
Abstract
Animal models of neurodegeneration induced by neuronal expression of truncated tau protein emerge as an important tool for understanding the pathogenesis of human tauopathies and for therapy development. Here we highlight common features of truncated tau models and make a critical assessment of possible pitfalls in their analysis. Particularly, the amount of soluble tau oligomers, which are suspected to be neurotoxic agents participating on the spreading of pathology inside the brain, may be overestimated due to a post-lysis oxidative tau oligomerization. Using a mouse brain lysate spiked with recombinant truncated and full length tau forms, we show that tau oligomers might inadvertently be produced during the isolation procedure. This finding is further corroborated by the analysis of brain lysates originated from a mouse model expressing truncated tau variant. Our results underline the necessity of thiol-protecting conditions during the analysis of tau oligomers involved in the etiopathogenesis of various tauopathies including Alzheimer’s disease.
INTRODUCTION
Neurofibrillary pathology is a defining hallmark of human tauopathies including Alzheimer’s disease (AD). Cognitive decline of affected individuals strongly correlates with the burden of neocortical neurofibrillary tangles (NFTs), which comprise tau protein as a constitutive component [1–3]. Faithful animal models reproducing neurofibrillary pathology of human diseases are important for getting insight into molecular underpinnings of tauopathy pathogenesis and for aiding therapy development. Despite extensive efforts, modeling of a complete neurofibrillary pathology in models with transgenic expression of a full-length tau requires almost exclusively introduction of a point mutation to tau [4]. However, hereditary mutations in human tau gene are involved in familial tauopathies, not in sporadic forms of AD. Tau belongs to a class of intrinsically disordered proteins [5, 6] and truncation of tau molecule imparts qualitative changes to its conformational ensemble [7, 8]. Noteworthy, post-translationally truncated tau protein is frequently encountered in AD [9, 10]. Truncated tau has been suggested to trigger neurofibrillary degeneration [10] and drive the pathological conversion of wild-type tau at neuritic plaques [11]. Fragments of tau thus represent an appealing candidate for development of transgenic animal models of tauopathy. The first transgenic rat line SHR318 establishing tau truncation as a factor sufficient to drive neurofibrillary degeneration in the absence of tau mutation was described eleven years ago [12]. Several transgenic models published since then further corroborated the gain of toxic function caused by tau truncation. Here we are reporting an overview of their prominent features.
There are several lines of evidence showing that soluble tau protein oligomers might be the neurotoxic agents participating on the spreading of pathology inside the brain (for review see [13]). Tau animal models can serve as an important platform for investigating oligomers’ toxicity. However, the process of oligomer analysis in general is vulnerable to artefact generation, as was shown for oligomers of amyloid-β peptide [14, 15]. Tau protein isoforms contain two cysteines, at positions 291 (only present in 4R tau) and 322 (present in 3R and 4R tau), which are highly prone to the formation of disulphides under oxidising conditions [16]. Therefore, the amount of disulphide-dependent tau oligomers detected in a tissue lysate might be influenced by post-lysis oxidation. In our previous study, we investigated the multimeric state of tau under non-reducing conditions and the mode to preserve tau thiols from dimerization. We found that in a preparation of tau the relative abundance of dimeric form can be extremely high, achieving nearly 70% of total tau protein [17]. Recently, it was reported that a co-expression of full-length tau isoforms 0N3R and 2N4R with truncated 3R tau151-421 leads to the generation of a high amount of reducible tau oligomers in the brain of transgenic mice [18]. However, the authors failed to protect free tau thiols from oxidation during isolation of oligomers. Hence we were interested in analyzing the possibility of a post-lysis tau oxidative oligomerization involving the above-mentioned tau proteins. We are demonstrating the easiness of disulphide-dependent tau oligomerization by action of the brain lysate, which may involve other tau molecules or even extraneous cellular proteins with a free sulfhydryl group.
MATERIALS AND METHODS
Expression and purification of tau proteins
Recombinant human tau proteins: Δtau (3R tau151-421), 0N3R and 2N4R (labelling of tau as in [18]) were prepared and stored as described previously [17]. Briefly, proteins were expressed in E.coli, released from cells by sonication, purified under reducing conditions with a three-step chromatographic procedure, and stored without any reducing agent in the monomeric state under argon atmosphere.
Preparation of mouse brainstem lysate
All animal experiments were approved by the local ethics and animal care and use committees. Brainstem lysate was prepared as previously published [18] with minor modifications. 6-week-old C57BL/6 mice or 11-week-old transgenic mice R3m/4, expressing 3R tau151-391 [22] were anesthetized intraperitoneally with a mixture of tiletamine (15 mg·kg–1), zolazepam (15 mg.kg–1) and xylazine (10 mg·kg–1) and then perfused transcardially with PBS. Following PBS perfusion, each hemisphere of the mouse brain was dissected into forebrain and brainstem and frozen in liquid nitrogen. Frozen tissue was homogenized 1:10 (w/v –mg per μl) in 20 mM Tris, pH 7.5, 137 mM NaCl, 1 mM EDTA supplemented with cOmplete™ EDTA-free protease inhibitor cocktail tablets in a glass homogenizer for one minute on ice. Where appropriate, lysis buffer were supplemented with a 125 mM of 2-aminoethyl-methanethiosulfonate (AEMTS, see below). The lysate was then centrifuged for 10 min at 32,000 g, at 2°C; supernatant was collected into a separate tube. Protein concentration in the lysate ranges from 5 to 6 mg/ml.
Experiments on post-lysis tau oxidation
All incubations were performed on ice. Recombinant tau proteins were mixed with the wild-type mouse lysate (25 ng of each tau form per 5 μg of lysate proteins) and incubated 10 min. Where appropriate, free thiols in the sample were blocked by 2-aminoethyl-methanethiosulfonate (AEMTS) added to a final concentration of 125 mM; the stock of AEMTS was prepared as a 250 mM solution in 10 mM Tris-HCl pH 8.0. AEMTS allows for a fast (5 min) and specific blocking of free protein thiols by modifying them to disulphides of 2-aminoethanethiol and might be superior to other blocking reagents in the terms of speed and selectivity [19]. Since the agent does not reduce the existing disulphide bonds, it preserves the status quo of the homogenized sample. Forebrain samples from the transgenic line R3m/4 were homogenized in parallel with and without the presence of AEMTS in the lysis buffer. Disulphide-dependent oligomers of tau were analyzed on a non-reducing western blot developed with HT7 antibody (diluted 1:2000, epitope between residues 159–163, Thermo Fisher Scientific cat.no. MN1000, Waltham, MA; secondary polyclonal goat anti-mouse immunoglobulins, HRP-labeled antibody diluted 1:2000, Dako, cat.no. P0447, Glostrup, Denmark).
RESULTS AND DISCUSSION
Neuronal expression of specifically truncated tau protein induces neurodegeneration in animal models
We performed a comprehensive literature search focusing on rodent animal models with developed neurodegeneration, transgenic for a truncated form of human tau protein. The number of models is continuously growing and currently comprises 14 independent lines, from which six have been published in the last year. Accumulated data allowed us to prepare an overview of neurodegenerative phenotype following expression of truncated tau, comparing the transgene expression, the forms of multimeric tau and neurotoxic/behavioral phenotype (Table 1). The truncated tau transgenes include the following tau fragments: tau296-390 similar to the tau fragment isolated from AD paired helical filaments [20]; 3R and 4R tau151-391 containing E391 truncation linked to the pathogenesis of AD [12, 22], 3R tau151-421 terminating at caspase-3 cleavage site D421 [18], two tau transgenes with intact N-termini, 1N4R tau1 -391 [23] and 0N4R tau1 -421 [24], and a fragment with intact C-terminus, 4R tau187-441, exploiting N-terminal truncation site associated with human four-repeat tauopathies [25]. All these models employ a non-mutated truncated tau, which makes them relevant to the sporadic form of disease. A “proaggregation” model expressing tau fragment 4R tau244 - 372 with the FTDP-17 (frontotemporal dementia with Parkinsonism linked to chromosome 17) mutation ΔK280 [26], addresses the truncation of mutated tau in a familial form of tauopathy. The majority of models adopt mThy1 promotor with the highest expression in the brainstem; the line SHR24 has the highest expression in the hippocampus. Two models adopt a forebrain specific promotor and one model has the human tau promotor. Expression occurs with a 1–10 and 0.5–4 fold molar excess over endogenous tau protein for mThy1 and forebrain or human tau promotors, respectively.
Rodent models of neurodegeneration expressing human truncated tau
MT, microtubule; N.A., no data available in published literature; m, months; NF, neurofilament; RIPA, radioimmunoprecipitation assay; SHR, spontaneously hypertensive rat; w, weeks; WKY, Wistar-Kyoto. aWestern blots in [18] show comparable level of all studied tau transgenes; at the same time, the level of 2N4R tau transgene in the spinal cord of line ALZ17 is at least 10-fold higher than that of endogenous tau [37]. bNo prevention of post-lysis formation of reducible tau oligomers accomplished. cProtocol for isolation of sarkosyl insoluble fraction adopted in [18] is more stringent than in other papers, i.e. less insoluble material is expected to be obtained. dRIPA buffer extracts tau associated with the cellular pellet after a medium-speed centrifugation; therefore, it may confer different population of tau oligomers than the sarkosyl procedure, in which the multimeric tau is pelleted by a high-speed centrifugation from the solution. ePost-lysis formation of reducible tau oligomers prevented. fProtein level in Tau35 line was not quantified, but can be estimated from the beta-actin signal on western blot after comparison with [18]. gWKY72 was stabilized by back-crossing the SHR72 line to the WKY genetic background.
Similarly to AD, tau pathology in truncated tau models starts with the formation of progressively phosphorylated tau oligomers (pretangle stage), which become later insoluble. Importantly, in most of the models, truncated tau transgenes sequester full-length tau (endogenous or transgenic) at some stage. Fully developed tau pathology including neurofibrillary tangles can be observed primarily in mouse and rat models expressing 3R and 4R tau151-391 (lines R3m/4 [22], SHR318 [12], SHR72 [27], WKY72 [28], SHR24 [21]) and in mice with forebrain-targeted transgene expression of 0N4R tau1 -421 (line TauC3 [24]), 4R tau187-441 (line Tau35 [25]) and 4R tau244 - 372 ΔK280 (line TauRD/ΔK280 [26]). Noteworthy, lines TauC3 and TauRD/ΔK280 exhibit an early NFTs formation even at a low transgene level [24, 26]. It results that forebrain targeted expression leads to an easier formation of NFTs.
Neurotoxicity of truncated tau is manifested mainly as chromatolysis [18, 29], dysregulation of synaptic proteins [18, 30–32], and deterioration of axons and synapses [18, 28–33]. Neurobehavioral phenotype depends on the site of expression and includes motor impairment due to neurogenic muscle atrophy [18, 32–34] and memory impairment in short-term and spatial learning tasks [18, 33]. Severity of the phenotype and the extent of neurofibrillary degeneration appear as a synergy of two events: (1) tau protein overexpression - the phenotype correlates with the site and the level of truncated tau expression similarly to the full-length mutant tau transgenic models [4], and (2) tau truncation - the phenotype becomes more prominent as the truncation proceeds from C- and N-termini generating shorter fragments. However, there are apparent boundaries of N- and C-terminal truncation: line L1 expressing tau296-390 exhibits only weak pretangle pathology [20], whereas line Tau4R expressing non-mutated 4R tau244 - 372 exhibits no tau pathology at all [11].
Importantly, the onset of neurobehavioral deficits in available models transgenic for truncated tau coincides with the first appearance of potentially toxic multimeric pre-fibrillary tau forms, before the observation of bona fide NFTs at the higher age of animals. This common denominator holds for all models but TauRD/ΔK280 line; however, in this case, the FTDP-17 mutation ΔK280 is a potent inductor of tau fibrillization per se, therefore the oligomeric-form-only state may be short-lived and hard to detect [30]. The distinctive position of oligomers in truncated tau models is in the line with their suspected role in AD and tauopathies [35].
Peculiarity of multimeric tau analysis in animal models
Summary of the truncated tau models (Table 1) revealed differences in the methodology used for the model characterization, specifically for the analysis of multimeric forms of tau. This gives rise to difficulties. First, different stringency of isolation procedures (see Table 1, notes c and d) may identify different subpopulations of oligomers, which creates difficulty in the comparison of results between the models and might lead to contradictory conclusions. Second, the post-lysis oxidative oligomerization of yet monomeric tau protein may artificially alter the monomer/oligomer ratio, which would lead to biased estimation of the extent of disulphide-dependent tau oligomerization. The latter risk can be efficiently eliminated by the protection of free thiols at the instant of tissue disintegration with suitable blocking agent, supplied in the lysis buffer [24, 38–40]. Unfortunately, this practice was not uniformly observed during the analysis of disulphide-dependent tau oligomers in truncated tau models (Table 1, notes b and e).
The risk of post-lysis oxidative oligomerization in models co-expressing truncated and full-length tau
In the work of Ozcelik et al. [18] they had described a strong correlation between disulphide-dependent tau multimers and nerve cell dysfunction in the models co-expressing 3R tau151-421 and full-length tau proteins, however, post-lysis disulphide formation was not prevented. Since the risk of post-lysis thiol oxidation is high, we were interested to what extent the oligomers detected in their work may originate from post-lysis oxidation of monomeric intracellular tau. We have designed an experiment in which we have modeled the behavior of monomeric transgenic tau 3R tau151-421, 0N3R and 2N4R after release from neurons to the bulk of brain lysate. The results were evaluated on a non-reducing western blot (Fig. 1A).
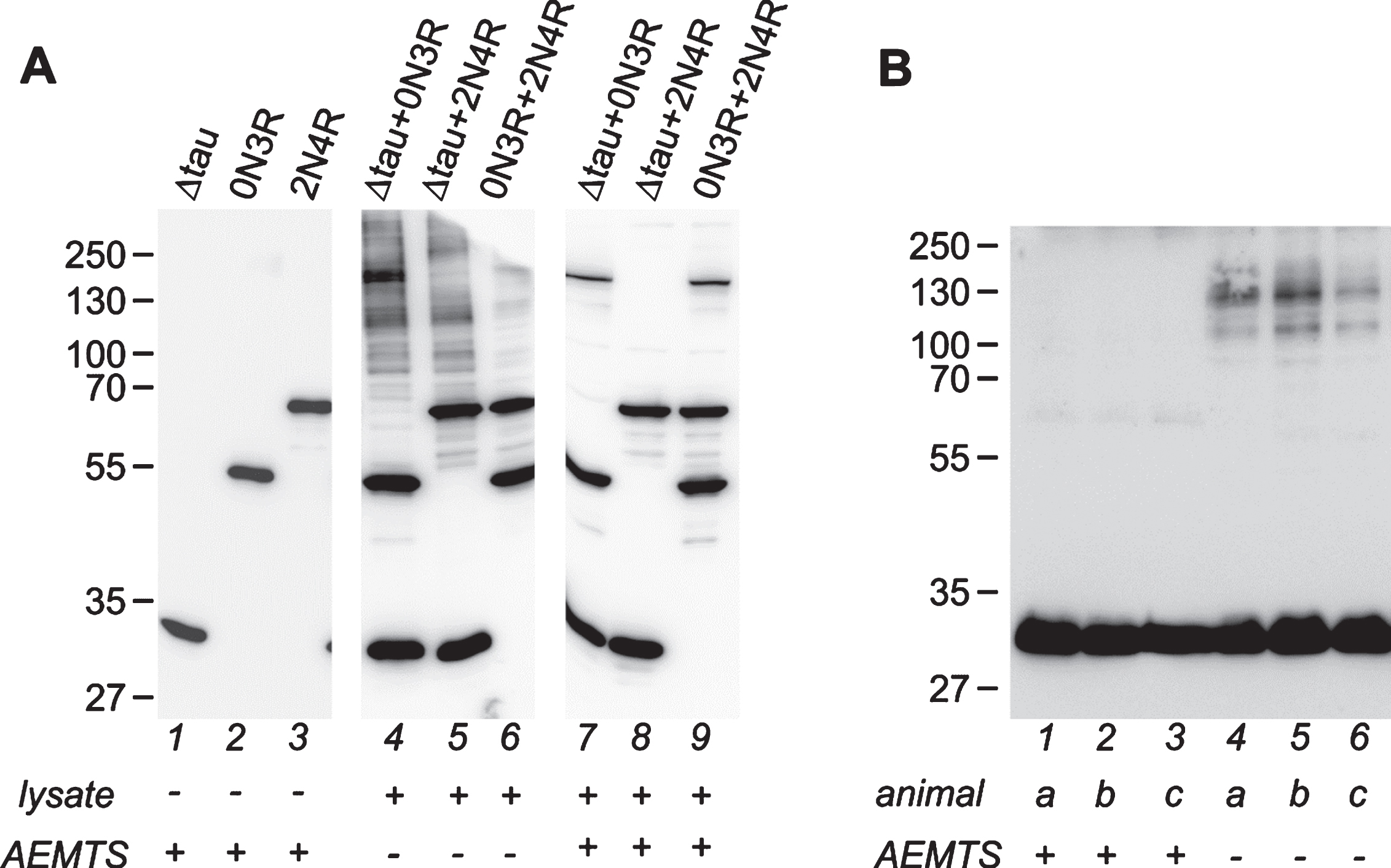
Mouse brain lysate is able to induce tau protein oxidative oligomerization. A) Western blot analysis of oligomerization of recombinant tau induced by brain lysate from a wild type animal. Lanes 1–3: Recombinant monomeric tau proteins used in the experiment, 50 ng per lane. Δtau is 3R tau151-421, the same truncated tau form as used in transgenic mice [18]. Lanes 4–6: combinations of tau proteins incubated with brainstem lysate in the absence of AEMTS. Lanes 7–9: tau proteins incubated with the lysate supplemented with AEMTS. Lanes 4–9 contain 50 ng of each tau protein and 10 μg of lysate proteins. Note the presence of prominent oligomeric tau ladder in lanes 4–6 in the absence of AEMTS. For an uncropped image of membranes used for composition of this panel, see Supplementary Figure 1. B) Artificial oligomerization of tau in brain lysate from transgenic line R3m/4. Lanes 1–3: forebrain tissues homogenized in buffer supplemented with AEMTS; lanes 4–6: forebrain tissues homogenized without AEMTS. N = 3; individual animals are labeled by lowercase italics. 20 μg of lysate proteins were loaded per lane. Forebrain lysate contains about 7.5 ng of truncated tau per one μg of total proteins, which is comparable to the concentration of recombinant tau used in the experiment on the panel A. Monomeric transgenic tau runs between 27–35 kDa. In the absence of AEMTS tau positive ladders are formed. Western blots were performed under non-reducing conditions and stained with human specific anti-tau antibody HT7. AEMTS was added to 125 mM final concentration, as indicated at the bottom of the figures.
A quality check of tau proteins used in the experiment showed that 3R Δtau151-421 and tau 2N4R were monomeric; tau 0N3R contained a trace amount of multimers, which did not interfere with subsequent analysis (Fig. 1A, lanes 1–3). When tau proteins were added to the brainstem lysate in the absence of free-thiol protecting agent (AEMTS), a significant amount of high molecular weight tau oligomers appeared (Fig. 1A, lanes 4–6). The most prominent oligomerization was observed in the presence of truncated tau 3R Δtau151-421. Strikingly, the oligomeric pattern resembled the profile of the brainstem lysates of transgenic mice co-expressing 3R Δtau151-421 and full-length tau isoforms published by Ozcelik and colleagues (compare with Figs. 4c and 5c in [18]). As expected, tau incubated in the brain lysate with simultaneous thiol blocking by the AEMTS remained monomeric (Fig. 1A, lanes 7–9). Further, we analyzed tau oligomers in the transgenic mice 3Rm/4. This line expresses human truncated 3R tau151-391 in the brainstem and forebrain [22] with the 1.2–2.4 fold excess over endogenous tau and accumulates sarkosyl-insoluble tau multimers at five months of age (Table 1). For preparation of brain lysate, we used lysis buffer with and without the AEMTS and transgenic tau was analyzed on western blot with a transgene-specific antibody HT7 (Fig. 1B). As seen in AEMTS treated samples, the forebrain of young transgenic animals contained monomeric transgenic tau and traces of tau dimers (Fig. 1B, lanes 1–3). However, in the absence of AEMTS, a significant amount of high molecular weight tau-positive species appeared in all analyzed samples (Fig. 1B, lanes 4–6). Interestingly, oligomers in all experiments form a ladder ranging from 70–90 kDa to 250 kDa, which strongly indicate the presence of tau hetero-oligomers with extraneous cellular proteins possessing a free sulfhydryl group, apart from tau-onlyoligomers.
These findings demonstrate again that without protection of free thiols, monomeric transgenic tau, which is abundantly present in the brain tissue of transgenic models (e.g., see [18]), could form artificial multimers after release from neurons during isolation. This raises the question to what extent the oligomers detected by Ozcelik and colleagues [18] may have originated from post-lysis oxidation of monomeric intracellular tau. Given the potential relevance of the models co-expressing truncated and full-length tau for unravelling the role of tau oligomerization in the pathogenesis of tauopathies, it would be highly needed to re-examine tau protein oligomerization using lysis buffer supplemented with a free-thiol protecting agent.
A rapid lysate-induced oligomerization of tau can be explained by enzymatic thiol oxidation catalysed by protein disulphide isomerases (PDIs), compartmentalized at endoplasmic reticulum membrane in intact cells [41]. It is likely that PDIs are being released into bulk solution following the brain tissue disintegration and may come into contact with soluble tau. As a consequence, free tau thiols may be rapidly oxidized during brain lysate preparation, creating (hetero-)multimers. Therefore, for a reliable analysis of oligomer content in neurofibrillary pathology a careful prevention of the post-lysis oxidation of monomeric tau has to be accomplished. This precaution holds for analysis of animal models as well as for human brain tissue.
Conclusions and outlook
Models transgenic for truncated tau reproduce pathological aspects of human tauopathies much easily than full-length tau models [18, 30]. This experimental evidence suggests tau truncation as a risk-determinant for the induction of tau pathology and encourages the use of truncated tau animal models for the development of a new generation of therapy testing and validation platforms. It emerges that the principle of truncated tau neurotoxicity lies in its enhanced propensity to form oligomers, which gradually sequester wild-type tau in the pathologic structures, impair transport and damage synaptic transmission. Future research should focus on a careful characterization of the first tau oligomers appearing in the models and on mechanism of their toxicity. Another aspect of future efforts should be the elucidation of pathways by which the full-length tau becomes sequestered into complexes with truncated tau and the identification of the risk factors that make the full-length tau vulnerable for such interaction. This will be essential for understanding the propagation and spreading of tau pathology in the sporadic form of AD and other tauopathies.
Future models transgenic for truncated tau should focus on a targeted insertion of a defined number of transgenes to specific genomic/cellular/brain regions [25], on balancing tau expression level to avoid potential artefacts due to excessive overexpression [42] and on the effect the truncation may have in the framework of various tau isoforms. Novel transgenic constructs could be derived from specific tau fragments found recently by mass spectrometry in AD patients [43, 44]. The methodologic pitfalls accompanying the analysis of tau forms in various transgenic models call for a unification of protocols used for isolation of multimeric tau fraction and for an appropriate determination of reducible tau protein oligomers. Such a protocol should preserve the oxidative status of the tissue by using thiol preserving agents like AEMTS.