
Abstract
Tauopathies are a specific type of slow and progressive neurodegeneration, which involves intracellular deposition of fibrillar material composed of abnormal hyperphosphorylation of the microtubule associated protein (MAP) tau. Despite many years of intensive research, our understanding of the molecular events that lead to neurodegeneration is far from complete. No effective therapeutic treatments have been defined, and questions surround the validity and utility of existing animal models. It is an urgent need to develop a novel animal model to study the underlying neurodegenerative mechanisms of tauopathies. Zebrafish models of tauopathies could complement existing models by providing an in vivo platform for genetic and chemical screens in order to identify new therapeutic targets and compounds, meanwhile zebrafish models have permitted discovery of unique characteristics of these genes that could have been difficultly observed in other models. Novel transgenic zebrafish models expressing wild-type or mutant forms of human 4R-tau in neurons have recently been reported. These studies show disease-relevant changes including tau hyperphosphorylation, aggregation and somato-dendritic relocalization. This review highlights the availability of transgenic tau zebrafish models that allow more detailed biochemical studies of tau in the zebrafish CNS to characterize solubility, fibril morphology and further clarify phosphorylation proceedings. Furthermore, a deeper knowledge of the zebrafish brain and a better characterization of tau caused by alterations in neurodegenerative disorders are needed.
INTRODUCTION
Tau is the major microtubule associated protein (MAP) of a normal mature neuron, alternative splicing of its pre-mRNA generates six molecular tau isoforms in human brain [1]. Alzheimer’s disease (AD) and a family of related neurodegenerative diseases are called as tauopathies (Table 1), which are the consequence of abnormal tau phosphorylation, abnormal levels of tau, abnormal tau splicing, or tau gene mutations [2–6]. In every one of these tauopathies, the neurofibrillary changes and their occurrence in the neocortex are associated with dementia. Although new substantial progress has been made in the tau pathology of tauopathies, the mechanisms underlying tau-induced neurodegeneration remain unclear. Thus, understanding the etiopathogenesis of the hallmark lesion of AD and related tauopathies is critical to develop rational therapeutic targets for human neurodegenerative diseases. Recently, tau-based strategies have received more attention; some are already in advanced clinical development [7–9], thus enlarging our range of potentially useful therapeutic tools to treat tauopathies. Consequently, investigations aimed at studying the role of tau in neurodegeneration and therapeutic targets based on this pathology are of great importance. In the current review, we review and summarize the biochemical properties of tau protein and the pathogenesis of AD and related tauopathies. Meanwhile, we also discuss the possible advantages of zebrafish models and recent developments concerning the possibility that zebrafish models of tauopathies might be useful for drug discovery and therapeutic targets in vivo.
Classification of tauopathies
PHYSIOLOGY, FUNCTION AND LOCALIZATION OF NORMAL TAU
Tau is encoded by a single gene, MAPT, which lies on human chromosome 17q21, containing 16 exons. Exons 1, 4, 5, 7, 9, 11, 12, and 13 are constitutive exons, and exon 0 or –1 is part of the promoter that is transcribed but not translated [10]. In adult human brains, exons 2, 3, and 10 are alternatively spliced to generate six isoforms of tau proteins [11]. The alternative splicing of exon 2 and exon 3 generates tau with no N-terminal insert (0N), one insert (1N) or two inserts (2N), whereas the alternative splicing of exon 10 produces tau proteins with three (3R) or four (4R) microtubule-binding repeat domains [12, 13] (Fig. 1). Only the 3R isoform is detected in fetal brain with the low expression level, whereas both the level and the number of tau isoforms increase during brain development. The ratio of 4R-tau to 3R-tau in the normal adult brain is about 1:1 [14, 15] and alteration of the ratio is involved in neurodegeneration (Table 1). The structure of tau is vital for its normal functions, the structure of tau proteins (2N4R, 441 amino acids) contains four regions: an N-terminal projection region (amino acids 1–150) contains two distinct alternatively spliced N-terminal inserts, a proline-rich domain (amino acids 151–243), a microtubule-binding domain (amino acids 244–369) and a C-terminal region (amino acids 370–441) [16, 17] (Fig. 2). As a primary neuronal protein, the biological activity of tau is promoting assembly and stability of microtubules, one of the major components of neuronal cytoskeleton that defines the normal morphology and structure of neurons as well as serves as tracks for axonal transport. Normal tau contains 2–3 mol phosphate per mol of the protein [18, 19], the level of phosphorylation is optimal for its activity. Hyperphosphorylated tau depresses its microtubule assembly activities and its binding to microtubules. It is well-known that abnormally hyperphosphorylated tau detaches from the microtubule and thus causes microtubule disassembles, and dephosphorylation can restore the microtubule binding and tau assembles, meanwhile hyperphosphorylation of tau also causes intracellular tau aggregates and reduces the anterograde and retrograde transport velocity of tau in axons [20–23]. Tau is mainly located in the axons bound to microtubules, it has also been found to bind other proteins such as actin [24, 25], spectrin [26], α-synuclein [27], presenilin1 (PS1) [28], PP1, PP2A [29, 30], and some kinases like cdk5 [31], GSK-3β [32], phospholipase C-γ [33, 34], and the fyn tyrosine kinase [35, 36]. Tau is also found to exist in the nucleus [37], plasma membrane [38, 39], synapses, dendrites and mitochondria [40, 41]. In several diseases, tau localization is altered. Especially the relocation of hyperphosphorylated tau from axons to the somatodendritic compartment is considered a pathological marker during an early development of tauopathies [42, 43] (Fig. 3).

A schematic diagram of the tau gene (MAPT) and protein isoforms in the human brain. The human tau gene is located on chromosome 17q21 and contains 16 exons. White boxes (Exons 1, 4, 5, 7, 9, 11, 12, and 13) represent constitutive exons, whereas orange exons (1, 4A, 6, 8, and 14) are not. Yellow exon 2, purple exon 3, and green exon 10 are alternatively spliced to generate six different tau isoforms depending on the presence or absence of exon 10 (4R or 3R) and 0 (0N), 1 (1N) or 2 (2N) N-terminal inserts encoded by exons 2 and 3.

A schematic diagram of the sub-regions and tau phosphorylation sites in the longest tau protein. Tau is divided into four regions: the acidic region in the N-terminal (Nt) projection region, the proline-rich region, the microtubule-binding repeats region contains four repeat domains (MBD), namely R1, R3, R4 and R2 (in green), and the C-terminal region. Approximately 45 phosphorylation sites on tau from AD brain have been identified, and these are found predominantly in the acidic, proline-rich, repeat, and C-terminal regions of tau.
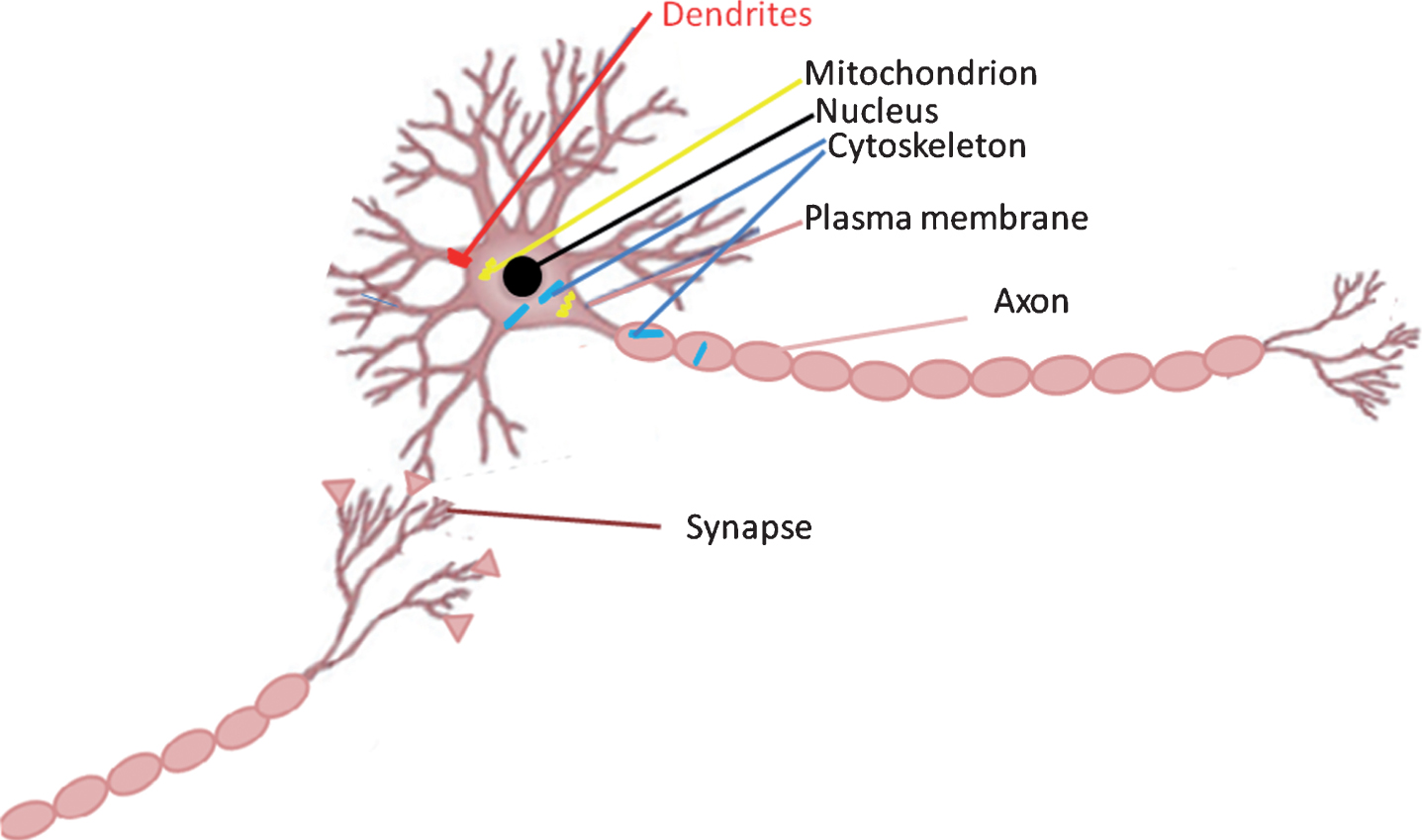
A schematic representation of tau localization in neurons. The majority of neuronal tau is associated with the microtubule cytoskeleton in axons. Tau also locates in various cellular compartments including in cytoskeleton (indicated in blue), mitochondria (indicated in yellow), the nucleus (indicated in black), plasma membrane, and synapses, Dendritic tau (indicated in red) is increased in the tauopathies.
ABNORMAL HYPERPHOSPHORYLATION AND AGGREGATION OF TAU
In many neurodegenerative disorders, including AD, tau proteins undergo several post-translational modifications. The post-translational modifications in tauopathies included tau hyperphosphorylation [19, 44], glycosylation [45], truncation [46], glycation [47], nitration [23], acetylation [48] and ubiquitination [49]. These modifications are highly associated with the aggregation of tau in tauopathies. Among the various post-translational modifications of tau, phosphorylation is the most extensively studied. Studies have reported that the largest brain tau isoform (namely tau40 or tau441) contains 80 serine or threonine and 5 tyrosine residue phosphorylation sites [50]. Despite the significant heterogeneity within the various tauopathies, the deposited tau in pathological lesions is perpetually and highly phosphorylated. Mass spectrometric analysis combined with specific antibody reactivity has shown that more than 10 phosphorylation sites can be detected on soluble tau purified from normal brain [50]. In contrast, approximately 45 different serine, threonine and tyrosine phosphorylation sites, representing more than 50% of all phosphorylatable residues, have been found in insoluble aggregated tau from AD brain [50–53] (Fig. 2). Hyperphosphorylation of tau not only loses its biological activity, but also regains-of-toxic functions [54]. Hyperphosphorylation reduces tau affinity for tubulin causing it to detach from microtubules, disrupts microtubule dynamics, and causes p-tau abnormal traffic from axons to form aggregates in the somatodendritic compartment, subsequently interfering with normal synaptic transmission [22, 55]. Tau hyperphosphorylation inhibits proteasome and promotes tau aggregation, thereby impairing synaptic function, proteosomal degradation, and autophagy. Normal tau is a small, unfolded, and short-lived cytosolic protein, which is a substrate for proteasomal degradation [56], hyperphosphorylated tau aggregation inhibits autophagy and subsequent lysosome cleavage [57].
SPREADING OF TAU PATHOLOGY
In recent years, a new concept has emerged in several neurodegenerative diseases including AD, cell-to-cell transmission of misfolded proteins in brain followed prion-like spreading mechanisms [58]. Misfolded tau would propagate from an affected neuron followed by its uptake in neighboring neurons. Consistent with this, both in vitro and in vivo studies demonstrated that tau could be secreted and endocytosed [59–64]. In vitro cells are stably transfected with human tau, then treated with tau oligomers that could produce tau seeding and cell-to-cell propagation of tau aggregation [65–68]. Moreover, injection of tau seeds from the brain interstitial fluid into the brains of P301S transgenic mice, which has shown an effective propagation of tau pathology in the mice brain [69]. In vivo studies showed that when AD-p tau was injected into the hippocampi of human wild type tau mice [70, 71], p-tau could be taken up by exposed neurons, then spread from the injected area to the contralateral hippocampus, and the morphology of the lesions depends on the pattern of hyperphosphorylated tau.
IMBALANCE IN REGULATION OF TAU PHOSPHORYLATION
Tau phosphorylation is regulated by a large number of different kinases and phosphatases, mainly at serine and threonine residues, therefore an imbalance of their activities is the direct cause of tau hyperphosphorylation [54]. Based on the kinase-recognized motifs, the serine/threonine kinases can be divided into two groups: proline-directed protein kinases (PDPKs) and non-PDPKs [12]. These PDPKs include glycogen synthase kinase-3 (GSK3) [32, 72], cyclin-dependent protein kinase-5 (cdk-5) [73], and mitogen-activated protein kinases (MAPK) family, further comprised of the extracellular signal-regulated kinases 1 and 2 (ERK1/2), p38, and c-Jun N-terminal kinases (JNK) [74–76]. The non-PDPK group includes casein kinase 1 (CK1) [50], tau-tubulin kinase 1/2 (TTBK1/2) [77], microtubule affinity-regulating kinases (MARKs) [78], cAMP-dependent protein kinase (PKA) [79], protein kinase B(PKB/Akt) [80], protein kinase C (PKC) [81], and Ca2 + /calmodulin-dependent protein, Kinase II (CaMKII) [82]. In AD brains, up-regulation of GSK-3β, CDK5, PKB, p38, and CK1 have been reported. Compared with protein kinases, a few phosphatases have been identified to dephosphorylate tau proteins in vitro and/or in vivo, including protein phosphatase-1 (PP1), PP2A, and PP5 [83]. Among these phosphatases, PP2A is the most effective phosphatase in dephosphorylating hyperphosphorylated tau from AD brains. Inhibition of PP2A and PP1 by OA (okadaic acid) or CA (calyculin A) causes tau hyperphosphorylation in vitro and/or in vivo. Inhibition of PP2A and PP1 in neuroblastoma culture cells leads to damage of axonal transport and retraction of the cell processes, PP2A accounts for at least 71% of the total tau phosphatase activity in human brain, and the activity of PP2A is significantly decreased in AD brains [85]. Therefore, maintaining a normal level of PP2A seems to be a promising strategy in preventing AD-like tau pathology [86, 87].
TAU DEPOSITS IN TAU-INDUCED NEURODEGENERATIVE DISEASES
In some tauopathies, like AD or Down’s syndrome, tau pathology is associated with other cerebral changes. AD is the most common and the best-studied tauopathies. Tau aggregates from AD are composed of the six hyperphosphorylated tau isoforms, which results in the appearance of three major tau immunoreactive bands of 68, 64, and 60 kDa by SDS-PAGE analysis [12, 88]. In Down syndrome, tau is also hyperphosphorylated sharing a pattern similar to that of AD. Corticobasal degeneration (CBD) has indicated frontoparietal atrophy and glial and neuronal tau inclusions; Tau is also present in hyperphosphorylated form, but only the 68 and 64 kDa [2, 12]. In frontotemporal lobar degeneration (FTLD)-Tau with MAPT mutations, tau inclusions were observed in neurons and glia cells. Some cases of the pattern are similar to that of AD, the 68, 64, and 60 kDa forms being found, meanwhile other cases of the pattern are like CBD, there is only the 68 and 64 kDa forms. Pick’s disease is characterized by the presence of cytoplasmic tau inclusions in neurons of the frontal lobe, well-known as Pick bodies; the granular cells of dentate gyrus are also affected. The appearance of inclusions presents at 64 and 60 kDa. In progressive supranuclear palsy (PSP), tau inclusions have been found in neuronal and glial cells, with both astrocytes and oligodendrocytes being affected. The pattern is like CBD, two major tau bands of 68- and 64 kDa are derived from hyperphosphorylated tau [89–92] (Table 1).
TRANSGENIC MOUSE MODELS OF TAUOPATHY
Several approaches have been built up in an attempt to model tauopathies in mice. The first transgenic tau mice expressing the longest human brain tau isoform was established in 1995 by Goedert’s group [93]. The first transgenic mice carried cDNAs encoding the largest human brain tau isoform with low levels (10%) of overexpression. In the transgenic mice, human tau protein was present in nerve cell bodies, axons, and dendrites. Tau was phosphorylated at sites that are hyperphosphorylated in paired helical filaments, although non-filamentous tau aggregates formed [93]. After that, several mouse lines with overexpression of different isoforms of wild-type tau have been reported [94–97]. In these mice, they shared a common character that filamentous tau aggregates were not observed. No neurofibrillary tangles were detected in the mice of young age. Additionally, the transgenic mice present large numbers of axonal spheroids in spinal cord, displaying accumulations of neurofilaments, mitochondria and vesicles, and most of the transgenic mice developed motor dysfunction [98]. The other approach to generate transgenic mice models of tauopathies is based on the mutations in tau associated with FTDP-17 [97, 99]. When the mutant tau detected in FTDP-17 patients was transfected or knocked in, neurofibrillary tangles were produced in different brain regions of the mice [100–103]. Transgenic mice expressed mutant human tau containing the P301L mutation [103, 104] that was located in the tubulin-binding domain, develop a spinal cord pathology, motor dysfunction, and neurofibrillary tangles that mainly localized in the spinal cord. Filaments were also found in transgenic mice expressing the human tau mutation R406W and V337M [105, 106].
USING ZEBRAFISH TO STUDY HUMAN NEUROLOGICAL DISORDERS
The zebrafish is a small freshwater fish that has been used extensively in laboratory models for a wide variety of human diseases, including cancer, cardiovascular disorders, osteoporosis, hemophilia, diseases of muscle, kidney, liver, and disorders of the central nervous system (CNS). The zebrafish genome and anatomy show only ∼420 million years of divergence from the human lineage, so most human genes have clearly identifiable orthologues in zebrafish [107–109]. The zebrafish is a robust fish, its cost is cheap, and large numbers are easily kept in the laboratory, where they breed all year. Zebrafish owns the faster development and longer lifespan, its life cycle include embryonic prehatching (0–72 hours post-fertilization (hpf)), larval (3–29 days post-fertilization (dpf)), juvenile (30–89 dpf), adult (90 dpf–2 years), aged zebrafish (>2 years), and death (4-5 years) (Fig. 4) [110]. Females can spawn every 2-3 days and own high reproductive rate, their eggs are larger (0.7 mm in diameter at fertilization) compare with other fish and the yolk is sequestered into a separate cell [111]. Zebrafish could be considered as an ideal model to study human neurological disorder since zebrafish shows conservation of basic brain organization [112], many important neuroanatomical structure [112–114] and neurochemical [113, 115] elements that are relevant to human disease. The adult zebrafish brain is relatively small (0.05 gram) but shares many organizational features with other vertebrates, they have forebrain, midbrain, and hindbrain, including diencephalon, telencephalon and cerebellum. Although zebrafish does not have a hippocampus, it has the lateral pallium, which is assumed to act similarly to the human hippocampus [116]. Moreover, humans primary neurotransmitter systems are found in zebrafish brains such as acetylcholine, dopamine, glutamate, γ-aminobutyric acid, serotonin, and histamine [117]. In addition, the zebrafish CNS also contains microglia [118], astrocyte [119], oligodendrocytes [120], myelin [121], and a blood–brain barrier [122], which have been implicated in the pathogenesis of neurological disorder. Zebrafish has large clutches of externally fertilized and transparent eggs (100 + embryos per spawning), which have developed rapidly large, with neurogenesis starting around 10 hpf, synaptogenesis and the first behaviors around 18 hpf. Within 1 day of development, many relevant features of the CNS appear, that allow direct observation of embryogenesis. In contrast with mammals, zebrafish shows a much greater potential in neurogenesis [123]. Neurogenesis continues along the entire adult life of zebrafish. About 6000 cells are born within every 30 min period in the adult zebrafish brain, the total estimated 107 cells in the zebrafish brain, mainly localized in 16 different proliferating regions, including regions equivalent to the subventricular zone of the lateral ventricle and the subgranular zone of the dentate gyrus in the hippocampus [123–126]. Zebrafish is considered as a simple and effective model which could be manipulated gene expression in its living embryos with relevance to human pathology for cell biological observations. Zebrafish embryos could be genetically microinjected with antisense oligonucleotides, mRNA, and transgenes [127]. These technologies make drastic changes in gene expression that can be tracked in the developing transparent embryos. For example, Tol2 transposase system is introduced into transgenic zebrafish as an efficient vector [128] to insert genes. Conditionally expressed transgenics also were generated using the GAL4-UAS [129] and Cre/loxP [130] systems for gene function analysis. Recently CRISPR [131] (clustered regularly interspaced short palindromic repeats)/Cas systems, transcription activator-like effector nucleases (TALENS), and zinc finger nucleases (ZFNs) have been developed for targeted gene sequences modification in the zebrafish [131]. These technologies have been validated for use in the zebrafish to develop zebrafish genetic models of neurodegenerative diseases such as AD, which would provide novel insights into molecular pathogenesis and potentially therapeutic targets. Specifically, the intact vertebrate CNS screening studies in vivo might discover drug targets, which cannot be present in cell models.

A schematic diagram of the life cycle of zebrafish. The life cycle of zebrafish includes embryonic prehatching (0–72 hours post-fertilization (hpf)), larval (3–29 days post-fertilization (dpf)), juvenile (30–89 dpf), adult (90 dpf–2 years), aged zebrafish (>2 years), and death (4-5 years).
TRANSGENIC ZEBRAFISH MODELS OF TAUOPATHIES
Most recently, there was a significant interest in the development of zebrafish models of tauopathies. This work has identified two paralogues of MAPT in zebrafish, called mapta and maptb. The two proteins encoded by the zebrafish genes have not yet been detected, but both mapta and maptb mRNAs were expressed in the developing CNS [132]. A complex pattern of alternative splicing of the mapta and maptb transcripts suggests that, such as human tau, zebrafish tau isoforms with different numbers of microtubule-binding repeats are expressed in the CNS, and larger forms of tau are expressed in the peripheral nervous system. Interestingly, mapta generates transcripts encoding 4–6 microtubule binding repeats, whereas maptb predominantly expresses a 3-repeat isoforms, raising the fascinating possibility that preserved functions of mammalian 3R- and 4R-tau are distributed between the two zebrafish genes (Fig. 5) [107, 132]. Moreover, the zebrafish genome contains highly conserved orthologues of each of the kinases implicated in tau phosphorylation. Several transgenic zebrafish models of tauopathies have been generated before. Tomasiewicz’s group had developed a zebrafish model transiently over-expressing FTDP-17 mutant human 4R-Tau. Zebrafish embryos were microinjected with the Tau-EGFP construct fused with the zebrafish neural specific GATA-2 promoter, showed mosaic neuronal expression of the fusion protein. 48 hours after microinjection, around 10–15% neurons demonstrated obvious disruption of cytoskeletal filaments in the axon and rearrangements in tau trafficking, including accumulation of tau in the cell body proximal to the axon and accumulation of fluorescent fibrillar tau staining throughout the cell body. Employing western blot, they had detected the human Tau-EGFP fusion protein was phosphorylated at phospho-S396/S404 in the zebrafish brain, as well as the expression of glycogen synthase kinase-3 in the zebrafish embryo and adult brain. FTDP-17 mutant human tau expressed in zebrafish neurons produced a cytoskeletal disruption that closely resembled the neurofibrillary tangles in AD [133]. Afterwards, Bai’s group had constructed a stable transgenic zebrafish expressing human 4R/0N Tau under transcriptional control of the zebrafish eno2 promoter [134]. The stable transgenic Tg (eno2: Tau) zebrafish showed widespread CNS neuronal expression of 4R/0N Tau, persisting through adulthood. Tau was expressed at∼8-fold higher levels in Tg (eno2: Tau) zebrafish brain than normal human cortex, and localized to axons, neuropil and ectopic neuronal somatic accumulations resembling neurofibrillary tangles seen in human tauopathies. In the study of Paquet et al. [135], the neuronal HuC promoter was employed to drive expression of a Gal4:VP16 fusion protein which was further bound to UAS sites in a bidirectional promoter transcribing the DsRed fluorescent marker protein and a mutant human MAPT associated with frontotemporal dementia (FTD), Tau-P301L. They have found that the stably transgenic Tg (HuC: Gal4/Tau-P301L-UAS-DsRed) zebrafish model was allowed to monitor its early pathology, including tau-specific hyperphosphorylation at T231/S235, T181, S262/S356, S396/S404, S422, and S202/T205 sites, conformational changes of tau, neuronal and behavioral abnormalities in the first 48 hpf. Especially the hyperphosphorylated tau, which has developed within 32 hpf, is assumed to play a key initiator of detachment of normal tau from microtubules and subsequent aggregation. Moreover, the larvae rapidly developed substantial neurodegeneration after a few days. After 5 weeks, the Tg zebrafish presented the appearance of neurofibrillary tangles. Cell death has been monitored in living stable transgenic Tg (HuC: Gal4/Tau-P301L-UAS-DsRed) zebrafish; they have detected significantly increased cell death in the whole spinal cord of 6-day-old transgenic Tg zebrafish. Finally, the use of novel high-potency GSK3β inhibitors (AR-534) reduced tau phosphorylation in this transgenic zebrafish, emphasizing that the stable zebrafish model of tauopathies can be used for the search of compounds that modify characters of human tauopathies. It should be noted that lacking of comparisons of phenotype between the nonmutant and mutant forms of human tau in the zebrafish. In Huang and coworkers’ study, they build up a transient expression system to express either zebrafish 3R-tau or human 4R-tau fusing with GFP proteins under the control of a neuronal HuC promoter. Around ten neuronal cells expressing tau-GFP in zebrafish embryos were directly imaged and traced by time-lapse recording to evaluate the neurotoxicity induced by tau-GFP proteins. Furthermore, they tried to detect whether zebrafish tau proteins truncated at Asp259 or Asp289 (equivalent to human tau truncation at Glu391 or Asp421) can induce neurotoxic effects. They generated zebrafish 3R-tau-Δ260-GFP or 3R-tau-Δ290-GFP, and human 4R-tau-Δ392-GFP or 4R-tau-Δ422-GFP constructs, respectively. The results indicated that either zebrafish or human truncated tau protein could cause similar high levels of neuronal death to that of zebrafish 3R-tau or human 4R-tau, and then the death was effectively protected by multiple signaling factors, such as Bcl2-L1, Nrf2, and GDNF. This zebrafish system may serve as an efficient in vivo imaging platform for discovering of novel drugs against tauopathies. To treat the zebrafish model with chemical compounds, they found that the chemicals exerted anti-oxidative or neurotrophic effects, and maintained human tau-GFP protein in a phosphorylated state [136]. More recently, Rubinsztein et al. had analyzed an independent multinational cohort consisting of 3,100 patients with neurodegenerative disease and 4,351 healthy control subjects, and found A152T associated with significantly higher risk for frontotemporal dementia and progressive supranuclear palsy syndrome. Furthermore, to assess the functional and biochemical consequences of a rare tau mutation A152T, they generated new transgenic zebrafish models exploiting the Gal4-UAS binary expression system. They employed responder constructs comprising wild-type or mutant A152T human tau (2N4R) fused to the photoactivatable protein Dendra, downstream of UAS. The wild-type human tau transgenic zebrafish caused no obvious morphological defects and no abnormalities in motor neurons, whereas mutant A152T-human tau zebrafish showed abnormal phenotypes and motility defects. They further investigated the evidence of tau-associated pathology in the two transgenic lines; Mutant A152T-human tau zebrafish demonstrated that A152T-tau expression is associated with greater tau associated pathology and neurodegeneration compare to wild-type human tau transgenic zebrafish. In addition, A152T variant was found to associate with disruption of proteasome function and delayed tau clearance in vivo, whereas autophagy function was unaffected. Hence, they increased A152T clearance by both pharmacological and genetic up-regulation of autophagy and improved tau pathology observed in mutant A152T-human tau zebrafish, suggesting a possible therapeutic strategy for these tauopathies [137]. Recently, Kizil and co-workers have developed cre/lox-based transgenic models of zebrafish that expressed human TauP301L under either her4.1 promoter or neural beta tubulin promoter [138]. They specifically chose her4.1 promoter since it is active in neural stem/progenitor cells with radial glial identity, and hypothesized that if the expression of TauP301L driving by her4.1 promoter started from the stem cell stage, and increased the number of neurons expressing the gene throughout the whole life of the zebrafish. In the zebrafish system, they found that chronic and abundant expression of TauP301L starting from early embryonic development led to tau hyperphosphorylation, but classical symptoms of tauopathies were absent in zebrafish such as neurofibrillary tangles, elevated apoptosis and active microglia. Additionally, cerebroventricular microinjection of Aβ42 into the adult TauP301L transgenic zebrafish found that TauP301L neither exacerbates the toxicity of Aβ42 nor initiates regeneration programs (Table 2). Together the transgenic zebrafish models of tauopathies could help researchers elucidate the biological mechanisms that underlie tau-related cognitive decline; however, the pathophysiological consequences of human tau gene expression in zebrafish CNS neurons are not clear. This is partly because several detailed phenotypes for transgenic tau zebrafish lines were absent, and the difference of transgenic tau zebrafish lines using different promoter elements is necessary to be clarified; additionally, it is controversial if the expression levels of the transgenes relative to the endogenous mapt genes of zebrafish are valuable to assess. Finally, neurogenesis in the adult zebrafish brain is much more abundant than in mammals [123], for example, zebrafish can continue to grow as adults [139] and in its adult brain have several progenitor zones [125]. On the contrary, mammalian brain has limited post embryonic neurogenesis [140], such as they have limited amount of proliferation in the adult brain, consequently making analysis of neuronal loss difficult. Despite these limitations transgenic zebrafish models provide a valuable system to explore whether chemical inhibitors can modulate the observed tauopathies-associated changes in vivo.

A schematic diagram of the zebrafish mapta and maptb mRNA transcript. The zebrafish mapta and maptb is located on chromosome 3 and chromosome 12, respectively. Orange and green boxes indicate exons that can be alternatively spliced. The orange boxes indicate exons encoding tubulin-binding motifs. The red arrows indicate microtubule-binding repeats region.
Tau transgenic zebrafish models and the characteristics
CONCLUDING REMARKS AND FUTURE DIRECTIONS
Despite remarkable advances in our understanding of tau biology, development, and functions over the past century, we are just beginning to decipher the enigmatic nature of these versatile players in order to best harness or modulate them in tauopathies. The zebrafish is rapidly emerging as an attractive model for tauopathies’ research. They are an ideal model for drug testing prior to clinical testing in mice. Herein we have discussed several transgenic zebrafish models, the overexpressed human tau in zebrafish CNS neurons results in similar biochemical and pathological changes to those found in human tauopathies. These changes included 1) tau hyperphosphorylation; 2) aberrant localization of tau to the somato-dendritic compartment; 3) adoption of abnormal conformations associated with tauopathies. The availability of transgenic tau zebrafish models allows more detailed biochemical studies of tau in the zebrafish CNS to characterize solubility, fibril morphology, and further clarify phosphorylation proceedings. However, there are still aspects of zebrafish models that need better understanding. For example, how to use the zebrafish system as models of tauopathies, we need to deeper and better understand zebrafish brain structure, physiology, and function. Whether the zebrafish could be employed to develop neurodegenerative models is controversial since zebrafish have an excellent capacity for regeneration and this ought to impact on the development of neurodegenerative phenotypes. Despite these limitations the recent availability of using genome editing technologies such as TALENs, ZFNs, and CRISP presents an exciting opportunity to develop zebrafish genetic models of neurodegenerative diseases such as AD [108]. Recent zebrafish models have revealed various aspects of tau biology and function that would otherwise be difficult to observe/analyze in other models in vivo. To develop effectively transgenic and mutant zebrafish models of tauopathies, we need to strengthen our understanding of the functions in zebrafish of some of the orthologs of the key genes implicated in human tau pathogenesis such as MAPT. The complexity of the human brain makes tauopathies particularly difficult diseases to model in animals. However, by employing a number of different models of tauopathies including the zebrafish, we can exploit the unique characteristics of each to unravel the molecular basis of these diseases in the future.
Footnotes
ACKNOWLEDGMENTS
This work was supported by the Chinese National Natural Science Foundation (81560241), the Foundation for Science and Technology projects in Guizhou ([2017]1014, LH [2017]7156, [2018]1009, [2018]5752), and the Foundation of the Education Department of Guizhou Province (KY [2016]035).