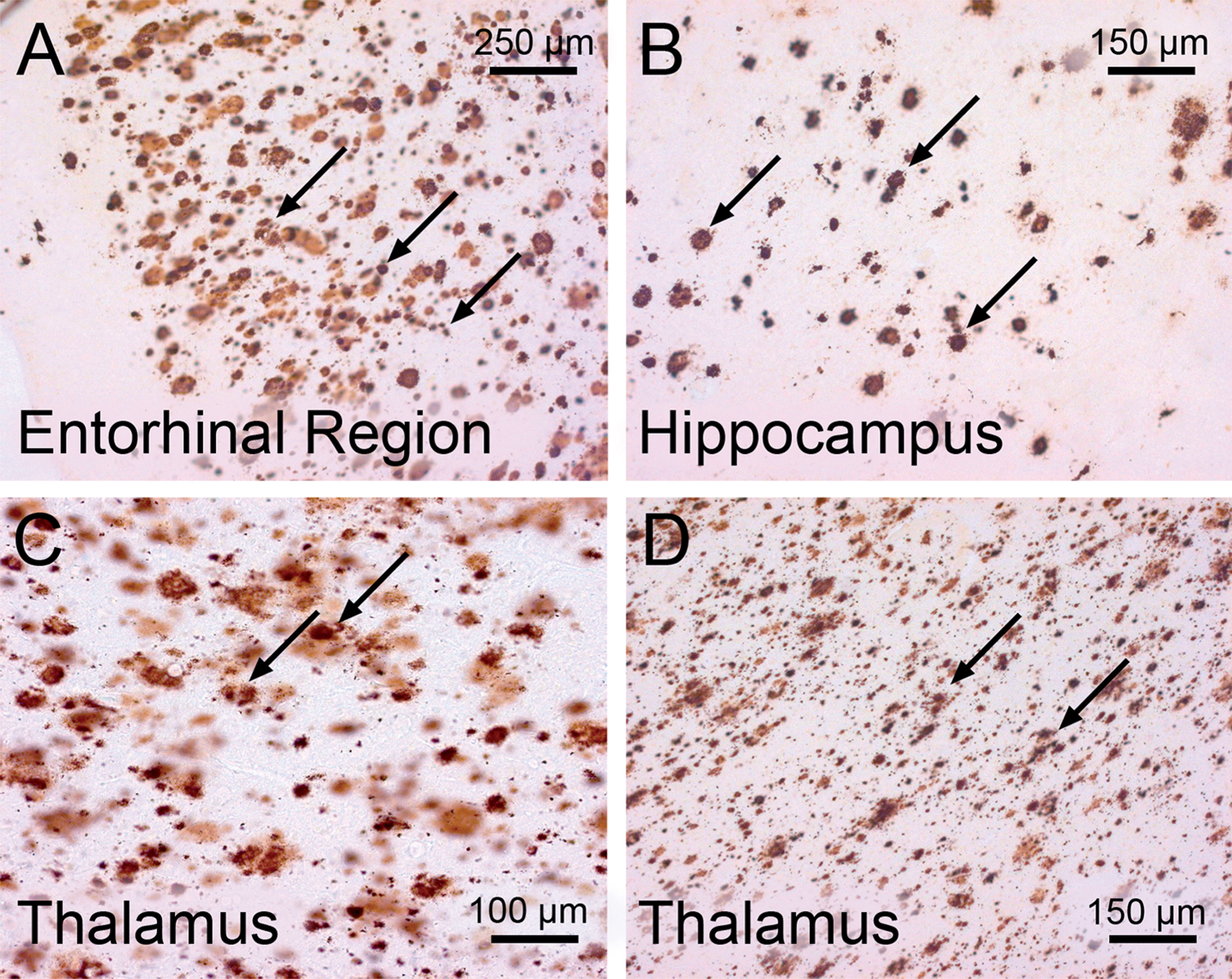
Extraneuronal depositions of the insoluble amyloid-β protein in the brain of a demented patient in the clinical phase of Alzheimer’s disease (AD). Immunoreactive amyloid-β deposits (arrows) in the cerebral cortex and thalamus of a demented AD patient (Braak stage V; Thal phase 4). A) Entorhinal region. B) Hippocampus. C) Mediodorsal nucleus of the thalamus. D) Ventrolateral nucleus of the thalamus. (A-D: 100 μm PEG sections, anti-amyloid-β immunocytochemistry with the 4G8 antibody).
AD-related brain β-amyloidosis commonly starts in the pre-dementia phase of AD and also spreads in the brains of patients in specific temporal and topographical sequences [3, 40], which were compiled by Thal et al. into five consecutive developmental phases. In the brains of demented AD patients, β-amyloidosis is eventually present in all parts of the allo- and neocortex, amygdala, basal forebrain, striatum, hypothalamus, thalamus, and in the cerebellum, but only in a few brainstem nuclei (Fig. 7) [3, 44]. Although the brain expansion of β-amyloidosis may already begin in the asymptomatic, pre-clinical phase of AD, the development of AD-related tau cytoskeletal pathology clearly precedes the onset of AD-related brain β-amyloidosis in the overwhelming majority of diseased brain regions [3, 18]. Additionally, the findings that the routes of AD-related tau pathology and β-amyloidosis do not coincide and lead to non-matching distribution patterns of AD-related neuronal and extraneuronal protein pathologies questions the universal validity of the amyloid cascade theory [3, 44].
THERAPEUTIC IMMUNOLOGICAL ANTI-β-AMYLOID APPROACHES
As a direct consequence of the prevailing amyloid cascade theory, disease-modifying therapies for AD have mainly focused on the reduction of amyloid-β depositions in the brain. These therapeutic strategies were aimed at either the enhancement of amyloid-β clearance by proteolytic enzymes or the prevention or reduction of aggregations of the amyloid-β protein. In order to prevent, reduce, or remove aggregations of amyloid-β proteins in the brain, passive and active immunization approaches were created
But until now, all anti-amyloid-β approaches most likely have been started too late during AD, were of limited efficacy, and produced serious side effects (e.g., meningoencephalitis, brain edema, brain microhemorrhage) [2, 36]. Unfortunately, more than 100 candidate treatment compounds have failed to prevent, reduce, or remove amyloid-β 1–42 deposits. Although active immunization was able to successfully clear amyloid-β plaques from the brains of AD patients, they did not, however, alter NFT load, and the treated patients did not show any decrease in the rate of clinical deterioration so far [2, 40–43]. Accordingly, many researchers in the field have started to search for new therapies related to alternative therapeutic targets in AD. In the course of these recent developments, the tau cytoskeletal protein has received increasing attention as a target candidate for therapeutic approaches in AD. Since abnormal tau phosphorylation and aggregation are early events in the development of AD-related neurofibrillary pathology, targeting the tau brain pathology has been considered to be more effective and is currently considered to represent the most promising therapeutic approach for AD. In animal models, antibodies generated were found to cross the blood-brain barrier, bind to phosphorylated tau, and reduce pathology without significant adverse effect thus providing strong support in favor of the idea that it is possible to reduce tau-related pathology with immunization [2, 36–38].
THERAPEUTIC ANTI-TAU APPROACHES
Following the anti-tau approach, therapeutic interventions should optimally target the hyperphosphorylated tau molecule during the very early pre-tangle phase rather than during its condensation to mature, metastabile, and argyrophilic NFT [16, 38]. In the hope that they can prevent the manifestation of cognitive changes or can at least slow down the cognitive decline in AD, various therapeutic approaches have been developed that target the tau protein at different stages of its posttranslational processing, self-aggregation, brain spread, or degradation [12, 43]. These approaches take different pathophysiological mechanisms into consideration. They aim (1) to influence tau hyperphosphorylation by engineering the enzymatic activity of the tau kinases or phosphatases (e.g., lithium and valproate as tau kinase inhibitors) [12, 43]; (2) to inhibit phosphorylated tau aggregation by interrupting tau-tau-interactions (e.g., methyleneblue and its derivates, phenothiazines) [12, 43]; (3) to achieve active immunization with tau phosphopeptides or passive immunization with tau antibodies to remove aggregations of the tau protein and to interrupt their brain spread [18, 38]; and (4) to promote tau degradation via the ubiquitin-proteasome-lysosomal pathway [36]. The attraction of antibody therapies lies in the high selective nature of antibodies and their comparatively low systemic toxicity. The active tau vaccine approach engages the cellular and humoral immune system, elicits an immune response that triggers the maturation of B and T cells, and generates high affinity antibodies against the administered antigen. In a passive approach, the triggering of the innate immune system is circumvented by infusing a specific antibody against the antigen or parts of it and the inherent clearance system then removes the antibody-bound ligand. The main hypothesis driving the development of anti-tau antibodies is that the antibody will enter the brain and bind the pathological species. The anti-tau antibody will then engage an extracellular and/or intracellular tau species, and thereby promote their clearance and prevent the further spread of pathology [37, 38].
THE BRAIN SPREAD OF THE TAU CYTOSKELETAL PATHOLOGY MAY OCCUR ACCORDING TO THE PRION HYPOTHESIS
Increasing empirical evidence favors the view that the anatomical interconnectivities play a crucialfactor for the pathophysiology of AD which determine the vulnerability or the resistance of select brain regions to AD-related tau aggregation pathology. Based on this hypothesis, AD-related tau aggregation pathology may spread transneuronally along interconnecting fibers in an inter-individually consistent pattern [7–9, 45–47] and result in a strikingly characteristic brain distribution pattern [3, 39]. Thus, AD can now be considered as a ‘connectivity disease’ (Fig. 8). This new concept conforms to the hypothesis that (1) AD may belong to the novel group of ‘prion-like’ neurodegenerative diseases and (2) that the stereotypical transneuronal brain spread of AD-related tau cytoskeletal pathology in fact could reflect transsynaptic transmission according to the new prion hypothesis [3, 48].
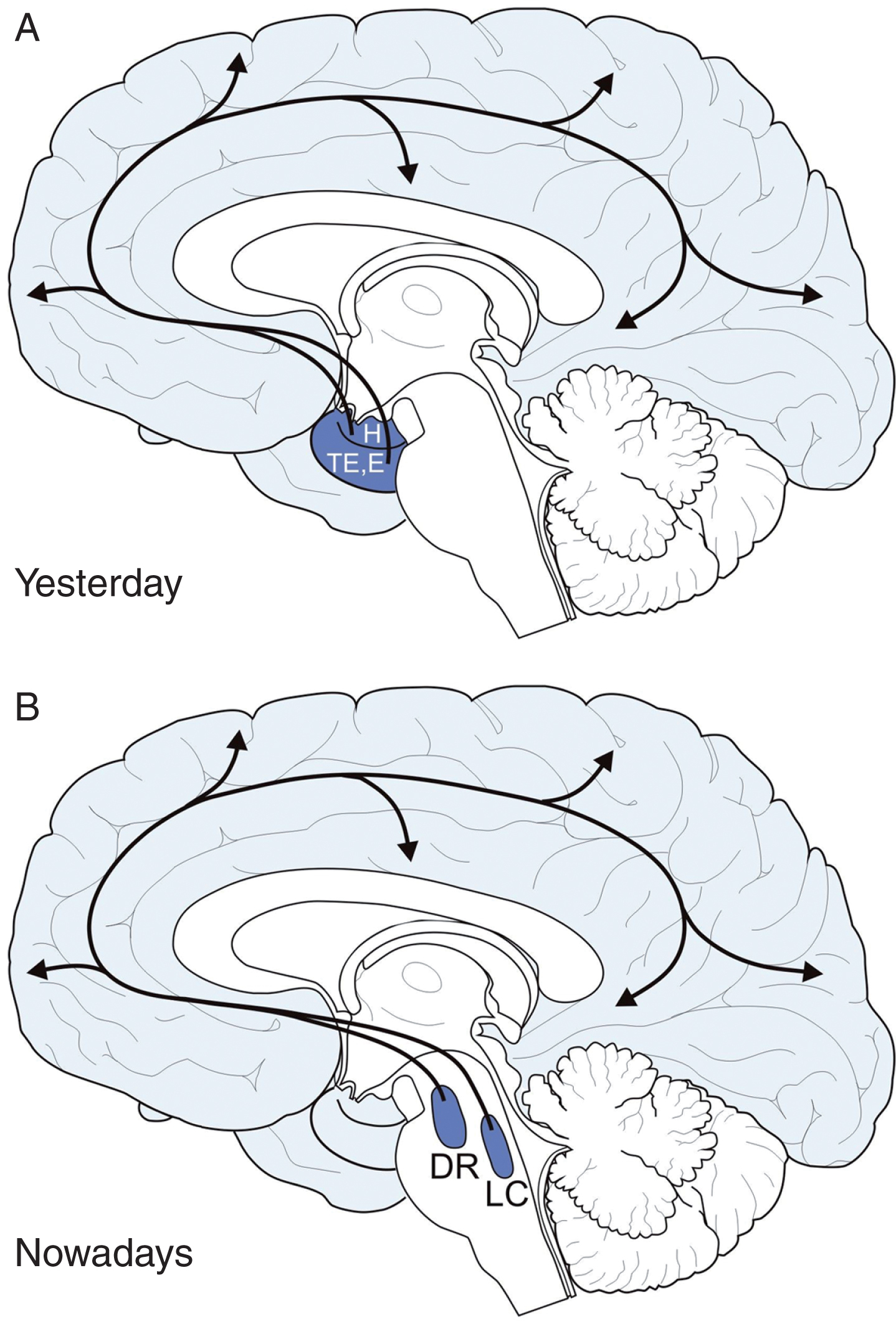
Origin of the Alzheimer’s disease (AD)-related tau cytoskeletal pathology in the brainstem. The evolution and brain expansion of AD-related tau cytoskeletal pathology is highly systematic. This pathology spreads in a highly systematic manner and in stereotypical temporal and spatial sequences throughout the brain. Owing to the transneuronal spread of this pathology along strictly defined anatomical pathways AD is currently regarded as a ‘connectivity disease’ of the human brain. A) AD has been for a long time regarded as a primary disease of the cerebral cortex. According to this outdated opinion the allocortical transentorhinal (TE) and entorhinal regions (E) and the hippocampus (H) for nearly twenty-five years have been believed to represent the sole brain regions which are initially affected by the tau cytoskeletal pathology. B) Recent pathoanatomical studies, however, have demonstrated that the initial tau cytoskeletal pathology occurs in the brainstem (i.e., midbrain dorsal raphe nuclei, DR, and or locus coeruleus, LC) in the absence of any cytoskeletal changes in the allocortex. DR, dorsal raphe nuclei; E, entorhinal region; H, hippocampus; LC, locus coeruleus; TE, transentorhinal region.
The concept that hyperphosphorylated tau protein may behave like a prion-like protein requires a variety of crucial assumptions: (1) a potential conformationally corrupted proteinaceous aggregates would enter the brain, (2) these aggregates would propagate in a transneuronal manner along anatomical pathways, (3) the local templated conversion of the native and soluble cytoplasmic tau proteins would be induced in the next nerve cell of the affected neuronal chain and leads to its self-aggregation, and (4) the newly synthesized prion-like aggregations would be released from host nerve cells and taken up by the next disease-prone nerve cell in the neural chain [24, 48].
Although the chronic disease spread according to the prion hypothesis is currently the prevailing pathophysiological idea of AD, further studies are required to support this new scientific idea and to prove unequivocally the prion-like nature and behavior of hyperphosphorylated tau protein [9]. In particular, the relationship between tau localization in neurons, tau release, and tau uptake needs to be established, as does the pathophysiological role of extracellular tau. In addition, more research is needed to identify the pathophysiological mechanisms that drive the release and propagation of pathogenic tau. Other open key questions include the nature of the tau species involved in the precise seeeding/templating mechanisms [49, 50].
According to the new prion hypothesis, tau immunotherapy may be a potentially effective strategy if initiated at early stages. With regard to anti-tau antibodies, these would have to enter the brain, bind to extracellular or intracellular tau species, promote their clearance, and eventually prevent the spread of the tau pathology to neighboring nerve cells. These considerations relate to the following preconditions: (1) The normally phosphorylated cytosolic tau protein is involved in the maintenance of axonal transport mechanisms [5, 42]. (2) The hyperphosphorylated tau protein may behave like corruptive prion protein (i.e., it is insoluble, relatively resistant to protease digestion, tends to aggregate, and seems to propagate by corruptive protein templating. (3) The hyperphosphorylated tau protein aggregates apparently may interact with the normally phosphorylated and soluble tau protein in the axons of susceptible nerve cells and may possibly act as seed units for the templated conformational change or corruption of the native tau protein. (4) The prion-like tau aggregates can be released from affected nerve cells into the extracellular space and can be taken up by the next interconnected susceptible nerve cells, where they again could serve as a seed. (5) The pathological tau exists outside of nerve cells and, therefore, provides an excellent access for future tau immunotherapies [15, 39].For an effective application of future anti-tau approaches, it will be highly beneficial to identify the brain sites which are firstly affected by the tau pathology because they may point toward the ‘port of entry’ of the pathogenetic agent.
IDENTIFICATION OF THE INITIAL BRAIN SITES OF THE TAU CYTOSKELETAL PATHOLOGY FOR EFFECTIVE EARLY IMMUNOLOGICAL ANTI-TAU THERAPIES
The allocortical transentorhinal and entorhinal regions in the mediobasal temporal lobe have been considered for nearly 25 years as the only brain regions affected during Braak stage I (Fig. 3, 5) [2, 21l]. However, a recent pathoanatomical study demonstrated that in Braak stage I tau immunoreactive aggregations are also present in all subcortical nuclei which send efferent projections to the transentorhinal and entorhinal regions and bear the brunt of the tau cytoskeletal pathology in advanced Braak stages V or VI (Fig. 6) [7–9, 51–57]. This shows that the allocortical transentorhinal and entorhinal regions are by no means the only regions of the human brain that become selectively affected during Braak stage I. At variance, this neuronal cytoskeletal pathology is already widely distributed in subcortical brain regions in Braakstage I.
Furthermore, the study of Stratmann et al. [9] demonstrated that hyperphosphorylated tau cytoskeletal pathology is also in the medial septal nucleus, nuclei of the vertical and horizontal limbs of the diagonal band of Broca, basal nucleus of Meynert; claustrum; hypothalamic dorsomedial, ventromedial, tuberomamillary and supramamillary nuclei, perifornical region and lateral area; thalamic central medial, paraventricular, laterodorsal, subparafascicular, and central lateral nuclei, medial pulvinar and limitans-suprageniculate complex; peripeduncular nucleus, dopaminergic substantia nigra and ventral tegmental area, periaqueductal gray, midbrain and pontine dorsal raphe nuclei, locus coeruleus, and parabrachial nuclei in Braak stage 0 [9] (Fig. 6). This indicates that subcortical nuclei intimately interconnected with the transentorhinal and entorhinal regions via ascending fiber tracts are affected by the immunopositive hyperphosphorylated tau aggregation pathology before their allocortical projection targets. Accordingly, the evolution of AD-related tau cytoskeletal pathology in the brain comprises not only the well-known ‘cortical phases’ reflected by Braak stages I-VI, but also preceding very early ‘pre-cortical phase’ (i.e., Braak stage 0), in which more subcortical brain sites are already involved than believed so far. These results also support the hypothesis that the AD-related tau cytoskeletal pathology originates in subcortical regions and that the tau aggregation pathology spreads during the asymptomatic, precortical phase transneuronally along anatomical pathways to the transentorhinal and entorhinal regions (Fig. 8) [8, 58–60]. This spread is anterograde and funnels all subcortical pathological influence more or less simultaneously to the transentorhinal cortex.
The initial findings on the very early involvement of subcortical nuclei helped to narrow down the brain sites of the very first AD-related tau cytoskeletal changes. However, further careful systematic re-investigations of the subcortical nuclei with efferent allocortical projections need to be performed in a large cohort of individuals in the pre-cortical Braak stage 0 in order to precisely characterize the brain area from which the AD-related cytoskeletal pathology originates. Among the candidates may be the midbrain raphe nuclei, the locus coeruleus, and the cholinergic nuclei of the basal forebrain (Fig. 8).
These subcortical brain sites that are very early affected during Braak stage 0 reach via their allocortical projections common targets (i.e., transentorhinal and entorhinal regions) [61, 62] and are also intimately interconnected with each other via reciprocal fiber tracts [63–77]. In addition, they are also reciprocally connected with the midbrain raphe nuclei [68, 70–72]. The precise characterization of the subcortical region with the first tau cytoskeletal alterations by means of postmortem studies and its confirmation by sophisticated in vivo imaging techniques appear as essential prerequisite to precisely navigate and position immunotherapeutic approaches (e.g., by chaperones or bi-specific antibodies to ferry the antibodies into brain, transient opening of the blood-brain barrier by chemical or radiological means or direct infusion of antibodies into the brain using a time-released pump) in the brain aimed at to eliminate the initial corruptive tau brain aggregates or seeding units and/or interrupt their seeded proliferation and possibly prion-like propagation [18, 78–80]. In view of the significant correlation of the AD-related brain tau cytoskeletal pathology with the evolution of the disease intrinsic dementia [14, 81], such a successful initial immunotherapeutic approach may be suitable to hold the disease progression, protect the remaining brain, and to effectively prevent dementia and other AD-related disease symptoms already in its pre-cortical neuropathological phase and pre-clinicalstages.
THE ENIGMATIC ASSOCIATION OF THE CEREBROSPINAL FLUID AND THE TAU CYTOSKELETAL PROTEIN
The extent of the tau aggregation pathology, neuronal injury, and neurodegeneration in AD is believed to be reflected by the ratio between the amounts of total (t-tau) and hyperphosphorylated tau (p-tau) in CSF. The change from normal to increased pathological levels of CSF t-tau or p-tau already occur during the asymptomatic preclinical phase of AD (i.e., Braak stages I-III) [82]. Although it is unclear whether and how the changes to the pathological CSF levels relate to the brain tau pathology, CSF t-tau and p-tau are widely recommended as biomarkers that may facilitate prediction of clinically manifested AD long before the onset of the typical clinical symptoms [82]. Since with the initial and circumscribed AD-related brain tau pathology is rather scanty and the associated neuronal loss only subordinate, it appears questionable that the increased pathological levels of CSF t-tau or p-tau in the asymptomatic preclinical phase of AD reflect the situation in the beginning diseased brain. We think that the pathological tau protein may be a primary and inherent constituent of the CSF of patients in the asymptomatic, pre-clinical phase of AD. From the CSF, tau may be transmitted to the brain parenchyma to affect the nerve cells adjacent to the ventricle system or the subararchnoidal space and thus continuously dispense the pathological tau material for the progressive and sequential brain spreading in AD. This may be supported by the fact that all of the very early affected brain regions are in close contact with the ventricle system or the subarachnoidal space.
The midbrain supratrochlear part of the dorsal raphe nuclei is in close contact with the CSF and ependyma of the fourth ventricle via its serotonergic fibers in the supraependymal plexus and its free nerve endings (Fig. 9) [70, 84]. This brainstem nucleus is very early affected by the tau cytoskeletal pathology and apparently is in a strategic position for the uptake of pathogenetic agents from the CSF and therefore could serve as the portal of entry of the brain for the AD-related disease process.
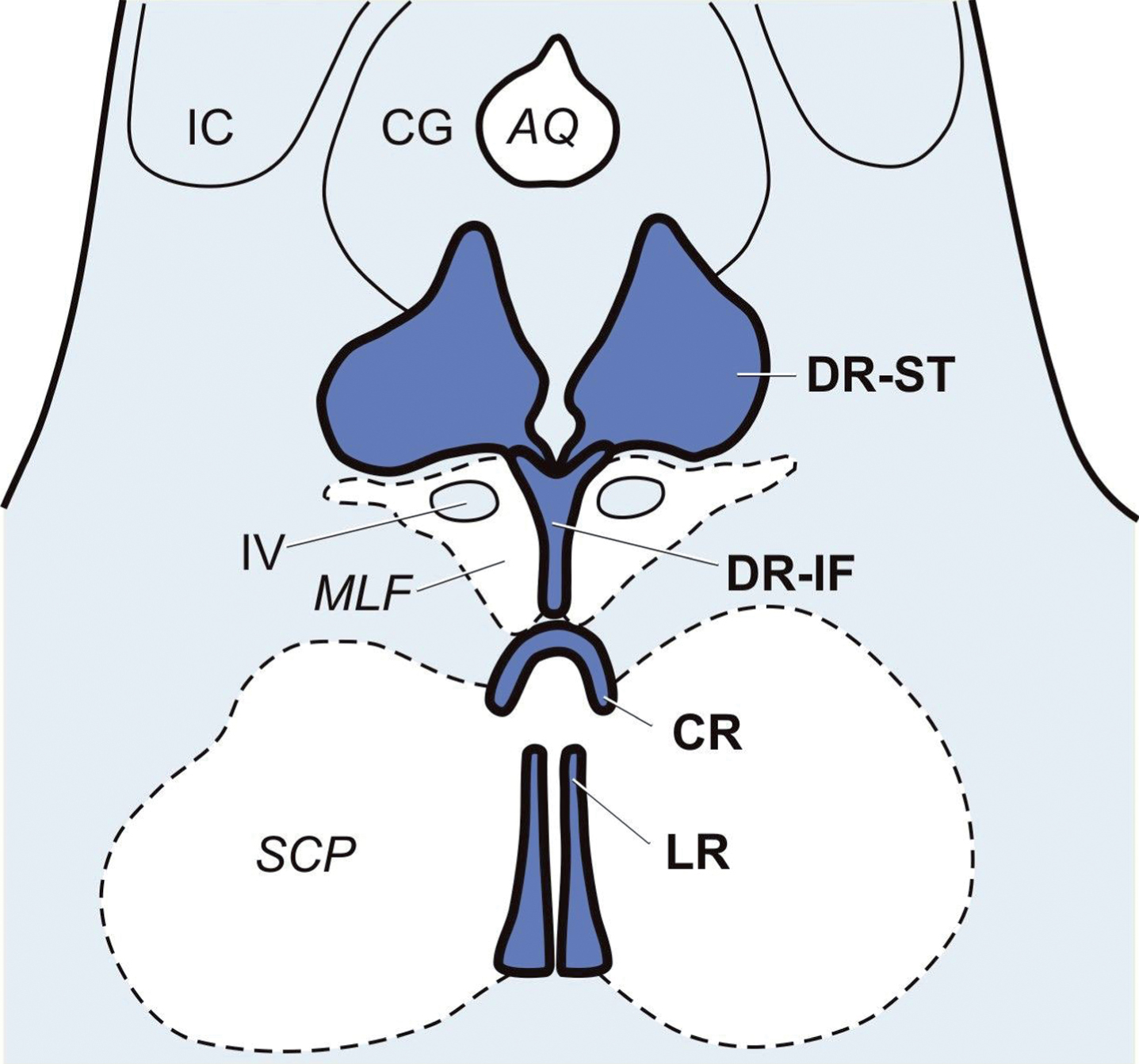
The midbrain raphe nuclei. Schematized horizontal section through the caudal midbrain at the level of the inferior colliculus (IC) with the midbrain raphe nuclei: supratrochlear (DR-ST) and interfascicular parts (DR-IF) of the dorsal raphe nuclei. AQ, aqueduct; CG, central gray; CR, central raphe nucleus; DR-IF, dorsal raphe nucleus, interfascicular part; DR-ST, dorsal raphe nucleus, supratrochlear part; IC, inferior colliculus; LR, linear raphe nucleus; MLF, medial longitudinal fascicle; SCP, superior cerebellar peduncle; IV, trochlear nucleus.
As a working hypothesis for future pathoanatomical studies aimed at elucidation of the suggested brain penetration of the tau disease process of AD, we therefore suggest the following evolutional scenario: Subsequent to their hypothesized transmission from the CSF and brain penetration of detrimental agents via the ependyma of the fourth ventricle (phase 1; Braak stage 0), the pathological process involves the supratrochlear part of the dorsal raphe nuclei and causes the formation of the first brain tau cytoskeletal pathology in its nerve cells (phase 2; Braak stage 0) (Fig. 6). Owing to its transneuronal spread via anatomical connections, this process then gradually affects specific subcortical nuclei and for the moment remains between these interconnected subcortical nuclei (e.g., medial septal nucleus, nuclei of the diagonal band of Broca, basal nucleus of Meynert, paraventricular thalamic nucleus, thalamic limitans-suprageniculate complex, dopaminergic substantia nigra and ventral tegmetal area, locus coeruleus, parabrachial nuclei) (phase 3; Braak stages 0 and I). From these subcortical nuclei, the pathological process then further ascends anterogradely using efferent projections and reaches the transentorhinal and entorhinal regions as the first regions of the cerebral cortex. The unexplained high vulnerability of these allocortical regions for the AD-related cytoskeletal pathology thus could be traced to the anatomical funneling and convergence of the ascending pathology from all of these very early affected subcortical brain sites (phase 4; Braak stages I and II). Starting at the allocortex, the cortical cytoskeletal pathology ultimately progresses along projection fibers to the hippocampus and areas of the neocortex (phase 5; Braak stages III-VI).