
Abstract
INTRODUCTION
In Alzheimer’s disease (AD), pathophysiological changes precede the clinical stages of mild cognitive impairment (MCI) and dementia [1]. There is some evidence that, in the course of preclinical AD, subjective cognitive decline (SCD) [2] may be an indicator of a stage termed “late stage” or “stage 3” preclinical AD [1], or “transitional stage to clinical AD” [3] with evidence of amyloidosis, tau-mediated neuronal damage, and “subtle cognitive decline”. Elderly individuals with SCD would, thus, form a high-potential target population for early disease-modifying therapies [1, 2].
This leads to an impetus to critically examine the prevalence of AD biomarker abnormalities, rates of clinical progression, and the predictive value of biomarkers for the latter, in SCD. Regarding these characteristics, a comparison of SCD versus MCI is of crucial importance to determine whether SCD is a useful and distinct at risk stage of early AD separate from or prior to MCI, respectively. Empirical data regarding the role of AD biomarkers for risk of progression in memory clinic patients with SCD is yet very scarce, and a comparison to MCI limited [4–6]. So far, the study of van Harten et al. [4] is the only memory clinic study on predictive value of cerebrospinal fluid (CSF) markers in SCD patients with conversion to MCI and/or AD dementia as outcome. No study so far has directly compared SCD versus MCI patients in a joint analysis. Importantly, different inclusion and exclusion criteria to distinguish SCD from MCI will impact biomarker prevalence and progression rates in the resulting samples. This is supported by reports of heterogeneous prevalence rates of abnormal CSF AD biomarkers in the range from ≈10 to 50% across memory clinic SCD patient samples with differences in age, methodology to distinguish SCD from MCI, and definitions of AD biomarker positivity [4–8].
In this regard, the exact definition of “objective cognitive impairment” required for MCI diagnosis is a critical variable. MCI is usually operationalized by “conventional” MCI criteria [9, 10]. However, these criteria are arguably prone to (mainly false-positive) diagnostic errors if the neuropsychological threshold of “objective cognitive impairment” within these criteria is not well operationalized, e.g., by only requiring a single or “one or more” impaired scores on a battery of selected tests [11]. This is emphasized by the fact that even in healthy individuals the base-rate of having ≥1 low/abnormal scores is 60–75% (depending on cutoffs used) when they are administered a comprehensive neuropsychological test-battery of ≈10 tests [12].
To address this problem, actuarial neuropsychological MCI criteria have been proposed [11]. These are designed to balance on the one hand sensitivity, by using a more liberal cut-off for impairment in a test score (–1SD instead of –1.5SD), and on the other hand reliability, by requiring consistent impairment of several scores within and/or across cognitive domains as opposed to single test score impairment. The idea behind this approach is to reduce spurious misclassifications due to the aforementioned base rate phenomenon, while still being sensitive to early cognitive impairment. This method has recently been proposed by the Subjective-Cognitive-Decline Initiative (SCD-I) Working Group as a favorable method to exclude objective impairment in the definition of SCD research samples in environments with extensive neuropsychological test data available (e.g., memory clinic studies) [13].
Given the aforementioned limited data, we sought to estimate prevalence of abnormal CSF biomarkers and their predictive value for clinical progression in memory clinic patients with SCD and compared results to MCI patients drawn from the same memory clinic study. To distinguish between SCD and MCI, we used the aforementioned actuarial, neuropsychological MCI criteria [11].
METHODS
Standard protocol approvals, registrations, and patient consents
The study protocol was approved by Institutional Review Boards of all participating study centers of the German Dementia Competence Network (DCN) [14]. All patients provided written informed consent.
Participants
We analyzed data from a multi-center longitudinal observational study of the German DCN, a collaborative study of German university memory clinics. Sample selection and assessment procedures of this study have been described previously [14]. All participants were referred to or sought help at the participating memory clinics for diagnostic work-up of subjectively experienced cognitive difficulties. A total of 813 non-demented memory clinic patients were included at baseline [14] from which we selected a subsample (n = 216, 26.6%) with available follow-up and CSF data. Comparison of the study sample with the group of patients excluded due to missing CSF, clinical, or neuropsychological data showed no differences regarding age, education, Mini-Mental-State-Examination (MMSE) score, gender or apolipoprotein E (APOE) ɛ4 status, suggesting that the assumption of a missing at random data pattern was not violated. The DCN cohort also collected cross-sectional data from n = 201 healthy control participants of 50 years and older who were cognitively normal and free of subjective cognitive deficits. This group consists of volunteering relatives (e.g., spouses) of participating patients with a mean age (M = 66.2, SD = 8.2) similar to the sample under study (Table 1). CSF-data was not collected for this group of the DCN cohort [14]. For the present study, we only used data of this group for illustration of patients’ neuropsychological performance relative to cognitive complaint-free healthycontrols.
Baseline characteristics of the whole study sample, MCI, and SCD patients
Values are presented as mean (SD), or number (%). Group differences based on independent sample t-test or χ2-test were appropriate; n.s., non-significant group difference, *p≤0.05, **p≤0.01; AD, Alzheimer’s disease. BADL, Bayer Activity of Daily Living Scale; CSF, cerebrospinal fluid; MADRS, Montgomery Asberg Depression Rating Scale; MMSE, Mini-Mental State Examination; MCI, mild cognitive impairment; SCD, subjective cognitive decline.
CSF measures
We collected CSF according to previously described standard operating procedures [15] by lumbar puncture from the L3/L4 or L4/L5 inter-vertebral region, into polypropylene test tubes with intermediate storage at site (–80°C). Samples were then aliquoted, deeply frozen, and shipped to a central biobank in Erlangen without undergoing any thawing/re-freezing cycles, where CSF-Aβ42, CSF-Tau, and CSF-pTau181 (Innogenetics, Ghent, Belgium) were analyzed with ELISA [16] in an ISO 9001-certified laboratory under routine quality control regime (intra-assay coefficients of variation: 2.3–5.9%; inter-assay coefficients of variation: 9.8–13.7%). We performed all analyses in duplicate and used the mean of the two.
Definition of preclinical AD by CSF pathophysiological markers
We defined preclinical AD by pathophysiological CSF markers of amyloidosis and tau-mediated neurodegeneration according to preclinical AD research recommendations proposed by the NIA-AA working group [1]. Evidence of amyloid pathology (A+) was defined by abnormally low CSF-Aβ42 (<600 pg/ml), while evidence of tau-mediated neurodegeneration (N+) was defined by presence of either abnormally high CSF-Tau (>300 pg/ml) or abnormally high CSF-pTau181 (>60 pg/ml). We applied our own, previously published cutoff values [15, 16], as is currently best practice. With regard to SCD, the concurrent presence of A+ and N+ is consistent with “stage 3” preclinical AD in the NIA-AA framework [1]. Accordingly, we defined CSF based preclinical AD as presence of A+ together with N+. However, we also looked at abnormal CSF-Aβ42, CSF-Tau, and CSF-pTau181, as individual markers respectively, to provide comparability to similar memory clinic studies on predictive value of CSF markers that included SCD patients [5, 4]. We also report results for a preclinical AD definition defined by an abnormal CSF-Aβ42/CSF-Tau ratio, as such ratios were found to be the best predictors for clinical progression from MCI to AD dementia [17]. Here, we used Hulstaert et al.’s [18] formula (Aβ42 / [240 + 1.18×Tau] <1) to define an abnormal Aβ42/Tau ratio.
Clinical, neuropsychological assessment
Standardized diagnostic procedures have been described previously [14]. Here, we only report on the parts relevant to the present study. Assessment included the German version of the neuropsychological assessment battery of the Consortium to Establish a Registry for Alzheimer’s Disease (CERAD) [19], which contains the MMSE [20] and is extended with the Trail-Making-Test (TMT) A and TMT-B [21]. In addition, the Wechsler-Memory-Scales [22] Logical Memory II subtest was applied. We assessed depressive symptomatology with the Montgomery-Asberg Depression Rating Scale (MADRS) [23] consisting of 10 items scored from 0 to 6 based on a clinical interview. A score ≥13 suggests at least mild depression [24]. Instrumental activities of daily living (IADL) were assessed with the Bayer-Activities of Daily Living Scale(BADL) [25].
Classification of patients into SCD and MCI based on the actuarial neuropsychological MCI criteria
Initially, the non-demented patients of the DCN cohort were recruited into the study based on a standardized diagnostic screening procedure to detect at least subtle amnestic or non-amnestic cognitive impairment: (1) subjective and/or informant report about cognitive decline (2) evidence of objective cognitive impairment (3) no or only minor IADL impairment (BADL score <4), and (4) not demented. Criterion (2) was met if patients’ test scores (cognitive domains in parentheses) fell more than one SD below age- and education-adjusted norms on the Wechsler Logical Memory II subtest (verbal memory) or on one or more of the following subtests of the CERAD battery: Word List Immediate Recall, Word List Delayed Recall (both verbal memory); Figure Copying (visuoconstruction); Verbal Fluency, Boston Naming Test (both language); Figure Recall (visual memory); TMT-A (processing speed); TMT-B (executive functions). This procedure was deliberately chosen by the DCN as a most liberal operationalization of conventional MCI criteria by Winblad et al. [10]. The base-rate of a low score in healthy controls in a test battery comprising nine scores is, however, very high (>70% for a z≤–1) [12].
For the present study, we therefore adapted the neuropsychological MCI criteria outlined by Bondi et al. [11] as follows: A patient was classified as MCI if at least one of the following two criteria were met: Criterion 1 was defined as impaired scores (>1 SD deficit) on two measures within at least one of the following three cognitive domains: 1) Verbal Memory, represented by Delayed Recall and Recognition score of the CERAD Word List; 2) Language, represented by Verbal Fluency and Boston Naming Test score of the CERAD battery. 3). Speed/executive function, represented by Trail-Making-Test (TMT) A and TMT B. Criterion 2 was defined as one impaired score (>1 SD deficit) in each of the three cognitive domains. A third criterion used by Bondi and colleagues, namely significant informant-rated difficulties in IADL in the absence of cognitive impairment (fulfilled in <5% of cases in their study [11]), was not applied here because the initial DCN algorithm based on Winblad et al.’s criteria [10] already required absence of significant IADL impairment.
Participants who did not meet the Jak-Bondi MCI criteria outlined above were classified as SCD. This SCD definition is fully in agreement with recently published recommendations to define pre-MCI SCD [13].
Statistical analysis
Statistical analyses were conducted with SPSS Version 22. Our main analysis focused on the rates of preclinical AD and clinical progression in the SCD patients. We determined risk for clinical progression to MCI (defined by the Jak-Bondi criteria outlined above) and AD dementia according to biomarker status with Cox-Proportional Hazard regression analyses. We conducted one separate analysis each for abnormality in CSF-Aβ42, CSF-Tau, and CSF-pTau181, and one each for the two alternative CSF preclinical AD definitions. In line with methodology of previous studies [4, 17], we report results of an unadjusted model (model 1), containing only the respective biomarker as a predictor, as well as for models adjusted for additional clinical variables/risk factors of AD (model 2, adjusted for gender and age; model 3, additionally adjusted for baseline MMSE, data missing for one SCD case; model 4, additionally adjusted for dichotomized APOE ɛ4 status, APOE data missing for 5 SCD cases). For comparison, we also report results of analogously conducted survival analyses in the MCI group with incident AD dementia as outcome.
RESULTS
Descriptive statistics
The Jak-Bondi MCI criteria were fulfilled in 134 (62.0%) patients of the study sample (50% met criterion 1, 3.7% criterion 2, 46.3% both criteria). The remaining 82 patients (38%) met neither criterion 1 or 2 and were classified as SCD. As the initial criterion of subjective cognitive difficulties at study entry could be reported by the patient and/or informant, we reviewed each case classified as SCD with regard to self-report of SCD. Self-report of cognitive difficulties was observed in each patient, confirming adherence to the recommended SCD research criteria [2, 13] in the entire SCD group.
Descriptive statistics of the whole sample and the two subgroups are given in Table 1. Figure 1 shows the neuropsychological (CERAD) battery z-score profile of both groups together with performance of the volunteer healthy controls of the DCN cohort. Mean performance of the SCD patients across all scores was significantly higher compared to MCI and well above the –1SD threshold, indicating unimpaired test performance on average. Further, it was worse compared to healthy controls whose mean performance, however, was consistently above the normative line of zero. Importantly, even these complaint-free healthy controls showed individualz-scores of < –1 in one or more of the nine tests depicted in Fig. 1 in 51.2% of cases.
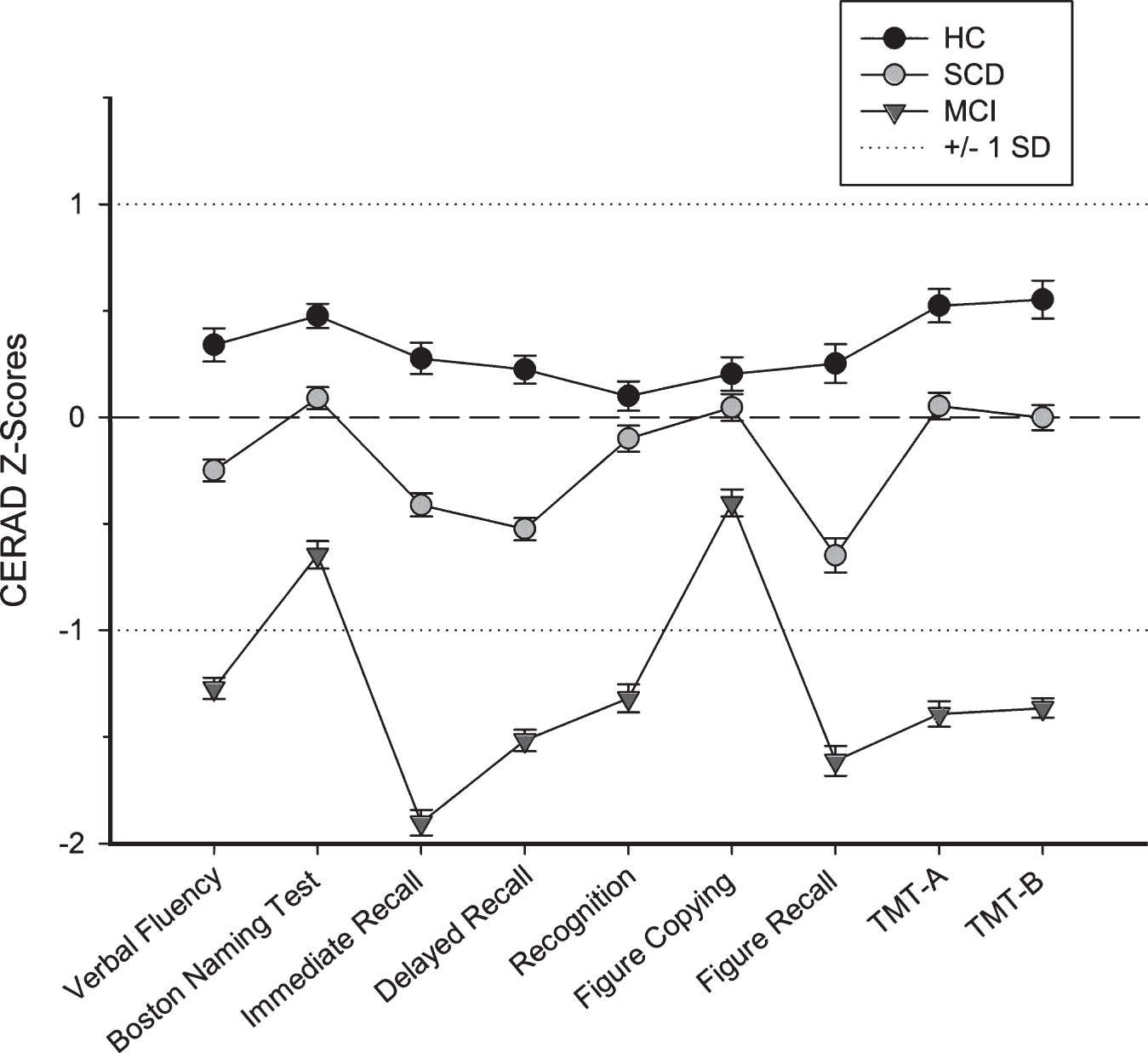
Neuropsychological profile in healthy controls, and patients with SCD and MCI. Test performance is shown as Z-scores (Means and Standard Errors), based on the German normative sample and represent age-, gender-, education adjusted scores. A Z-score of lower than –1 corresponds to a one standard deviation deficit compared to the normative sample. CERAD, German Version of the Consortium to Establish a Registry for Alzheimer’s Disease neuropsychological assessment battery. This battery includes the Trail-Making-Test (TMT) A and B as additional tests. The Subjective Cognitive Decline (SCD) and Mild Cognitive Impairment (MCI) group differ significantly on all variables (p < 0.001; p < 0.05 for Figure Copying). Scores of SCD also differ significantly from that of healthy controls (HC) for all variables except Recognition and Figure Copying (p < 0.001; p < 0.01 for TMT-B, p < 0.05 for TMT-A).
No group differences were observed regarding CSF-Aβ42, both for absolute mean values and number of patients below the cutoff. However, MCI patients showed more abnormal CSF-Tau and CSF-pTau181 values. Groups did not significantly differ in frequency of a CSF AD profile when defined as “A+N+” [1]. However, the MCI group showed significantly higher prevalence of a CSF AD profile, when defined as an abnormal Aβ42/Tau ratio. Clinical progression to incident AD and other dementia was significantly higher in MCI compared to SCD.
Predictive value of biomarkers for clinical progression
SCD sample
Eighteen (22%) SCD patients progressed to MCI and 10 (12%) to AD dementia. Three individuals (3.7%) progressed to a non-AD dementia. For the survival analysis models, we treated these cases as non-progressors to MCI/AD dementia as a competing risk model was not feasible with this few alternative outcome events (analogous procedure in the MCI group analysis). Abnormal CSF-Aβ42 was associated with a 2.7-fold risk for clinical progression, while there was no risk increase observed in participants with abnormal tau or pTau181 values already after adjustment for age and gender. Both definitions of CSF preclinical AD were predictive for clinical progression with higher hazard ratios than for CSF-Aβ42 abnormality alone (Table 3, Fig. 2a).

Survival curves for clinical progression (to MCI/AD dementia) in patients with subjective cognitive decline. (a) shows Kaplan-Meier curves corresponding to the unadjusted models of the Cox-Proportional Hazard Regression analyses (see also Table 3a) for each biomarker predictor in the SCD patient sample with clinical progression to incident MCI/AD dementia as outcome. Number of SCD patients at risk: n = 82 at 0 months; n = 42 at 24 months.
Baseline characteristics of stable SCD versus SCD with clinical progression to MCI or AD dementia
Values are presented as mean (SD), or number (%). #Three individuals converted to non-AD dementia. They are not listed in this table as a separate subgroup and were excluded for group comparison statistics between non-converters and those with clinical progression to MCI/AD dementia. Group differences based on independent samplet-test or χ2-test were appropriate; n.s., non-significant group difference, *p≤0.05, **p≤0.01, ***p≤0.001; AD, Alzheimer’s disease; BADL, Bayer Activity of Daily Living Scale; CSF, cerebrospinal fluid; MADRS, Montgomery Asberg Depression Rating Scale; MMSE, Mini-Mental State Examination; MCI, mild cognitive impairment; SCD, subjective cognitive decline.
Cox proportional hazard models for clinical progression in the SCD patients (n = 82) and in those classified as MCI with the Jak-Bondi criteria (n = 134)
Data presented as hazard ratio (95% CI) for individuals with a positive (abnormal) result in the respective single biomarker / AD profile versus those with normal values. *p≤0.05, **p≤0.01, ***p≤0.001. Three individuals converted to non-AD dementia. They were included in the computation of the models as non-progressors to MCI/AD dementia (i.e. together with the stable SCD group) as a competing risk model was not feasible with only these three individuals forming a group with alternative outcome. Model 1: unadjusted model; Model 2: adjusted for age, gender; Model 3: adjusted for age, gender, MMSE; Model 4: adjusted for age, gender, MMSE, ApoE status.
MCI sample
Overall, the risk increase (hazard ratios) for biomarker positive vs. negative MCI patients was more pronounced compared to that of positive versus negative SCD patients. Further, in contrast to results in the SCD group, tau markers alone were better or (after adjustment for cognitive performance) equally good predictors then CSF-Aβ42. However, similar to the results in SCD, a combination of CSF-Aβ42 and tau markers was more predictive than any individual marker (Table 3, Fig. 2b).

Survival curves for clinical progression (to AD dementia) in patients with MCI according to the Jak-Bondi criteria. (b) shows Kaplan-Meier curves corresponding to the unadjusted models of the Cox-Proportional Hazard Regression analyses (see also Table 3b) for each biomarker predictor in the MCI patient sample with clinical progression to incident AD dementia as outcome. Number of MCI patients at risk: n = 134 at 0 months; n = 66 at 24 months.
DISCUSSION
In the present study, we investigated a relatively large memory clinic sample of SCD patients with regard to prevalence of CSF biomarker abnormalities and the predictive value of these for clinical progression, and compared results to patients with MCI from the same cohort. As a novel approach and major strength of this study, we distinguished SCD from MCI by application of actuarial neuropsychological MCI criteria to define objective cognitive impairment. These criteria have been shown to be more accurate and reliable compared to conventional MCI criteria in a large cohort study (ADNI [11]) and are also recommended in novel SCD research criteria guidelines [13]. The rates of conversion to dementia in individuals with a diagnosis of MCI defined through these criteria in our study (41%) were quite similar to that in ADNI (49%) [11]. This speaks to the robustness of these criteria across studies despite differences in recruitment and setting[14, 26].
Memory clinic SCD patients in particular, form a high-potential target population for early disease-modifying therapies. These patients actively seek medical advice and may, thus, present high commitment to participate in early intervention/prevention studies. They are also the ones who would most likely receive future treatment. It is therefore critical to further characterize these patients, in contrast both to those patients with objective cognitive impairment at the MCI level and to cognitively normal elderly without SCD. The present study makes important contributions in this regard.
We showed that the SCD patients in our memory clinic sample had average neuropsychological test performance well above the –1SD threshold in all CERAD tests (not just those used in the Jak-Bondi algorithm). This confirms that these patients are indeed “cognitively unimpaired” and therefore correctly classified as non-MCI. However, due to the fact that the sample is one of referred, help-seeking individuals who are concerned about self-perceived memory decline [27], we still expected, and observed a relatively high number of individuals who are presumably on the disease trajectory of AD, albeit at an earlier (pre-MCI/preclinical) stage of the disease [1, 2]. This is supported by evidence of amyloid pathology (reduced CSF-Aβ42 levels) in 35% of the SCD patients which was not different from that seen in MCI and is around 10–15% higher compared to meta-analysis estimates in cognitively normal individuals without SCD of comparable age [28]. This is also the case for frequency of positive ApoE4 status [28, 29]. Abnormal CSF-Aβ42/Tau ratios indicative of preclinical AD were present in 34% of SCD patients in our study. These numbers align well with results of a recently published neuroimaging study [30] that compared help-seeking SCD patients (“SCDclinic”), with community volunteers that either scored high (“SCDcommunity”) or low (“controls”) on a self-report cognitive difficulties questionnaire. The authors found similarly enriched rates of brain amyloid burden in “SCDcommunity” (34%) and “SCDclinic” (29%) as compared to controls (9%). However, the “SCDclinic” group further distinguished itself from the “SCDcommunity” subjects at the group level by reduced gray matter volume in AD typical temporal and parietal regions. While this converging evidence from CSF and neuroimaging studies supports the concept of “SCDclinic” as an early at risk stage of AD, we also acknowledge that conflicting findings regarding imaging markers of AD in SCD have been reported (see Table 1 of [30] for a comprehensive overview).
Clinical progression to MCI or AD dementia was observed in every third patient (34.2%) of the SCD group within an average follow-up time of 27 months. This clinical progression rate was increased to 60% (28% to MCI, 32% to AD dementia) in those SCD patients with biomarker evidence of preclinical AD (abnormal CSF-Aβ42/Tau ratio). This emphasizes the viability of the SCD concept to detect patients at risk for cognitive decline and AD dementia and the additional benefit of biomarker assessment for further risk enrichment. The reported rates of clinical progression are informative for the design of clinical trials that would apply an SCD definition and MCI exclusion criteria similar to our study.
Comparison of our study with the few existing studies on CSF biomarkers and clinical progression in SCD highlight the potential impact of differences in MCI exclusion criteria for the definition of SCD samples as well as the necessity to report these in detail in research reports [13]: Visser et al. [6] reported on the predictive value of CSF AD biomarkers for clinical progression in memory clinic SCD, non-amnestic and amnestic MCI patients, using the same Aβ42/Tau ratio as in the present study [18]. Their SCD group was very similar in terms of age, education, and also ApoE4 frequency. Further, while the prevalence of a CSF AD profile was higher in both SCD (52%) and MCI (75%) in that study, the enrichment factor of MCI versus SCD (≈1.5 times more likely in MCI) was identical in both studies. Within comparable follow-up time, the conversion rate to AD dementia in MCI patients with a positive CSF AD biomarker profile was also almost identical (47.8% here versus 46.7% [6]). However, we also observed progression to AD dementia in the CSF AD biomarker positive SCD group while this was not the case in [6]. Different criteria to distinguish SCD from MCI in both studies may explain this discrepancy. Visser et al. applied conventional MCI criteria to define non-amnestic and amnestic MCI by impairment in “one or more” tests across memory and non-memory domains (using a –1.5SD threshold). The Jak-Bondi approach applied in our study is more conservative regarding the consistency of impairment within and across cognitive domains while the deficit-threshold for each test is more liberal (–1SD). This means that some patients classified as non-amnestic or amnestic MCI in the study of Visser et al. are classified as SCD in our study. As a result, the SCD sample of the present study is, on average, slightly more progressed toward the MCI threshold. This may explain the discrepant finding regarding clinical progression in SCD given similar follow-up periods. A slightly lower MMSE mean score in our SCD group (27.9 points) which lies between that of Visser et al.’s SCD (28.8) and amnestic MCI (25.9) group supports this assumption. MCI conversion was not an outcome in Visser et al., so we cannot compare our results in this regard.
Rates of preclinical AD and clinical progression in our SCD sample were considerably higher than what was reported from a memory clinic study of SCD patients with comparable sample size and follow-up time, similar outcome of clinical progression (i.e., to MCI and/or AD dementia), but younger mean age (60 versus 67 years) [4]. A recent Spanish memory clinic study reported abnormal CSF Aβ42/Tau ratio in 20% of SCD cases but evaluated CSF predictive performance only in a mixed SCD/MCI sample with AD dementia as outcome [5]. In our SCD sample, combined Aβ42/Tau profiles were more predictive than CSF-Aβ42 alone which was by far the best predictor in van Harten et al.’s study [4]. One apparent reason for this discrepant finding may be the younger age and that the positive predictive value of amyloid for AD decreases with age [5]. The lower prevalence of preclinical AD and conversion rates in SCD patients of van Harten et al.’s study may also be due to their application of Petersen’s conventional criteria to exclude MCI [31], while we applied the stricter, actuarial neuropsychological Jak-Bondi method. In both studies, markers of tau pathology alone were not associated with clinical progression in SCD. Thus, despite the discrepant finding regarding the most predictive biomarker profile, both studies highlight the important role of amyloid compared to tau markers in pre-MCI at risk stages of AD.
Finally, our results confirm that a combination of CSF biomarkers of neurodegeneration (Tau/pTau181) and amyloidosis (Aβ) provides a more optimal prediction of progression to AD dementia than single parameters [5, 32]. We demonstrated this for MCI and SCD independently. Further, our results suggest that, across the whole at risk spectrum of SCD and MCI, the Aβ/Tau ratio is a more robust predictor than the categorical combination of “A+N+” as the ratio performed only slightly worse in predicting clinical progression in SCD but, in line with recent findings [17], far outperformed the A+N+ combination in the MCI group.
It is important to note that a 4-fold categorical predictor (i.e. A-N-, A-N+, A+ N-, A+N+) may bear additional predictive information not captured in this study [8, 34]. However, we refrained from this approach due to sample size restrictions, leading to only a few cases falling into the A+N- (n = 9) and A-N+ (n = 18) category in the SCD subgroup, which precluded a robust Cox-regression analysis with adjustment for covariates. Future studies with larger sample size should also evaluate whether the distinction between biomarkers of tau pathology (“T”, measured by CSF-pTau or tau PET) and biomarkers of neurodegeneration or neuronal injury (“N+”, measured by FDG-PET, structural MRI, or CSF total tau) bears additional predictive information regarding risk of clinical progression toward AD dementia. This distinction has recently been proposed in the “A/T/N system” by Jack and colleagues [35].
For the present analyses, we had to select a subsample of patients with available follow-up and CSF biomarker data which bears the risk of selection bias. However, we found only slight baseline differences between excluded and included patients, speaking to the representativeness of our results for the DCN memory clinic cohort.
In summary, the findings of the present study support the SCD concept in AD clinical research. The neuropsychological, biomarker, and clinical progression characteristics of this patient group indicate that SCD is an early at risk stage of AD, distinct from and prior to MCI. The study further highlights the usefulness of the neuropsychological MCI criteria to distinguish between SCD and MCI in a comprehensible, reproducible, and arguably more reliable manner, and the importance of such aspects for preclinical AD sampling strategies.
Footnotes
ACKNOWLEDGMENTS
This study has been supported by a grant from the German Federal Ministry of Education and Research (BMBF): Kompetenznetz Demenzen (01GI0420). The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript.