
Abstract
Mitochondrial impairment is a feature of neurodegeneration and many investigators have suggested that epigenetic modifications of the mitochondrial DNA (mtDNA) might be involved in late-onset Alzheimer’s disease (LOAD), but evidence in humans is limited. We assessed the methylation levels of the mtDNA D-loop region in blood DNA from 133 LOAD patients and 130 controls, observing a significant 25% reduction of DNA methylation levels in the first group (2.3 versus 3.1%). Overall, the present data indicate that there is a decreased methylation of the D-loop region in LOAD peripheral blood DNA, suggesting that mtDNA epimutations deserve further investigations in AD pathogenesis.
INTRODUCTION
Late onset Alzheimer’s disease (LOAD) is a complex disorder resulting from the interaction between genetic and non-genetic factors [1]. Epigenetic mechanisms are able to change gene expression under the influence of environmental factors such as diet, hazardous exposures, and life events, and their deregulation could contribute to LOAD onset [2]. In fact, a growing number of studies performed in vitro, in vivo, and ex vivo in human tissues, showed that aberrant DNA methylation, histone modifications, and deregulated microRNA (miRNA) expression play a pivotal role in LOAD pathogenesis [2].
Particularly, DNA methylation represents one of the most studied epigenetic modifications in Alzheimer’s disease (AD) [3]. In mammals, this reaction is carried out by DNA methyltransferases (DNMTs) and occurs mainly at CpG sites located throughout the genome. When methylation occurs in the promoter region, it usually induces repression of gene expression, while methylation of non-CpG islands is associated with the prevention of genomic instability phenomena, such as the movement of transposable elements [4]. DNA methylation changes at both gene-specific and global levels have been detected in AD [3]. For example, the analysis of postmortem brain regions of monozygotic twins discordant for AD revealed reduced global levels of DNA methylation in the temporal neocortex of the AD twin [5], and increased methylation of repetitive elements such as LINE-1, often used as a surrogate of global DNA methylation, has been found in blood DNA of AD patients compared to healthy controls [6].
Although mitochondrial DNA (mtDNA) impairment is a feature of AD, little attention was given to the mitochondrial epigenome itself, and until now only one study searched for methylation modifications in mtDNA of AD patients [7]. Particularly, Blanch and coworkers investigated the methylation levels of the mitochondrial displacement loop (D-loop) region and of MT-ND1 and MT-ND6 genes (mitochondrial NADH dehydrogenase subunit 1 and 6), observing differential methylation levels, mainly in the D-loop region, in the entorhinal cortex of individuals in preclinical stages of AD respect to healthy control individuals, suggesting that the D-loop region could be a sensitive epigenetic target in AD pathology.
Previous studies conducted in colorectal cancer and adjacent tissues revealed that D-loop methylation could regulate the transcription of mtDNA genes, such as MT-ND2 coding for a subunit of NADH (ND2) [8]. Particularly, a reduced D-loop methylation positively correlated with increased ND2 expression in colorectal cancer tissues [8]. Furthermore, D-loop methylation was found to be sensitive to particulate matter <2.5μm (PM2.5) in blood DNA of exposed people [9], as well as in placental DNA of pregnant women [10]. This is particularly interesting given the increasing amount of studies linking air pollution to epigenetic changes and dementia risk [11, 12].
In the present study, we assessed DNA methylation levels of the mtDNA D-loop region in blood DNA of a large cohort of LOAD patients and healthy matched controls in order to search for differences associated with the disease status detectable in an easily available tissue.
MATERIALS AND METHODS
Study population
A total of 133 LOAD patients and 130 matched controls were collected at the Department of Neuroscience, University of Pisa (Table 1). Diagnosis of probable AD was performed according to DSM-IV [13] and NINCDS-ADRDA criteria [14]. Based on age at onset above 65 years and absence of a family history of dementia, all the subjects were assumed to be sporadic LOAD cases. As normal controls, we recruited healthy volunteer subjects matched to LOAD patients for age and gender, as well as for ethnicity and geographic origin (both LOAD and control individuals were Italian Caucasians, resident in northern Tuscany, and were recruited simultaneously). All the LOAD patients had a Clinical Dementia Rating (CDR) scale of 1 or 2. Cognitive functions and family history of dementia were ascertained in controls after a rigorous neurological examination, including only healthy subjects with no presence of cognitive impairment (Mini-Mental State Examination >26; CDR = 0) and of relatives who developed AD or other dementias. DNA samples were collected in the frame of a recent study approved by the ethics committee of the Pisa University Hospital and aimed at addressing epigenetic changes in AD [15]. All the individuals signed a written informed consent before enrollment in the study. Individuals taking drugs, vitamins or supplements known or suspected to interfere with DNA methylation were excluded from the study. Further details about the study design can be found in our previous papers [15, 16].
Demographic characteristics of the study population
aStudent’s t Test. bFisher Exact Test.
Extraction of genomic DNA and bisulfite modification
An aliquot of blood was collected from each subject in EDTA tubes and stored at –20°C until assayed. Genomic DNA extraction was performed using the QIAmp DNA blood Mini Kit (Qiagen, Milan, Italy, Catalog N° 51106) following the manufacturer’s protocol. The extracted DNA was quantified using a Nano Drop ND 200c spectrophotometer (NanoDrop Thermo scientific). 200 ng of DNA from each sample were treated with sodium bisulfite in order to convert all unmethylated cytosines into uracil. The EpiTect Bisulfite Kit (Qiagen, Milan, Italy, Catalog N° 59104) was used for this purpose, following the manufacturer’s instructions. A sample of genomic DNA completely unmethylated (Amplified human genomic DNA, completely unmethylated, Qiagen, Cat No./ID: 59568) was used as control assay to check the bisulfite conversion efficiency that resulted to be of 99% in average. Moreover, to avoid potential batch effects, each kit was used to treat simultaneously the same number of LOAD and control samples. Furthermore, samples derived from different bisulfite treatments were analyzed independently on separate occasions to verify the inter-assay variability, observing a very goodreproducibility.
Methylation sensitive-high resolution melting analysis
Methylation of the D-loop region was assessed by means of methylation sensitive-high resolution melting (MS-HRM) technique using a CFX96 Real-Time PCR detection system (Bio-Rad). The D-loop region (GenBank: J01415.2) was analyzed for the presence of CpG islands by CpG plot software (http://www.ebi.ac.uk/Tools/seqstats/emboss_cpgplot/). We developed a MS-HRM protocol according to literature criteria [17], using methylation independent primers (MIP) designed by means of the software MethPrimer [18]. The sequences of the primers were as follows: forward 5’-GGAGTTTTTTATGTATTTGGTATTTT3’, reverse 5’- ACAAACATTCAATTATTATTATTATATCCT-3’. These primers amplified a D-loop amplicon of 222 bp which comprises the nucleotides 35-256 based on the sequence deposited in Genbank accession no. J01415.2, and that included 10 CpG sites. MS-HRM analyses were performed as described elsewhere [15, 19] and further details are available in the Supplementary Material.
Statistical analyses
Demographic data, such as age at sampling and gender, were compared between groups by means of Student’s t Test and Fisher Exact Test, respectively. Differences in D-loop methylation levels between AD patients and controls were compared by one-way multifactorial ANOVA correcting for age at sampling and gender. Statistical analyses were performed with STATGRAPHICS 5.1 plus software package for Windows. The statistical power of the study was calculated with the free sample size calculator (http://clincalc.com/Stats/SampleSize.aspx). We designed a study with an a priori power >80% to detect DNA methylation differences of 1% or higher between groups.
RESULTS
MS-HRM analysis showed the existence of an inter-individual variability in D-loop methylation levels ranging from 0 to 9% (Fig. 1). D-loop methylation levels (%) were lower in AD patients (2.3±0.2) respect to control subjects (3.1±0.2) in a statistically significant manner (p = 0.04) (Table 2). No significant correlation between D-loop methylation levels and age at sampling was observed (r = –0.09, p = 0.14), and no difference in methylation levels between males and females was detected (p = 0.55) (data not shown).
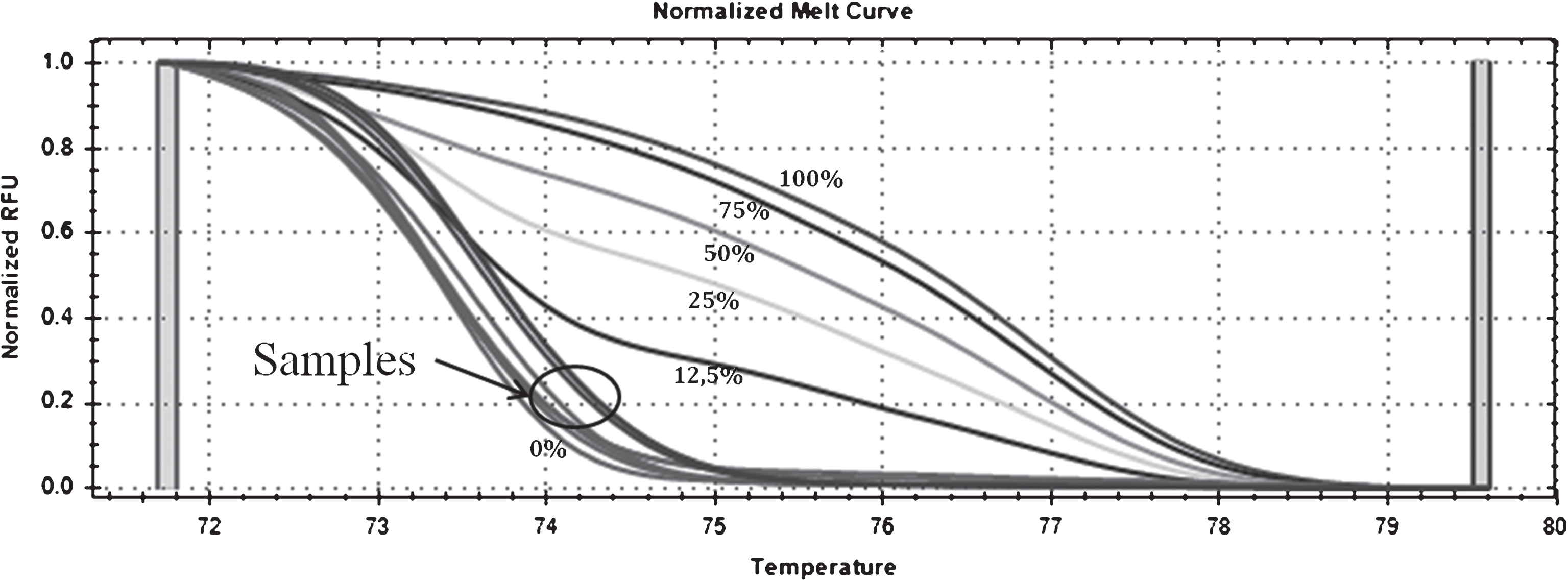
MS-HRM curves of D-loop methylation. Standard curves generated by mixing methylated and unmethylated standard DNAs (0%, 12.5%, 25%, 50%, 75%, and 100%) and curves of the samples ranging from 0 to 9% are shown with an arrow.
Mean D-loop region methylation levels (%) in AD patients and matched healthy controls
aMultifactorial analysis of variance, corrected for age at sampling and gender.
DISCUSSION
In the current study, we investigated the methylation levels of the mitochondrial D-loop region in blood DNA of a relatively large cohort of LOAD patients and healthy matched controls. D-loop is a non-coding region of the mtDNA of about 1.1 Kb, critical for both mtDNA replication and transcription [20]. We observed significantly lower D-loop methylation levels in LOAD patients respect to healthy controls (p = 0.04). Until now, only one study investigated mtDNA methylation changes in neurodegenerative diseases [7]. The authors found increased methylation levels of the mtDNA D-loop region in the entorhinal cortex of eight patients with AD-related pathology (stages I/II and III/IV of Braak) respect to controls, and the degree of methylation was higher in early than in later disease stages. Furthermore, a dynamic pattern of methylation of this region was observed in transgenic AD mice (APP/PS1 mice) along with disease progression [7]. The same study also revealed decreased D-loop methylation levels in the substantia nigra of ten Parkinson’s disease patients with respect to healthy matched controls [7], suggesting that the degree of methylation of this region might characterize different tissues, or disease stages, of individuals suffering from neurodegeneration. In the present study, which is the first addressing this issue in peripheral tissues, we observed a decreased methylation of the D-loop region in the mtDNA extracted from blood DNA of LOAD patients with respect to healthy matched controls, strengthening previous evidence suggesting that this region could be susceptible to epigenetic modifications in neurodegenerative diseases. As previously demonstrated by Blanch and coworkers using cells depleted of the mtDNA, this region is peculiar of the mitochondrial genome [7]. Differences between present data and previous observations in AD specimens [7] could be due to the different tissues analyzed or to different disease stages. Unfortunately, we have no repeated sampling from patients at different disease stages to address if the methylation levels of this region are subjected to a dynamic regulation with disease progression, as previously suggested in animal models of the disease [7].
The search of epigenetic modifications has grown exponentially during the past decade in AD blood samples [21]. An important advantage in the use of peripheral tissues, of easier availability in the study of AD, is the opportunity to investigate the molecular events associated with the different stages of the disease. Otherwise the study on postmortem brains only provides a snapshot of the final result of the pathogenetic processes of the disease, which does not necessarily reflect the mechanisms that lead to it [22]. Both gene specific and global DNA methylation analyses have been performed in peripheral blood of AD patients, but the studies were limited to nuclear DNA [6 , 24]. However, mitochondrial impairment is a feature of the AD condition and both mitochondrial alterations and changes in mitochondrial gene expression have been detected in peripheral lymphocytes of AD patients [25, 26], or in patients affected by other neurodegenerative diseases [27, 28]. Moreover, mitochondrial dysfunction through the over production of reactive oxygen species is able to induce epigenetic modifications [29, 30]. Therefore, the analysis of epigenetic modifications in the mtDNA, that is more susceptible to oxidative stress than nuclear DNA, is gathering interest in AD and other neurodegenerative diseases [7]. Noteworthy, both present and previous studies [9] revealed that on average D-loop methylation levels in peripheral blood DNA are of about 2–4%, so that more than 100 patients and 100 controls should be required to detect with enough power a difference of about 1% as we observed in the present study. Indeed, another advantage of using blood samples rather than postmortem tissues is that they can be easily collected from hundreds of living subjects. Further investigation is now required to clarify how D-loop methylation changes affect the expression of mtDNA genes in AD, as well as to clarify the environmental factors able to induce thosechanges.
In summary, the present data indicate that there is a reduction in the percentage of D-loop methylation in peripheral blood of LOAD samples, suggesting that mtDNA epimutations deserve further investigations in AD pathogenesis.