
Abstract
Alzheimer’s disease (AD) is a neurodegenerative disorder of the elderly. As the prevalence of AD rises in the 21st century, there is an urgent need for the development of effective pharmacotherapies. Currently, drug treatments target the symptoms of the disease and do not modify or halt the disease progress. Thus, natural compounds have been investigated for their ability to treat AD. This review examines the efficacy of curcumin, a polyphenol derived from turmeric herb, to treat AD. We summarize the in vivo and in vitro research describing the mechanisms of action in which curcumin modifies AD pathology: curcumin inhibits the formation and promotes the disaggregation of amyloid-β plaques, attenuates the hyperphosphorylation of tau and enhances its clearance, binds copper, lowers cholesterol, modifies microglial activity, inhibits acetylcholinesterase, mediates the insulin signaling pathway, and is an antioxidant. In conclusion, curcumin has the potential to be more efficacious than current treatments. However, its usefulness as a therapeutic agent may be hindered by its low bioavailability. If the challenge of low bioavailability is overcome, curcumin-based medications for AD may be in the horizon.
INTRODUCTION
Alzheimer’s disease (AD), accounting for 60 to 80% of dementia cases, is an irreversible, progressive brain disorder characterized by behavioral changes and loss of cognitive functions [1]. Common symptoms include short-term memory loss, cognitive deficits, and an inability to perform tasks of daily living [2]. As the disease progresses, afflicted individuals often withdraw from society, lose bodily functions, and die. The life expectancy following diagnosis ranges from an average of three to nine years [3]. Globally, approximately 46 million people are afflicted with dementia and the population ages, the number is projected to increase to 131.5 million by 2050 [4]. The worldwide cost of dementia is US $818 billion and is expected to be a trillion dollar disease in 2018 [4]. Given the economic cost and the increasing prevalence of the disease, it is essential to explore drug therapies.
Presently, there are a limited number of synthetic drugs that are available for the management of the disease. However, several natural compounds have been investigated for their efficacy in treating AD [5]. Curcumin is a natural compound derived from the herb turmeric [6]. Recent research has focused on its mechanisms of action by which it can modulate AD progression.
This review examines the efficacy of curcumin to treat AD. It describes the pathophysiology of AD, current pharmacotherapies for AD, and the problems with existing therapies. Then, the review delineates the major mechanisms of action by which curcumin acts on the multi-faceted disease process of AD. Finally, the challenges associated with curcumin treatment are discussed and directions for future research are outlined.
PATHOPHYSIOLOGY OF ALZHEIMER’S DISEASE
The main histological features of AD include neuronal loss, senile plaques, neurofibrillary tangles, and accumulation of cytosolic lipids [7, 8]. The loss of neurons and synapses results in atrophy of the cerebral cortex and subcortical regions, principally in the temporal lobe, parietal lobe, and frontal cortex [9]. Loss of cholinergic neurons, cells that release the neurotransmitter acetylcholine (ACh), has been observed in the nucleus basilis of Meynert (nBM), an area that projects to the frontal cortex [10]. The other disease mechanism hypotheses are contingent on two proteins: amyloid-β (Aβ) peptide and tau, the former forming plaques and the latter formingtangles [8].
The irregular folding and aggregation of Aβ peptides into senile plaques is implicated in the initial development of neurodegeneration in AD [8]. Various isoforms of Aβ peptides, the most common being 40 and 42 amino acids in length, are generated from sequential proteolysis of the transmembrane amyloid-β precursor protein (AβPP) by β-APP-cleaving enzyme 1 (BACE1) and γ-secretase. Aβ peptides easily aggregate into oligomers around cells and have a crucial role in pathogenic events. These events include increased calcium influx, oxidative stress, inflammatory processes, altered kinase and phosphatase activities, irregular regulation of transcription factors, and disruptions in ion channel and receptor gene expression [11].
In addition to Aβ plaques, intracellular tau tangles may initiate the disease cascade. Hyperphosphorylated states of tau destabilize microtubules, compromising the integrity of the cell’s cytoskeleton and disrupting the intracellular transport system. However, genetic and pathological evidence suggests that Aβ accumulation precedes neurofibrillary tangles, which makes Aβ an ideal target for pharmacotherapies [12].
CURRENT PHARMACOTHERAPIES FOR ALZHEIMER’S DISEASE
The oldest hypothesis for the etiology of the disease, on which four present drug treatments are founded upon, is the cholinergic hypothesis. It proposes that AD is caused by loss of cholinergic neurons [13], and a decrease in ACh levels results in short term memory loss and confusion. Acetylcholinesterase (AChE) inhibitors (tacrine, rivastigmine, galantamine, and donepezil) are a class of drugs that reduce the rate at which ACh is degraded in the synapse, thus increasing the concentration of ACh in the brain [14].
Excitotoxicity due to glutamate, another neurotransmitter, has been observed in AD. Physiologically, glutamate stimulates the N-methyl-D-aspartate (NMDA) receptor on cells, resulting in calcium influx and the activation of intracellular signals for learning and memory. However, excessive glutamate results in overstimulation over the receptor, toxic amounts of intracellular calcium, and eventually cell death [15]. Memantine is a NMDA receptor antagonist that inhibits overstimulation by glutamate and has also be indicated for the treatment of AD [16]. While AChE inhibitors and NMDA antagonists are prescribed for AD, none of these drugs are curative: they do not delay the onset of AD or slow its progression. The available treatments target the symptoms of the disease, have a small benefit, and are palliative in nature. The efficacy of AChE inhibitors declines as cholinergic neurons degenerate during the progression of the disease. Current therapies also have other detriments such as cost, side effects, the need for a prescription, and lack of accessibility [17]. As such, there is a need to explore other pharmacotherapies that are more accessible and alter the disease process.
HISTORY OF CURCUMIN
In light of the need for more efficacious treatment options for AD, natural compounds, such as curcumin, are being explored as potential pharmacotherapies. Curcumin is a polyphenol derived from turmeric (Curcuma longa), an herb native to southern Asia [18]. It is primarily used as a spice in southeastern Asian cuisine, as a food-coloring agent, and has been utilized in traditional medicine practices [19]. In Ayurveda, an Indian system of medicine, curcumin has been documented for centuries to treat respiratory conditions, liver disorders, anorexia, cough, rheumatism, among other disorders [20]. The past decade has shown a surge in research in exploring the compound’s antifungal, antiviral, antioxidant, and anti-inflammatory properties [19]. Pharmacologically, curcumin’s effects are extensive: it has multiple sites of action, including modulation of inflammatory molecules, transcription factors, enzymes, protein kinases, growth factors, cell cycle regulatory proteins, metal ions, DNA, lipids, and proteins [21]. Animal studies have indicated that curcumin may be a therapeutic option for a wide range of human diseases such as diabetes, obesity, cancer, systemic chronic diseases, and psychiatric and neurological disorders [21].
In epidemiological studies, India has been established to have one of the lowest prevalence rates of AD in the world [22, 23]. Since India has widespread turmeric consumption, with individuals consuming 21.7 to 28.6 grams per month [24], the neuroprotective role of curcumin in AD is a possibility. Since the relationship between curcumin consumption and a lower prevalence of AD been observed, basic scientific research has focused on the mechanisms of action in which curcumin may act in AD to corroborate its potential benefit.
MECHANISMS OF ACTION OF CURCUMIN IN ALZHEIMER’S DISEASE
Curcumin’s mechanisms of action are pleiotropic (Fig. 1). It targets the two histological markers of AD, Aβ and tau. Additionally, curcumin modulates other aspects of the disease process. It also binds copper, lowers cholesterol, modifies microglial activity, inhibits acetylcholinesterase, enhances the insulin-signaling pathway, and is an antioxidant.
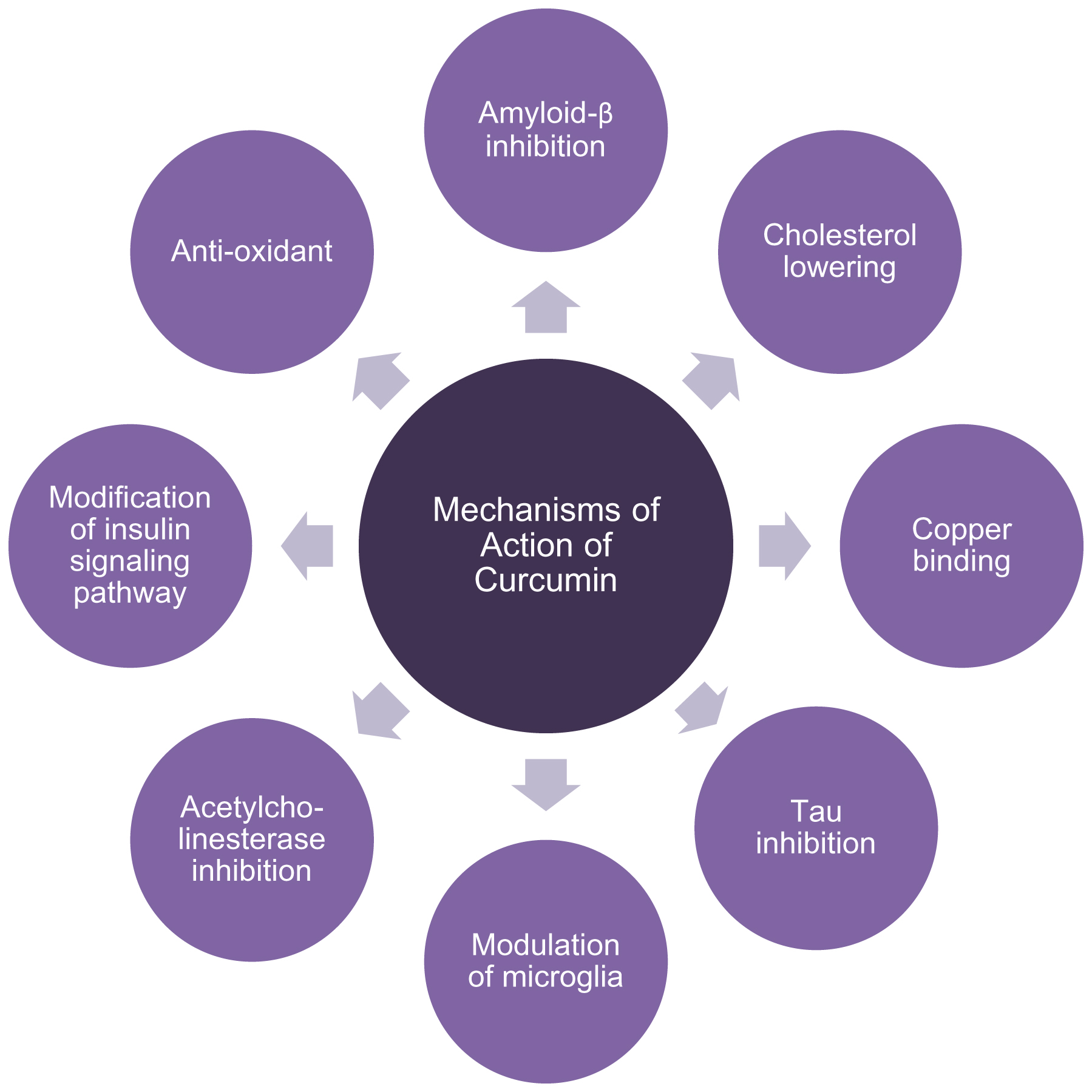
This flowchart illustrates the diverse mechanisms of action by which curcumin offers neuroprotection in AD. The compound inhibits the production and neurotoxicity of the two histological markers of AD, amyloid-β and hyperphosphorylated tau. Additionally, curcumin modulates other aspects of the disease process. It binds copper, lowers cholesterol, modifies microglial activity, inhibits acetylcholinesterase, enhances the insulin-signaling pathway, and is an antioxidant.
Aβ inhibition
Since the deposition of Aβ plaques is the characteristic feature of AD, curcumin has been studied for its ability to prevent the formation and accumulation of Aβ. Intragastric curcumin administration to a mice model of AD reduced Aβ formation by downregulating BACE1 expression, the enzyme that cleaves AβPP to Aβ. The curcumin-administered rats were alleviated from synaptic degradation and had improved spatial learning and memory outcomes [25]. Similarly, Di Martino et al. [26] identified curcumin as inhibitor of BACE1 in vitro. Another enzymatic target for the production of Aβ is presenilin-1 (PS-1), a protein in the γ-secretase complex and a substrate for glycogen synthase kinase-3β (GSK-3β). γ-secretase and GSK-3β are both implicated in the generation of Aβ. When human neuroblastoma SHSY5Y cells were treated with curcumin, there was a marked reduction in the production of Aβ. There was also a decrease in PS-1 and GSK-3β protein levels in a dose- and time-dependent manner, suggesting that curcumin decreased Aβ production through inhibition of GSK-3β-dependent PS1 activation [27]. In a rat model of AD, oral administration of curcumin was shown to reduce hippocampal Aβ accumulation, along with improvement of cognitive impairment in a passive avoidance task and the Morris water maze, a test of spatial learning and memory [28]. Promisingly, curcumin has shown to inhibit the formation and accumulation of Aβ in vitro and in vivo, suggesting the possibility of comparable results in a clinical context.
In addition to inhibiting Aβ production, curcumin has been demonstrated to inhibit aggregation and promote disaggregation of fibrillar Aβ in vivo and in vitro [29, 30]. This mechanism may be due to the structure of curcumin: an in vitro study postulated that the the hydrophobicity of curcumin or the interactions between the keto or enol rings of curcumin and aromatic rings of Aβ dimers destabilized the attractions requisite for the formation of beta-sheets in Aβ plaques [31]. In addition, curcumin’s polar hydroxyl groups on the two aromatic rings of the molecule interact with polar pockets of the Aβ peptide, rendering it suitable to destabilize beta-sheets. The inflexible linker between the two aromatic rings is conducive to this binding property [32].
The neuroprotective effects of curcumin are not limited to its prevention of Aβ fibril formation, but may also be implicated in its prevention of Aβ-mediated neurotoxicity. A study of human neuroblastoma SHSY5Y cells showed that curcumin protected against Aβ membrane-mediated neurotoxicity by reducing the rate of Aβ insertion into the plasma membrane [11]. Curcumin attenuated Aβ-membrane interactions and reduced Aβ-induced membrane disruption in artificial lipid bilayers, thereby potentially circumventing high calcium influx and cell death [11]. Curcumin may also prevent intracellular calcium elevation by mediating the Aβ-induced phosphorylation of the NMDA receptor [33]. Moreover, curcumin may shift the Aβ aggregation pathway to the formation of nontoxic conformers. Thapa et al. [34] demonstrated that curcumin promoted the formation of nontoxic, “off-pathway” soluble oligomers and prefibrillar aggregates. The same study demonstrated that curcumin also reduced the toxicity induced by a variety of Aβ conformers, including monomeric, oligomeric, prefibrillar, and fibrillar Aβ. However, these studies were conducted in vitro, and do not necessary have in vivotranslation.
Tau inhibition
Hyperphosphorylated tau and its aggregation into neurofibrillary tangles are crucial to the pathogenesis of AD, and numerous studies have shown that curcumin to prevent tau hyperphosphorylation and neurotoxicity [35–38]. GSK-3β is an enzyme that adds phosphate groups onto serine and threonine amino acid residues and regulates the phosphorylation of tau [39]. Accordingly, inhibition of GSK-3β may protect cells from tau-induced neurotoxicity and curcumin has been identified as such an inhibitor [26]. Huang et al. [37] demonstrated that curcumin inhibited hyperphosphorylation of tau through another mechanism. In human neuroblastoma SHSY5Y cells, curcumin inhibited tau hyperphosphorylation through the phosphatase and tensin homologue (PTEN)/protein kinase B (Akt)/GSK-3β pathway, a cellular signaling pathway induced byAβ [37].
Curcumin may also have a role in tau tangle clearance and alleviating tau-induced neurotoxicity. BCL2 associated athanogene 2 (BAG2) is a molecular chaperone that delivers tau to the proteasome for degradation [35]. When rat primary cortical neurons were treated with curcumin, BAG2 was significantly upregulated and hyperphosporylated tau levels decreasesd. Importantly, curcumin doubled BAG2 levels at low concentrations that were clinically relevant [36]. However, this study did not use neurons in a pathological state, so curcumin’s clinical applicability in the context of BAG2 and AD may be limited. In a nematode model of tauopathy curcumin was effective in alleviating tau-induced neuronal dysfunction [38]. In the curcumin-treated nematodes, the amount of acetylated α-tubulin, an indicator of microtubule stabilization, was significantly greater than the non-curcumin treatment group. This result suggests that curcumin may have mitigated the neurotoxicity of tau by improving microtubule stabilization [38]. This result indicates that curcumin has the potential to modulate tau-induced toxicity even in the absence of Aβ, implying that curcumin can act downstream of the Aβ neurotoxic cascade.
Copper binding
In addition to tau and Aβ, metal ions, particularly copper (Cu2 +), may contribute to the pathology of AD [40]. A shift in metal homeostasis is linked to AD [41], and in fact, serum copper levels are elevated in AD compared to healthy controls [42]. Copper interacts with Aβ to produce neurological decline, though the exact mechanisms are unclear. Copper may promote Aβ plaque formation through a variety of mechanisms [43–46]. The metal has been shown to bind Aβ peptide to create an inter-strand histidine brace, enabling the formation of the beta-sheet structure seen in plaques [46]. In addition to altering protein structure, copper can be coupled to the Fenton reaction to produce toxic reactive oxygen species (ROS) such as hydroxyl ion radicals and superoxide anions [47]. Oxidative stress has been demonstrated to regulate the expression of AβPP and lead to overproduction of Aβ [45]. Studies have also suggested a role of copper in AβPP processing. BACE1 has been shown to interact with copper and exposure to metal ions also increased both AβPP and BACE1 mRNA [43, 44]. In addition, it is noteworthy to mention that cellular prion protein, which has been recently identified as a receptor for misfolded Aβ oligomers, binds to copper as well [48]. Because copper has been associated with AD, metal chelation may be a therapeutic mechanism for AD.
Indeed, curcumin has metal chelation properties. Picciano and Vaden [49] demonstrated that curcumin chelated copper in the presence of Aβ peptide. Furthermore, curcumin inhibited spontaneous fibril formation from Aβ peptide in the presence of copper and zinc [50]. However, curcumin’s chelation may work best at low concentrations. In primary rat cortical neurons, curcumin at a low dosage prevented copper-induced oxidative stress whereas a high dosage of curcumin failed to decrease copper-induced oxidative stress. Rather, a high dosage caused chromosomal aberration and cell damage [51].
Cholesterol lowering ability
An increasing amount of epidemiological, animal, and cellular studies support the association between hypercholesterolemia and AD [52–55]. Plasma cholesterol levels are approximately 10% higher in AD patients in comparison with healthy controls [56], and high cholesterol levels alter AβPP metabolism to increase Aβ production [57]. Furthermore, individuals with the ɛ4 allele of ApoE are at an increased risk of developing AD [58]. The ApoE protein transports cholesterol in the brain and aids in the aggregation of Aβ. Accordingly, lipid-lowering therapy has the potential to reduce the risk of AD [59]. In a population-based case-control study, it was discovered that early statin use, a drug used to lower cholesterol, was significantly associated with a reduction in AD progression in mild to moderate AD patients compared to those without statin use [60]. Although it is somewhat controversial, like statin, curcumin may also be employed for its ability to lower cholesterol levels.
Curcumin has been shown to inhibit sterol regulatory element binding proteins (SREBPs). These are transcription factors that upregulate the synthesis of enzymes involved in glycolysis, energy production, lipogenesis, and cholesterol production. The activation of SREBP-1 not only contributes to cholesterol synthesis, but has also been shown to be neurotoxic [61]. In vitro, curcumin inhibited SREBP expression in hepatocytes, thereby decreasing the biosynthesis of cholesterol and fatty acid [62, 63]. Similarly, in vascular smooth muscle cells, curcumin inhibited cholesterol accumulation by inhibiting the nuclear translocation of SREBP-1 [64]. These inhibitory results have also been reflected in vivo. In a mice model of obesity, curcumin ameliorated serum lipid levels and insulin sensitivity, another risk factor for AD which will be discussed below [63].
In addition to mediating cholesterol production, curcumin has been shown to have an effect on cholesterol uptake. Peschel et al. [65] demonstrated that curcumin lowered cholesterol levels by altering hepatic gene expression to increase low-density lipoprotein (LDL) receptor expression, a receptor that mediates the endocytosis of cholesterol-rich LDL. However, Tai et al. [66] found that curcumin did not alter LDL receptor transcription, but enhanced LDL receptor levels on the cell surface, in addition to LDL receptor activity. Curcumin downregulated proprotein convertase subtilisin/kexin type 9 (PCSK9) gene expression, a protein involved in LDL receptor breakdown. Thus, when curcumin lowered PCSK9 expression, more LDL receptors were available to uptake cholesterol-rich LDL.
As well as hepatic proteins, curcumin also exerts an effect on intestinal proteins. Curcumin inhibited uptake of dietary cholesterol through suppression of Niemann-Pick C1-like-1 (NPC1L1) expression, an intestinal cholesterol transporter [67]. When rats were fed a high fat diet supplemented with curcumin, the curcumin group had significantly decreased serum triglyceride, total cholesterol, and LDL-cholesterol as compared to the control group, respectively. The curcumin group also had higher fecal triglyceride and cholesterol levels. Researchers attributed the increased fecal lipid levels to curcumin’s ability to upregulate cholesterol 7a-hydroxylase enzyme, the rate-limiting step in the synthesis of bile acid from cholesterol [68].
Anti-inflammatory and modulation of microglia
Microglia, the resident macrophage of the central nervous system (CNS), have a critical role in the innate immune response of the CNS and are implicated in AD [69]. They can either be classified as M1 or M2, based on their activated phenotypes. M1 microglia secrete neurotoxic inflammatory cytokines (such as tumor necrosis factor-alpha (TNF-α), interleukin-1β (IL-1β), IL-6), prostaglandins (COX-2), ROS, and nitric oxide. M2 microglia, in contrast, release neuroprotective anti-inflammatory mediators, secrete proteins that maintain the extracellular matrix, and phagocytize toxic protein aggregates and cellular debris [70]. Recent studies have demonstrated that Aβ diverts microglia from their neuroprotective M1 phenotype to their neurotoxic M2 phenotype [71]. A positive feedback loop of inflammation has been formed between Aβ accumulation, activated microglia, and microglial inflammatory mediators, which promote further Aβ accumulation and neuroinflammation. Accordingly, inhibition of this loop may be a therapeutic target to halt the disease progression.
Curcumin may be an anti-inflammatory agent with efficacy in AD through its modulation of M1 microglial activation, signaling pathways, and product secretion. Shi et al. [72] demonstrated that curcumin blocked extracellular signal-regulated kinase 2 (ERK1/2) and p38 kinase signaling in Aβ-activated microglia in vitro, leading to the reduced production of TNF- α, IL-1β, and IL-6 mRNA and protein levels. Curcumin also improved microglial viability against Aβ and suppressed Aβ-induced microglial activation. Another study showed that supplementation of curcumin to lipopolysaccharide-activated microglia significantly attenuated the release of nitric oxide and inflammatory cytokines, as well as reducing the expression of inducible nitric oxide synthase [73]. The alteration of microglial activity was likely through curcumin’s ability to suppress phosphoinositide 2 kinase (PI3K)/Akt phosphorylation and nuclear factor κB (NF-κB) activation, signaling molecules important in the microglial activation pathway and neuroinflammation [73]. Liu et al. [74] also demonstrated curcumin’s ability to reduce activation of microglia, inhibit the NF-κB signaling pathway, and reduce cytokine production. Curcumin directly bound to the peroxisome proliferator-activated receptor gamma (PPARγ), increasing protein levels of PPARγ. PPARγ is anti-inflammatory as it downregulates the NF-κB and ERK pathways responsible for neuroinflammation. Thus, the data suggested that PPARγ was a potential target of curcumin’s anti-inflammatory effects. Notably, the effects occurred both in vivo and in vitro.
The neuroprotective benefits of M2 microglia may be enhanced by curcumin. When microglia of AD patients were treated with curcuminoids in vitro, Aβ phagocytosis by 50% of the microglia was increased compared to the control group [75]. However, this study was limited by its sample size: macrophages were obtained from 6 patients with AD and 3 controls. Thus, in addition to preventing M1 microglial inflammatory, curcumin may inhibit plaque accumulation through its enhancement of neuroprotective microglial activity, though research in this field is limited.
Acetylcholinesterase inhibition
Curcumin has also been revealed to modulate AChE in a mechanism similar to the first-line prescribed drugs for AD, AChE inhibitors. Akinyemi et al. [76] investigated the effects of curcumin the cadmium-induced memory impairment of rats. Cadmium poisoned rats had higher levels of AChE activity, and curcumin treatment reversed this activity to control levels. However, da Costa et al. [77] reported that curcumin did not prevent the effect of chronic cadmium administration on AChE activity in the cerebral cortex synaptosomes of rats, but was able to inhibit AChE in the cortex and striatum. Similar to cadmium, monosodium glutamate (MSG) also causes neurotoxicity. Rats orally administered MSG had a significant elevation in AChE level compared to the control group. However, when rats were administered MSG and curcumin, they showed a significant decrease in AChE activity when compared to the MSG only group [78]. Furthermore, curcumin prevented the cognitive deficits associated with chronic alcohol consumption in rats, partially by attenuating alcohol-induced activation of AChE activity [79]. While curcumin has been shown to inhibit AChE in various studies, no research has been conducted in the context of AD cell or animal models.
Modification of insulin signaling pathway
Emerging data has established that brain insulin resistance is closely associated with cognitive decline and promotes biological abnormalities in AD [80]. Peripherally, insulin is a hormone that mediates glucose uptake into tissues. In the brain, the hormone regulates functions that are also disrupted in AD: cerebral blood flow, inflammatory responses, oxidative stress, tau phosphorylation, apoptosis, lipid metabolism, synaptic plasticity, and memory formation [81]. When insulin binds to the insulin receptor, it activates intracellular insulin receptor substrate 1 (IRS-1), which consequently activates PI3K, Akt, and further downstream targets [82]. Postmortem studies on AD patients has shown that brain tissues exhibited decreased insulin receptor binding [83], lower levels of activated insulin receptor [84], and increased inhibitory phosphorylation of IRS-1 [82]. More recently, a study confirmed the insulin resistance present in AD brains. Via ex vivo stimulation, a physiological dose of insulin was administered to brain tissue from AD patients and healthy controls. In the hippocampal formation, prefrontal cortex, and cerebellar cortex, insulin induced significantly less activation of its signaling pathway in AD tissue compared to healthy tissue [82]. Because brain insulin resistance is a common and early characteristic of patients with AD, developing treatments that target the hormone’s signal transduction pathway may be efficacious for ameliorating the progression of AD.
Curcumin has been shown to regulate the insulin-signaling pathway and cognitive deficits associated with insulin resistance in AD. Feng et al. [85] used a transgenic mouse model of AD to investigate the mechanisms and effects of curcumin in AD. When the mice were administered curcumin for six months, the hippocampal CA1 tissue expressed lower levels of insulin receptor and IRS-1, while the expression of PI3K, phosphorylated PI3K, Akt, and phosphorylated Akt increased compared to control. The results demonstrated that curcumin upregulated the PI3K/Akt signaling pathway to reduce insulin resistance in AD. In addition to modulating the insulin-signaling pathway, curcumin has been shown to improve memory deficits in a rat model of diabetes [86]. Streptozotocin is a compound administered to rodents that exerts preferential toxicity toward pancreatic beta cells, inducing diabetes. When rats were injected with streptozotocin, learning and memory acquisition was impaired in behavioral tasks. Streptozotocin treated rats that were also administered with curcumin had better performance in the tests than streptozotocin and vehicle. Furthermore, less neuronal loss was observed after curcumin treatment compared to the control group [86]. The behavioral and histological results, along with the elucidation of curcumin’s modulatory effect on the insulin signaling pathway, indicate that curcumin treatment may be effective at reducing the cognitive impairment and pathophysiological markers caused by insulin resistance in AD.
Antioxidant
Oxidative stress has an important role in the development and progression of AD [87]. It occurs when there is an imbalance in the redox state, involving the accumulation of ROS or a decrease in antioxidant defense. The brain is especially sensitive to oxidative stress as it has a high oxygen consumption rate, less enzymatic defense against oxidative stress, and is composed of lipids that are easily oxidized [88]. In addition to lipids, ROS can interact with proteins, nucleic acids, and other molecules to detrimentally modify their structures and functions [47]. Ultimately, oxidative stress can cause cellular apoptosis [89]. In the case of AD, an excess of reactive oxygen species may be produced by mitochondrial dysfunction and/or anomalous buildup of transition metals, and furthermore, Aβ and tau proteins may potentiate this redox imbalance. Oxidative stress may increase the production and aggregation of Aβ and expedite the phosphorylation and polymerization of tau, consequently creating a cycle that fosters the progression of the disease [87]. Given the detrimental role of ROS in AD, restoring redox balance may be a potential method for treating AD.
Curcumin has been investigated for its antioxidant properties: directly through scavenging free radicals and indirectly through upregulating the cytoprotective response [90, 91]. In numerous in vitro antioxidant assays, curcumin demonstrated ability to scavenge free radicals, reduce ferric ions (Fe(3+)), and chelate ferrous ions (Fe(2+)) [90]. Curcumin also inhibited lipid peroxidation, that is, the oxidative degradation of lipids, at a level comparable the standard antioxidants butylated hydroxyanisole, butylated hydroxytoluene, α-tocopherol, and trolox [90]. Indirectly, curcumin has been shown to upregulate the expression of genes encoding for antioxidant proteins, such as heme oxygenase-1, superoxide dismutase, catalase [91]. Additionally, curcumin upregulated the transcription of proteins that replenish the antioxidant glutathione, including glutathione reductase, glutathione peroxidase, and glutathione S-transferase [91]. These observations, however, were not in the context of neuronal cells.
Curcumin has been demonstrated to circumvent the detrimental effects of ROS in the context of AD. When SH-SY5Y neuronal cells were incubated with hydrogen peroxide, a potent source of oxidative stress, they experienced lipid peroxidation and elevated cytosolic calcium concentrations, and expressed the pro-apoptotic protease enzymes caspase-3 and –9. However, when the cells were incubated with hydrogen peroxide and curcumin, the levels of lipid peroxidation, cytosolic calcium concentration, and caspase-3 and –9 concentrations were lower [92]. Additionally, the cells exposed to curcumin had higher levels of reduced glutathione and glutathione peroxidase, consistent with previous studies on non-neuronal tissue [92]. Similar results were observed in neuronal PC12 cells. Curcumin inhibited Aβ-induced mitochondria-mediated apoptosis through regulation of the B-cell lymphoma 2 (Bcl-2) protein family, a group of proteins involved in apoptosis [93]. The compound significantly blocked poly ADP ribose polymerase (PARP) cleavage, caspases activation, and ROS-mediated DNA damage [93]. In addition to Aβ induced mitochondrial toxicities, curcumin also has protective affects against synaptic toxicities. Reddy et al. [94] demonstrated that SHSY5Y cells pre- and post-treated with curcumin and incubated with Aβ had reduced mitochondrial dysfunction, maintained mitochondrial biogenesis and dynamics, and had normal synaptic activity. Interestingly, the effects of curcumin were stronger in pretreated than post-treated cells, suggesting that the compound may be better for prevention than treatment in AD like neurons [94].
BIOAVAILABILITY
Despite the wealth of evidence favoring curcumin’s ability to combat AD, a major challenge exists in curcumin’s low bioavailability, thereby significantly reducing its potential to reach the brain to supply therapeutic benefits. Compared to oral dosages, blood plasma levels of curcumin are very low or negligible. Poor absorption, rapid metabolism, and rapid systemic elimination are contributing factors to the low plasma and tissue levels of curcumin. Diet may also modulate the absorption of curcumin [95]. Dietary fiber, divalent minerals, and viscous and high protein meals are likely to decrease the bioavailability of polyphenols like curcumin, whereas digestible carbohydrates, dietary lipids, and antioxidants improve polyphenol bioavailability [96]. Additionally, its insolubility in aqueous solutions poses a challenge for administration and its short-half life renders the compound ineffective over long periods of time [97].
The two recently published systematic reviews of curcumin clinical trials revealed the discrepancy between between in vitro and animal models findings and human trials outcome [97, 98]. Poor bioavailability may explain the lack of success in clinical trials in using curcumin as a treatment for AD. Baum et al. conducted a six-month randomized, placebo-controlled, and double-blind clinical trial of oral curcumin in patients with AD [99]. No differences were detected between treatment groups in measurements of cognitive impairment or Aβ levels. Ringman et al. investigated the use of oral curcumin for AD in a 24-week randomized, placebo-controlled, and double-blind study [100]. Similar to Baum et al.’s results, there were no significant differences in cognitive function, Aβ levels, or tau levels between the intervention group and the placebo group. Aligning with previous research that demonstrated curcumin’s low bioavailability, Ringman et al. also reported bioavailability as a limitation of the study [100]. More recently, Cox et al. using a more bioavailable solid lipid curcumin formulation in a double-study demonstrated the therapeutic effects of curcumin on cognition and mood in a healthy older population [101].
To be effective as a drug therapy, new delivery strategies for curcumin are being researched. The first of these approaches include the development of curcumin analogs. Analogs that resemble the active site of the compound and analogs that mimic the anti-amyloid and anti-AChE effect have been designed to improve the bioavailability, water solubility, stability, and delivery of curcumin [102]. Second, curcumin can be conjugated to metal oxide nanoparticles or encapsulated in lipid nanoparticles, dendrimers, nanogels and polymeric nanoparticles to improve its solubility [103]. Third, the use of micelles and phospholipid complexes can improve the absorption of curcumin from the gastrointestinal tract, resulting in higher plasma levels [104]. Fourth, the use of adjuvants like piperine interferes with hepatic and intestinal glucuronidation, thereby inhibiting the rapid metabolism of curcumin in the liver and intestinal wall [105].
LIMITATIONS AND FUTURE RESEARCH
Despite the success of in vivo and in vitro studies, the positive results have not fully translated to clinical studies [99, 100]. The key obstacle to utilizing curcumin as a therapeutic compound in humans is its low oral bioavailability. Accordingly, future research should focus on improving the bioavailability of curcumin. Presently, curcumin analogs, conjugation to nanoparticles, the use of micelles, cyclodextrins, and phospholipid complexes, and consumption with an adjuvant can increase serum concentrations, improve solubility, reduce metabolism, and increase bioavailability [102–106]. Clinical studies that aimed to decipher the effect of curcumin in AD did not use any of the aforementioned methods to improve bioavailability. Plausibly, the low bioavailability of unformulated curcumin hindered its ability to exert its effects, contributing to the studies’ lack of success. Thus, an area for further research is the analysis of different oral formulations in the context of a clinical study to treat AD. In addition, both toxicological and pharmacokinetic profiles should be required for any new curcumin formulations including the oral ones [107].
Another method to improve curcumin bioavailability could be to couple its consumption with certain foods. Currently, no clinical research has been conducted investigating the efficacy of curcumin in AD when its consumption is accompanied with high-lipid and/or digestible carbohydrate-rich foods, so this is also an area for future investigation. Furthermore, improving bioavailability through dietary factors may be more appealing than the previously listed mechanisms. It has the potential to be cost-effective, affordable, and accessible across geographic or socioeconomic barriers.
Another area needing further investigation is the analysis of biomarkers and behavior as a means of evaluating AD progress when treatment with curcumin is applied in vivo. The cholesterol lowering ability of curcumin has been extensively characterized, but not in the context of AD. Further studies could investigate the levels of SREBPs, LDL receptors, or transporters alongside behavioral outcomes in animal models of AD. Equivalently, while research has shown curcumin to be an AChE inhibitor in various animal models of neurotoxicity, no studies have been conducted in an AD model. Future research is required to confirm curcumin’s AChE inhibition in the context of AD. Finally, certain mechanisms of action of curcumin need additional confirmation or exploration in vivo. While evidence exists to suggest curcumin’s ability to enhance microglial uptake of Aβ, the current research is constrained by small sample size and lack of in vivo research.
Finally, Nelson et al. [108] argues that curcumin exerts false activity in vitro and in vivo due to its classification as a pan-assay interference compound (PAIN). PAINs are compounds that demonstrate apparent activity in various assays not through specific compound/target interactions, but rather through assay readout interference [109]. Curcumin exhibits all known PAIN modes of interference including fluorescence interference, redox activity, compound degradation, and membrane disruption, implicating that its activity in an assay that does not exclude nor account for these behaviors may not be legitimate [108]. Thus, future research should be prudent to carefully evaluate potential interference mechanisms to ensure that the activity observed is due to a specific compound/target interaction, rather than interference. A summary of the areas for further research is shown in Fig. 2.
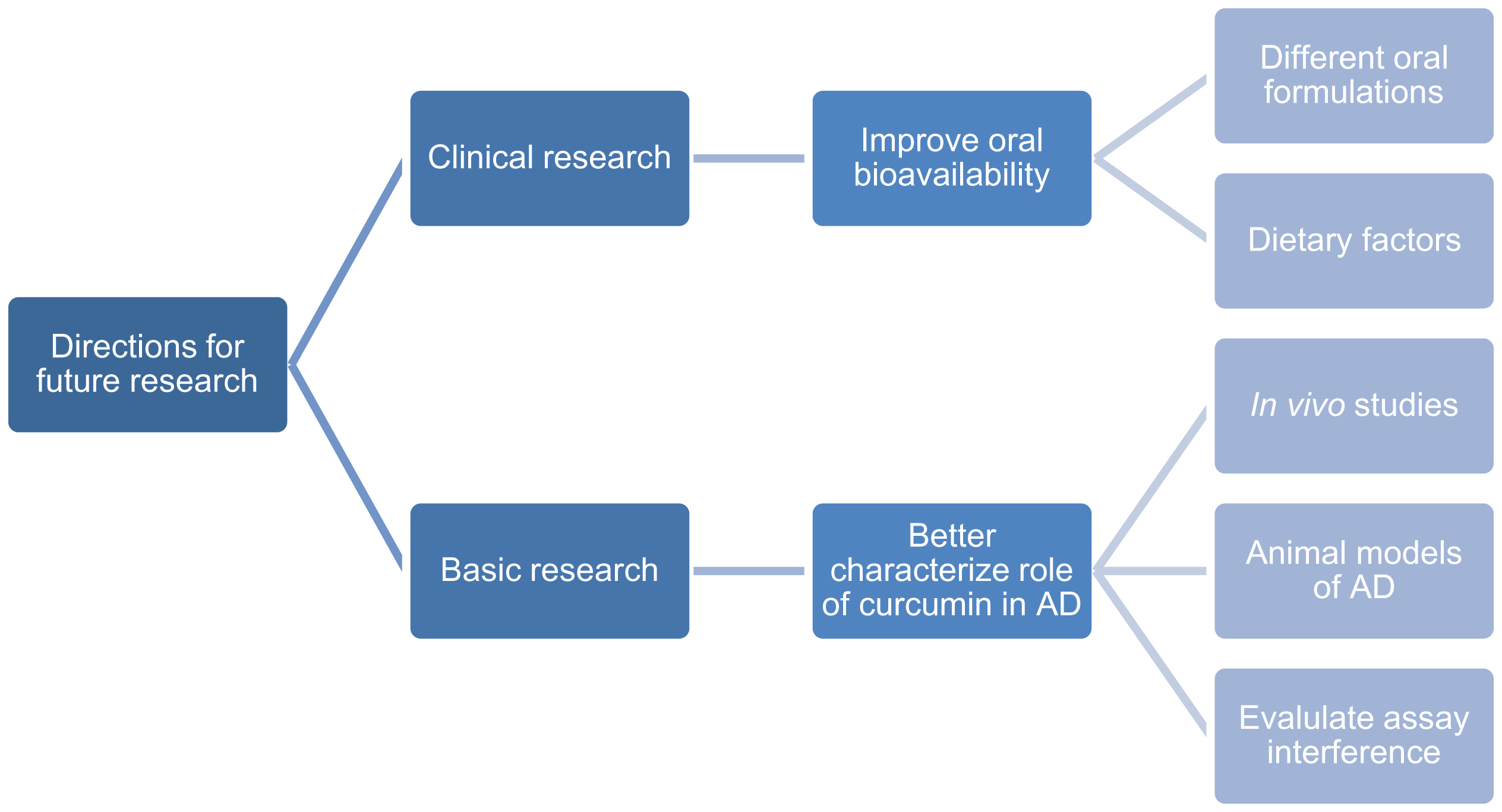
This flowchart categorizes the directions for future research of curcumin’s effects in AD into two categories: clinical research and basic research. The key barrier to clinical utility is curcumin’s lack of oral bioavailability. While oral formulations that improve bioavailability are available, there exists a need to apply these formulations in a clinical trial. Additionally, future clinical trials should investigate the efficacy of curcumin in AD when it is consumed with high-lipid and/or digestible carbohydrate-rich foods, two dietary factors that enhance curcumin absorption. In basic research, certain mechanisms of action of curcumin need further elucidation. As such, more in vivo studies need to be conducted, especially in animal models of AD. Furthermore, curcumin has shown to interfere with assay readouts, so researchers should be prudent to exclude or account for such interference to avoid reports of false activity.
CONCLUSION
Curcumin has been speculated to be able to prevent and treat AD, but research elucidating its specific mechanisms of action has only been performed in the past several decades. Foremost, curcumin inhibits the two key histological features of AD, Aβ plaques and tau tangles. Its other mechanisms of action relate to other risk factors, physiological activities, or biomarkers associated with AD. Curcumin binds copper, lowers cholesterol, prevents inflammatory microglial activity while enhancing Aβ microglial phagocytic activity, inhibits AChE, mediates the insulin signaling pathway, and restores redox balance [110].
The diverse array of the effects of curcumin and its disease modifying properties may have a notable impact on the future of drug treatment for AD. AChE inhibitors and NMDA antagonists are the current mainstay treatments, but these drugs only target a single disease pathology and do not delay the onset of the disease or alter its progression. Moreover, their efficacy declines as neurons degenerate. In contrast, the effects of curcumin are multi-targeted and it has the ability to modify the disease process. More research needs to be done to improve the bioavailability of curcumin so that the success of pre-clinical studies can be translated to clinical outcomes. If curcumin is confirmed to show the same efficacy in humans as in in vitro and in vivo studies, the disease-modifying treatment of AD is a worthwhile possibility.