
Abstract
In the study, we examined whether the silent information regulator 1 (SIRT1) can attenuate oxidative stress in the brains of mice carrying the APP/PS1 double mutation and/or in primary neonatal rat neurons exposed to oligomers of amyloid-β peptide (AβOs). Starting at 4 or 8 months of age, the transgenic mice were treated with resveratrol (RSV, a stimulator of SIRT1) or suramin (an inhibitor) (each 20 mg/kg BW/day) for two months. The primary neurons were exposed to AβOs (0.5 μM) for 48 h and thereafter RSV (20 μM) or suramin (300 mg/ml) for 24 h. Cell viability was assessed by the CCK-8 assay; SIRT1 protein and mRNA determined by western blotting and real-time PCR, respectively; senile plaques examined immunohistochemically; ROS monitored by flow cytometry; and the contents of OH-, H2O2, O2·-, and MDA, and the activities of SOD and GSH-Px measured by standard biochemical procedures. In comparison to wild-type mice or untreated primary neurons, the expression of SIRT1 was significantly lower in the brains of APP/PS1 mice or neurons exposed to AβOs. In these same systems, increased numbers of senile plaques and a high level of oxidative stress were apparent. Interestingly, these two latter changes were attenuated by treatment with RSV, but enhanced by suramin. These findings indicate that SIRT1 may be neuroprotective.
INTRODUCTION
Alzheimer’s disease (AD), one of the most devastating neurodegenerative diseases with significant cognitive deficit [1], is characterized by gradual, but irreversible cognitive decline leading to functional signs of aphasia (loss of speech and poor word recognition), apraxia (inability to make voluntary movements), and agnosia (lack of recognition of objects) [2]. It has been indicated that age remains the greatest risk factor for AD and is thus a fundamental driver for development of the disease [3]. Systems either driving brain aging are contributors to risk of AD and include glucose hypometabolism and mitochondria dysfunction, innate immune and inflammatory reactions, and amyloid-β (Aβ) processing.
Extracellular plaques containing Aβ, intraneuronal neurofibrillary tangles, brain atrophy, and loss of neurons are the pathological hallmarks of this disease [4, 5], with clear evidence that the accumulation of Aβ, a 4-kDa polypeptide formed by proteolytic cleavage of the precursor protein, AβPP, is a primary pathogenic event [6]. The data suggest that Aβ oligomers (AβOs) play substantive roles in neurotoxicity, reflected in synaptic and memory impairments [7–9]. Moreover, it has been demonstrated consistently that Aβ alters energy homeostasis, mainly by disturbing mitochondrial function [10]. Aβ may contribute to a neurotoxic environment in AD [11–13].
The seven known members (SIRT1-SIRT7) of the silent information regulator 2 (Sir2) family of proteins, or sirtuins, are unique histone deacetylases (HDACs) whose activity is dependent on the level of NAD+ and thus the cellular metabolic status [14]. The potential role of sirtuins in different disease states has been examined extensively [15] and these proteins have attracted considerable attention as epigenetic regulators of aging [16].
Among the mammalian sirtuins, SIRT1 has been most extensively reviewed [15] and treatment of neuronal cultures with NAD+ and resveratrol (RSV), activators of SIRT1, reduces Aβ binding to amyloid fibrils and enhances cellular α-secretase activity, thereby activating the protective non-amyloidogenic pathway [17]. In cell-based models for AD and amyotrophic lateral sclerosis, SIRT1 and RSV promote neuronal survival [15].
Oxidative stress involves an imbalance between production of reactive oxygen species (ROS) and defense systems [18]. Superoxide free radicals, hydrogen peroxide, singlet oxygen, nitric oxide (NO), and peroxynitrite can be generated in elevated quantities during the reduction of oxygen and lead to cellular injury [19]. A great deal of evidence suggests that oxidative stress may be involved in the pathogenesis of AD [20, 21]. In vivo experiments suggest that Aβ increases oxidative damage.
Interestingly, in transgenic animals with Aβ amyloidosis, markers of oxidative stress were distributed in a manner similar to that in brain tissues of patients with AD, suggesting a relationship to Aβ deposition [22]. Expression of CuZn-superoxide dismutase (SOD) and heme oxygenase-1, markers of oxidative stress, is increased in the brains of a transgenic mouse model of AD, providing strong support for the hypothesis that Aβ is neurotoxic in vivo and that such toxicity is mediated by free radicals [23]. Although oxidative stress thus appears to play an important role in the pathogenesis of AD, the mechanism(s) by which the redox balance is disturbed and the source of free radicals have not yet been elucidated in detail.
There are indications that the activity of SIRT1 declines with age, potentially due to ROS-mediated depletion of NAD+, and that a compensatory mechanism elevates the level of SIRT1 protein [16, 24]. In the present study, we employed mice with mutations in both the genes encoding APP and presenilin 1 (APP/PS1) or primary hippocampal neurons exposed to AβOs and an activator or inhibitor of SIRT1 to explore potential effects on AβOs and oxidative stress.
MATERIALS AND METHODS
Materials
RSV, Suramin, and Aβ1-42 (Sigma-Aldrich, USA); Cell Counting Kit-8 (Dojindo, Japan); Neurobasal-A Medium, Hibernate-E Medium, B-27 Serum-Free supplement, GlutaMAX Supplement (Life Technologies, USA); mouse anti-NeuN antibody (Merck Millipore, Germany); rabbit anti-glial fibrillary acidic protein (GFAP) antibody (Dako, Denmark); anti-β-Amyloid, 1–16 antibody (BioLegend, USA); mouse monoclonal anti-SIRT1 antibody (Abcam, Britain); anti-rabbit and -mouse IgG conjugated with horseradish peroxidase, rabbit monoclonal anti-β-actin antibody (Sigma-Aldrich, USA); anti-mouse IgG labeled with CY-3, and anti-rabbit IgG labeled with 488 (Thermo scientific, USA); Hyper Performance Chemiluminescence film and Electrochemiluminescence (ECL) Plus reagent (Amersham, Sweden); Fluorometric Intracellular ROS Kit (Sigma-Aldrich, USA); kits for measuring OH-, H2O2 and O2·-, malondialdehyde (MDA), SOD, superoxide dismutase1 (SOD1), superoxide dismutase2 (SOD2) and glutathione peroxidase (GSH-Px) (Nanjing Jiancheng Bioengineering Institute, China); Universal TaqMan 2×PCR mastermix (Applied Biosystems, USA); and all other general chemicals (Sigma-Aldrich, USA) were purchased from the sources indicated.
Cell cultures
Primary hippocampal neurons were prepared from the brains of neonatal Sprague-Dawley rats by a published procedure with minor modifications. In brief, the hippocampal regions were dissected out within 2–3 min after sacrifice and thereafter maintained in Hibernate A Medium on ice. After removing the meninges, the hippocampus was washed 3 times with Hank’s buffered saline solution and then digested with 0.25% trypsin for 10 min at 37°C. Subsequently, the incubation medium was discarded and DMEM containing 10% FBS added to terminate digestion. After washing twice more with Hank’s buffered saline solution, the digested tissue was resuspended in 2 ml Neurobasal/B27 complete medium (Neurobasal A Medium with 2% B27, 1% GlutaMAX Supplement, 100 U/ml penicillin and 100 mg/mL streptomycin) and disrupted by repeated suction through fire-polished glass pipettes. The upper suspension of single cells was transferred into a new tube and the cells counted by trypan blue exclusion and thereafter placed onto 96-, 12-, or 6-well poly-L-lysine-coated plates at a density of approximately 5.0×104/cm2. The neurons were maintained under a humidified atmosphere containing 5% CO2 at 37°C, with replacement of half of the medium once every 3 days. The purity of these primary neurons was evaluated by immunofluorescent double-staining with mouse anti-NeuN antibody (diluted 1:300) followed by anti-mouse IgG labeled with CY-3 (red) and with rabbit anti-GFAP antibody (diluted 1:300) followed by anti-rabbit IgG labeled with 488 (green). After 10 days of incubation, the medium was replaced with neurobasal medium lacking B27 and the neurons then prepared for various treatments.
Experimental animals
Five 4-month-old B6.Cg-Tg (APPswe, PSEN1dE9) mice with a 85Dbo/Mmjax background (all males with a body weight of 20–30 g) and 15 wild-type (WT) mice (all females with body weight of 20–30 g) of the same strain were purchased from Shanghai Nanfang Biological Technology Development Co., Ltd. All animals were acclimatized at a humidity of 30–55% and temperature of 22–25°C for one week, following which each female APP/PS1 mouse underwent natural mating with three WT males. At a postnatal age of 12–20 days, the tip of the tail of each pup was cut off and DNA extracted for genotyping by the polymerase chain reaction (PCR), with detection of the products obtained by 1.5% agarose gel electrophoresis. The PCR primers for target transcripts (Table 1) were designed on the basis of the complete cDNA sequences deposited in GenBank.
Sequences of the primers used to detect the APP and PS1 genes by PCR
Starting at the ages of 4 and 8 months, respectively, the APP/PS1 and WT mice received RSV, suramin or physiological saline (20 mg/kg), by gavage once daily, for two months [25, 26]. All experiments described here were pre-approved by the Ethical Committee of Guizhou Medical University, China (No. 1702110).
Treatment of cultured neurons with AβOs, RSV, and suramin
AβOs were prepared by a new procedure [27]. The primary neurons were seeded onto 96-, 12-, or 6-well PLL-coated plates and B27 drawn with from the culture medium 2 h prior to treatment.
To find the optimal conditions for cell survival in starvation media, cells were first exposed to different concentrations of AβOs (0–2 μM), RSV (0–150 μM), and suramin (0–600 mg/ml) for 48 or 24 h and 10 μl Cell Counting Kit then added to each well. Incubation was then continued for 1–4 more h and absorption at 450 nm determined. On the basis of the results of this test for viability, suitable concentrations of and incubation times with AβOs, RSV, and suramin were chosen.
Quantification of SIRT1 protein by western blotting
The primary neurons cultured on 6-well plates or brain tissue (0.05 or 0.1 g) were homogenized in PBS buffer containing a complete mixture of protease inhibitors in a glass homogenizer. This homogenate was then centrifuged at 12,000 g for 40 min at 4°C and the protein concentration of the resulting supernatant determined with the BCA protein assay kit (Thermo scientific, USA). Next, these proteins were subjected to 10% SDS-PAGE and then blotted onto polyvinylidene difluoride (PVDF) membranes with a transfer unit (Bio-Rad Inc.). For quantification of SIRT1, these PVDF membranes were incubated thereafter with anti-SIRT1 antibodies (1:1000 dilution) at 4°C overnight. After washing, the membranes were incubated with horseradish peroxidase (HRP)-conjugated anti-mouse IgG (1:5000) for 60 min. Finally, these membranes were incubated in ECL Plus reagent for 30 s-5 min and the signals thus obtained visualized by exposure to Hyper Performance Chemiluminescence film.
Determination of SIRT1 mRNA by quantitative real-time PCR
Total RNA was isolated from primary neurons on 6-well plates or brain tissue with Trizol reagents (Invitrogen, USA), using DNase I to remove residual genomic DNA. Thereafter, 3 μg total RNA was converted with the first-strand cDNA synthesis kit (Promega, USA) and oligo-d (T)18 primers following the protocol recommended by the manufacturer. The real-time PCR primers for target transcripts were designed on the basis of the complete cDNA sequences in GenBank (accession numbers: NM_006256146.2, NM_001159589.2 for SIRT1 and NM_031144, NM_007393.5 for β-actin) (Table 2).
Sequences of the primers used to detect mRNA encoding SIRT1 and β-actin by real-time PCR
Quantitative real-time PCR was carried out utilizing the ABI PRISM 7300 Sequence Detection System (Applied Biosystems, USA) and analyzed with GeneAmp7300 SDS software. In brief, the 10-mu l reaction mixture contained 1 μl first-strand cDNA, 5 μl 2×SYBR Green Master (Rox) Mix, 0.5 μl each of the forward and reverse primers (10 μM), 3 μl DNase and RNase-free H2O. The thermal cycling conditions were 2 min at 50°C and 10 min at 95°C, followed by 40 cycles at 95°C for 15 s and then 1 min at 60°C. The levels of the SIRT1 and β-actin transcripts were calculated as 2–ΔΔCT, where ΔCT represents the difference between the cycle threshold (CT) values for the target gene and β-actin.
Examination of senile plaques in the mouse brain by immunohistochemistry
Immunohistochemical staining for senile plaques in the mouse brain was performed as described previously [28]. In brief, the sections were treated with blocking buffer (DAKO) for 30 min at RT and thereafter incubated with anti-β amyloid 1–16 antibody (1:100 dilution in PBST, BioLegend, USA), overnight at 4°C. Following a thorough rinse in PBST, these sections were then incubated with the secondary antibodies, i.e., biotinylated goat anti-mouse IgG or biotinylated goat anti-rabbit IgG (diluted1:200 in PBST) for 60 min at RT. Subsequently, sections were incubated with the avidin-biotinylated enzyme complex and DAB and then dehydrated with increasing concentrations of ethanol, cleared with xylene, and mounted in Permount. Negative controls were incubated with non-immune serum instead of the primary antibodies, which resulted in no detectable staining.
Levels of ROS in primary neurons
Primary neurons were seeded onto 96- or 6-well PLL-coated plates and those on the 6-well plates collected into 1.5-ml Eppendorf tubes. 100 μl Master Reaction Mix was added into each well of the 96-cell plate or each tube and the neurons placed in an incubator under 5% CO2 at 37°C for 1 h. Finally, the fluorescence intensity atλex=490/λem=525 nm was determined.
The contents of OH-, H2O2, O2·-, and malondialdehyde (MDA) and activities of GSH-Px, SOD, Zn/Cu- SOD1 and Mn- SOD2 in cerebral tissue, primary neurons, and mitochondria
The primary neurons seeded onto 6-well plates or brain tissue (0.05 g) were homogenized in 9 ml normal saline, followed by centrifugation. The protein concentration in the resulting supernatant was determined using the BCA kit (Thermo scientific, USA) and the levels of OH-, H2O2, O2·-, and MDA and activities of SOD, SOD1, SOD2, and GSH-Px were measured employing appropriate kits (Nanjing Jiancheng Bioengineering Institute, China).
Statistical analyses
The values for the different groups are expressed as means±SD. The data were compared by analysis of variance (ANOVA), followed by Student-Newman-Kenl’ s test or the two-paired Student’s t test, utilizing the SPSS17.0 software (SPSS Inc., USA).
RESULTS
Purity of the primary cultured neurons and confirmation of the APP/PS1 murine genotype
Immunostaining of the primary cultured neurons prepared from the hippocampal region of the brains of newborn rats with antibodies directed toward NeuN (a marker for neurons), GFAP (a marker for astrocytes), and DAPI (a marker for nucleus) revealed that approximately 90% of these cells were neurons (Fig. 1A-E).
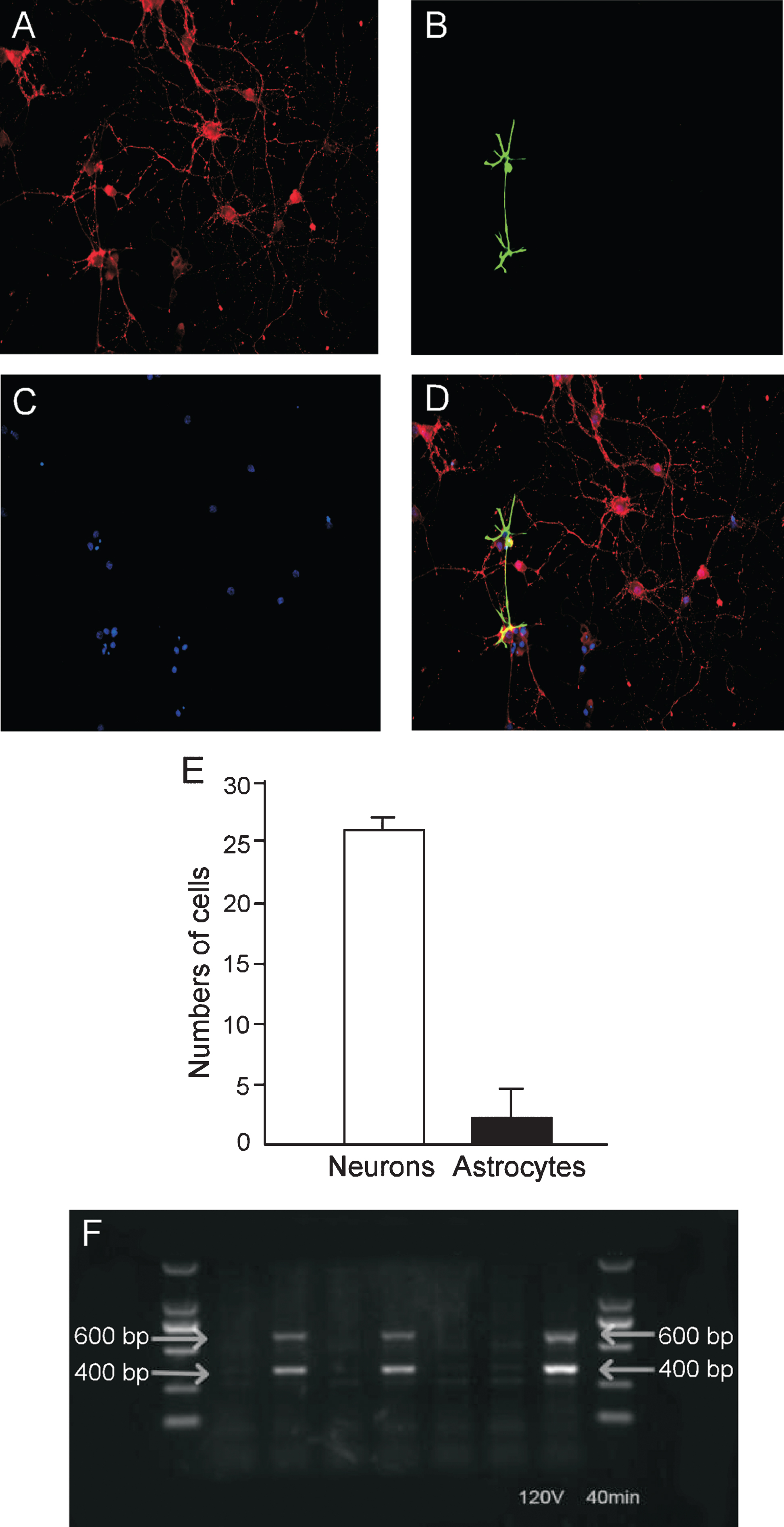
Immunostaining of primary cultured neurons prepared from the neonatal rat hippocampus and confirmation of the APP/PS1 double mutation in mice. The neurons were immunostained with mouse antibody directed against NeuN (labeled with red color) and rabbit antibody against GFAP (labeled with green color), and cell nuclei (stained blue with DAPI). A) NeuN-positive neurons (red); B) GFAP-positive astrocytes (green); C) nuclei (blue); D) merging the staining for neurons (red), astrocytes (green) and nuclei (blue); and E) the percentages of neurons and astrocytes. F) The genotype of the APP/PS1 double-transgenic mice. Bands 2, 4, and 7: the transgenic mice; bands 1, 3, 5, and 6: wild-type mice. M = BioDL100 DNA marker.
Using genomic DNA as a template for amplification, bands about 400 bp and 600 bp in size, consistent with the APP and PS1 genes, respectively, were seen in the transgenic, but not WT mice (Fig. 1F).
Viability in the primary neurons exposed to AβOs, RSV or/and suramin
As detected by the CCK-8 test, the viability of the primary hippocampal neurons was significantly reduced upon treatment with ≥0.5 μM AβOs (Fig. 2A) for 48 h, or ≥20 μM RSV (Fig. 2B) or ≥300 mg/ml suramin (Fig. 2C) for 24h. Interestingly, this decline induced by 0.5 μM AβOs was attenuated by 20 μM RSV (Fig. 2D) and enhanced by 300 mg/ml suramin (Fig. 2E). Therefore, these latter concentrations of RSV and suramin were chosen for exposure without inducing significant cytotoxicity.
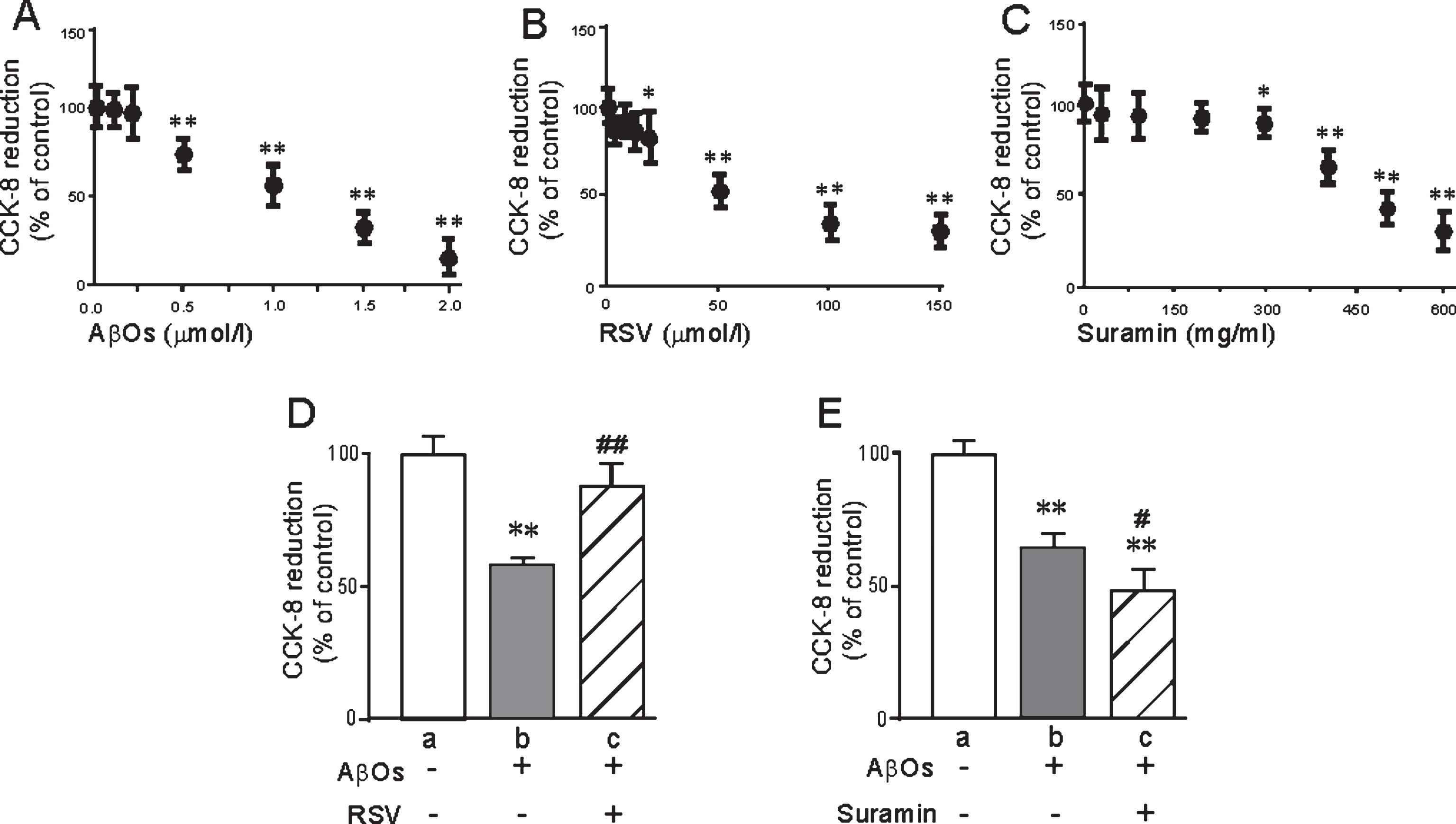
The viability of primary neurons exposed to AβOs, RSV and/or suramin, as determined by CCK8 reduction. A) AβOs (0–2 μM); B) RSV (0–150 μM); C) suramin (0–600 mg/ml); D) AβOs (0.5 μM) and/or RSV (20 μM); and E) AβOs (0.5 μM) and/or suramin (300 mg/ml). The values presented are means±SD of six independent experiments. *p < 0.05 and **p < 0.01 in comparison to untreated cells (controls), and #p < 0.05 and # #p < 0.01 in comparison to D-b or E-b, as determined by analysis of variance (ANOVA), followed by Student-Newman-Keul’s test.
Expression of SITR1 protein and mRNA in mouse brain and primary neurons
As determined by western blotting and real-time PCR, respectively, the levels of SIRT1 protein and mRNA in the brains of the APP/PS1 mice were significantly decreased at 6 months (Fig. 3A, D) and 10 months (Fig. 3B, E) of age compared to WT. These same levels were obviously reduced by exposure of primary neurons to AβOs (Fig. 3C, F). Furthermore, the protein level was significantly enhanced in the mouse brain or primary neurons exposed to RSV (Fig. 4A, B) and decreased by suramin (Fig. 4C, D). At the same time, the level of SIRT1 mRNA in both cases was significantly elevated by RSV and lowered by suramin (Fig. 4C, D).
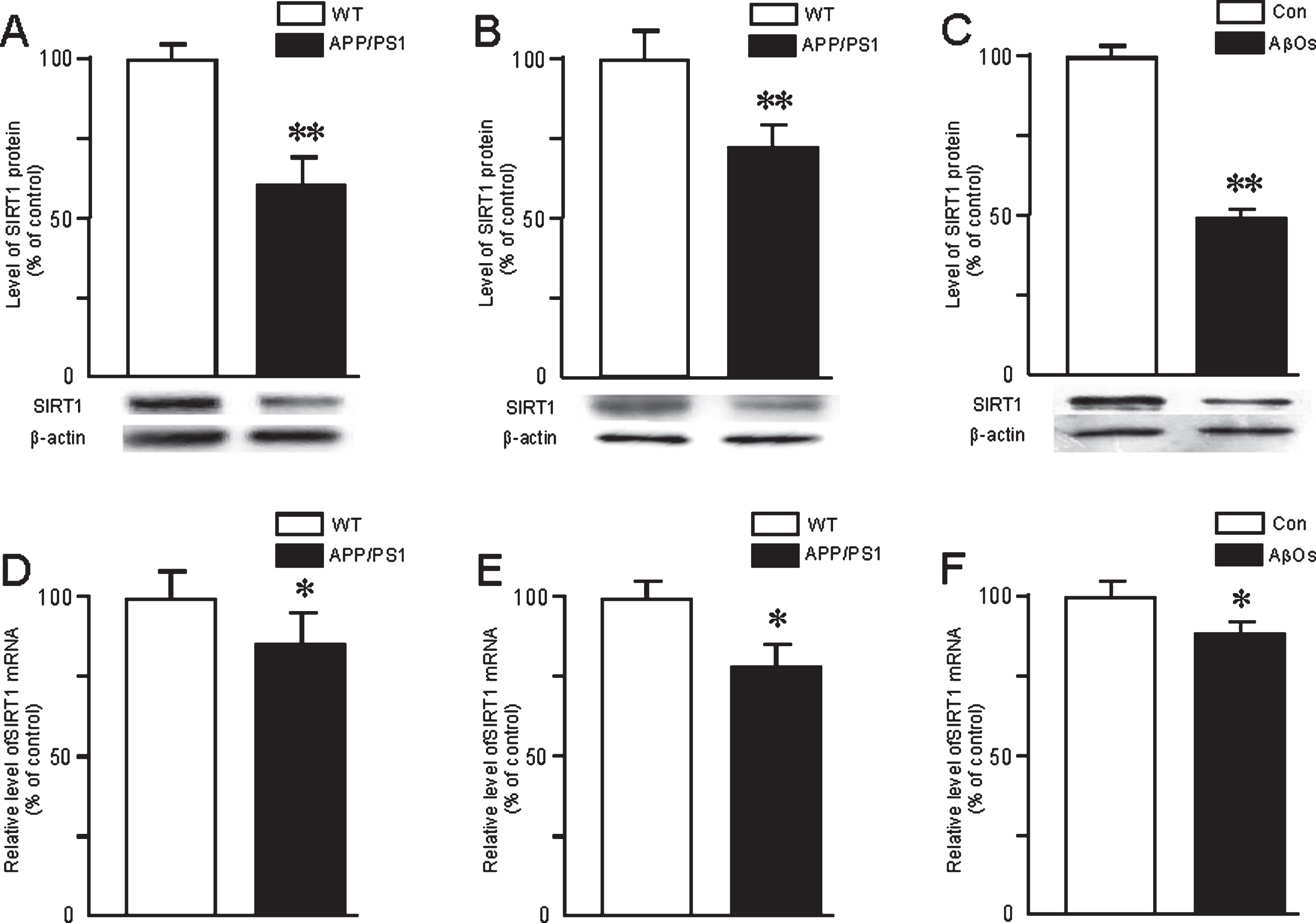
Expression of SIRT1 protein and mRNA in the brains of APP/PS1 mice at 6 (A, D) or 10 (B, E) months of age and in the primary neurons exposed to AβOs (0.5 μM) (C, F). The protein level was determined by western blotting and mRNA by real-time PCR. The values shown are the means±SD of 8 samples. *p < 0.05 and **p < 0.01 in comparison to the corresponding controls, as determined by analysis of variance (ANOVA), followed by Student-Newman-Keul’s test. Representative western blots are shown A, B, and C.

Expression of SIRT1 protein and mRNA in the brains of 4-month-old APP/PS1 mice (A, C) and primary neurons (B, D) treated with RSV (20 mg/kg or 20 μM) or suramin (20 mg/kg or 300 mg/ml). The protein levels were determined by western blotting and mRNA by real-time PCR. The values shown are the means±SD of 8 samples. *p < 0.05 and **p < 0.01 in comparison to the corresponding controls, and #p < 0.05 and # #p < 0.01 in comparison to treatment with RSV (b), as determined by analysis of variance (ANOVA), followed by Student-Newman-Keul’s test. Representative western blots are shown beneath A and B.
Immunoreactivity of the senile plaques in the brain of APP/PS1 mice
Examination of the brains of 6- (Fig. 5) or 10-month-old (Fig. 6) APP/PS1 mice revealed Aβ-immunoreactive senile plaques distributed throughout the cortex and hippocampus. Furthermore, the numbers and size of these plaques were markedly reduced in both the cortex and hippocampus by exposure to RSV and increased by suramin. No amyloid plaques were detected in the brains of WT mice with or without treatment by RSV or suramin (Figs. 5 and 6).
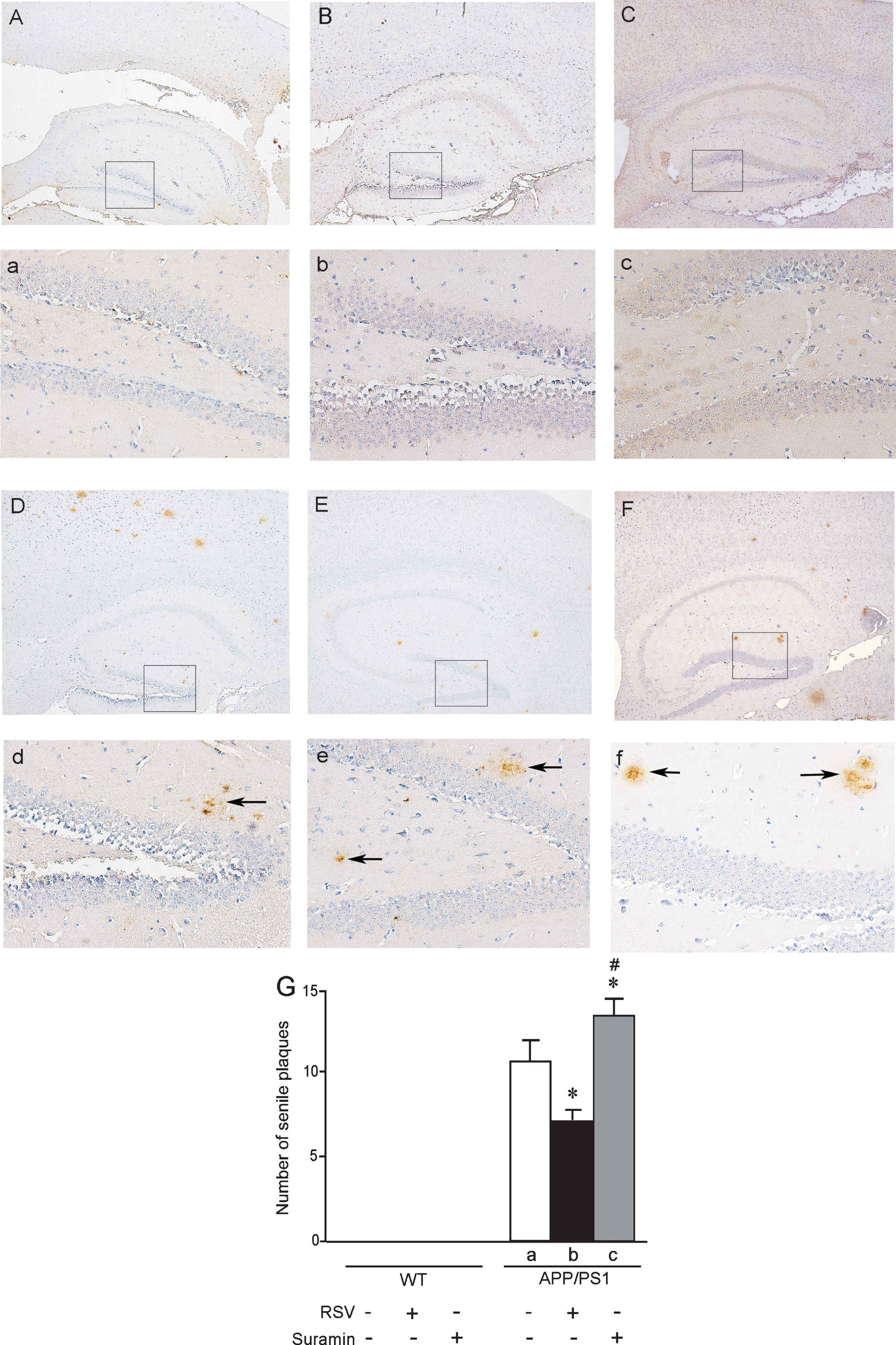
Senile plaques in the hippocampus of APP/PS1 mice at 6 months of age. The wild-type (WT) and APP/PS1 mice received RSV, suramin, or physiological saline (PS) by gavage once each day for two months. A and a) WT mice given PS; B and b) WT mice given 20 mg RSV/kg; C and c) WT mice given 20 mg suramin/kg. D and d) APP/PS1 mice given PS; E and e) APP/PS1 mice given 20 mg RSV/kg; F and f) APP/PS1 mice given 20 mg suramin/kg. Magnification: A-F, 40 X; a-f, 200 X (magnification of the squares in A-F). The solid arrows indicate senile plaques. G: the numbers of senile plaques detected in the hippocampus. The values shown as the means±SD of 8 samples. *p < 0.05 compared to APP/PS1-a and #p < 0.05 in comparison to APP/PS1-b, as determined by analysis of variance (ANOVA), followed by Student-Newman-Keul’s test.
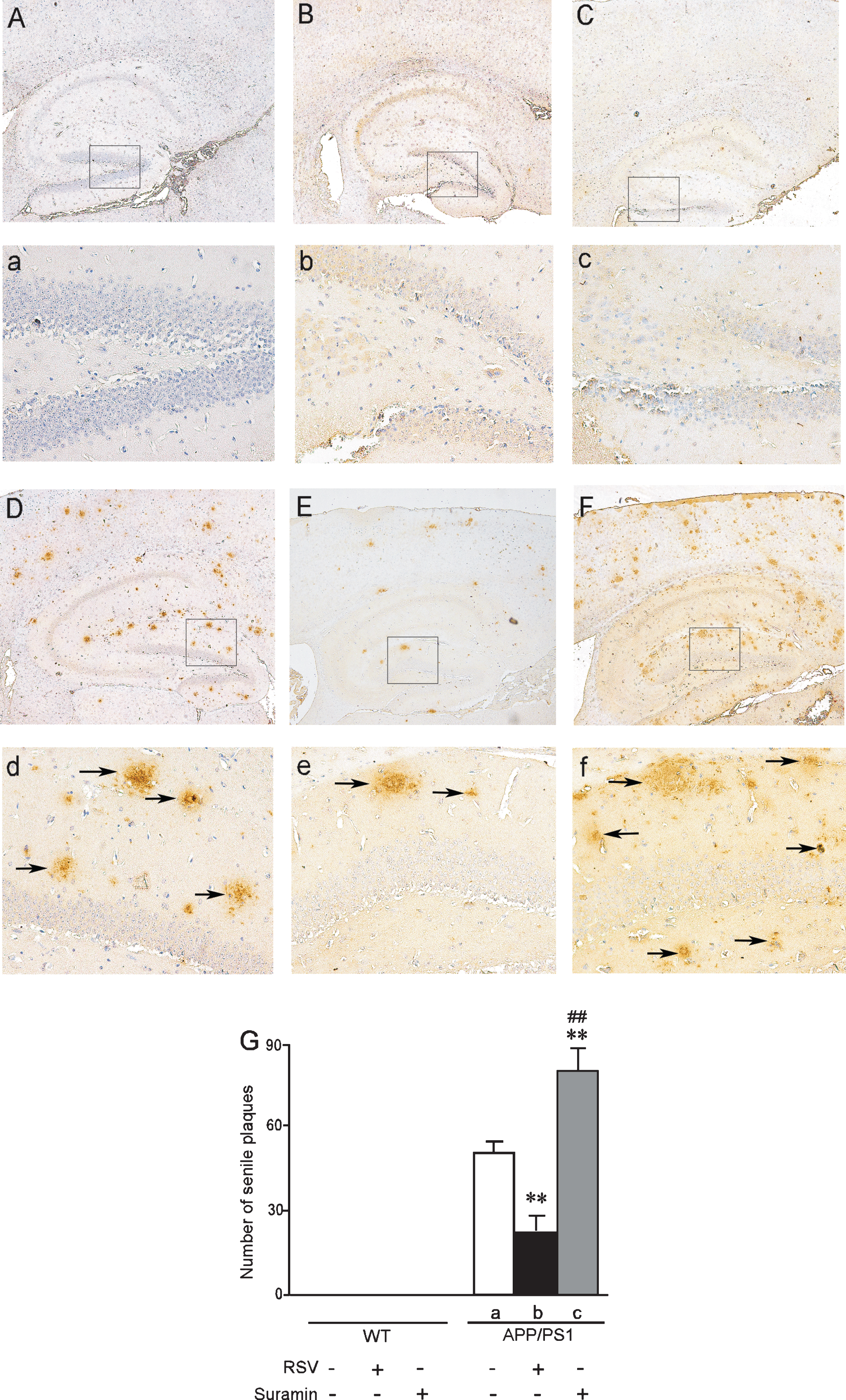
Senile plaques in the hippocampus of APP/PS1 mice at 10 months of age. The wild-type (WT) and APP/PS1 mice received RSV, suramin, or physiological saline (PS) by gavage once each day for two months. A and a) WT mice given PS; B and b) WT mice given 20 mg RSV/kg; C and c) WT mice given 20 mg suramin/kg. D and d) APP/PS1 mice given PS; E and e) APP/PS1 mice given 20 mg RSV/kg; F and f) APP/PS1 mice given 20 mg suramin/kg. Magnification: A-F, 40 X; a-f, 200 X (magnification of the squares in A-F). The solid arrows indicate senile plaques. G: the numbers of senile plaques detected by Aβ immunohistochemistry. The values shown are the means±SD of 8 samples. **p < 0.01 compared to APP/PS1-a, # #p < 0.01 in comparison to APP/PS1-b, as determined by analysis of variance (ANOVA), followed by Student-Newman-Keul’s test.
Activities of SOD and GSH-Px and levels of MDA and free radicals in the brains of APP/PS1 and WT mice, as well as in primary neurons
The activities of SOD and GSH-Px in the brains of APP/PS1 mice were higher than in the controls (WT) at both 6 (Fig. 7A, C) and 10 months of age (Fig. 7B, D). Interestingly, treatment of the transgenic mice with RSV attenuated these differences (Fig. 7), whereas suramin augmented them (Fig. 7).
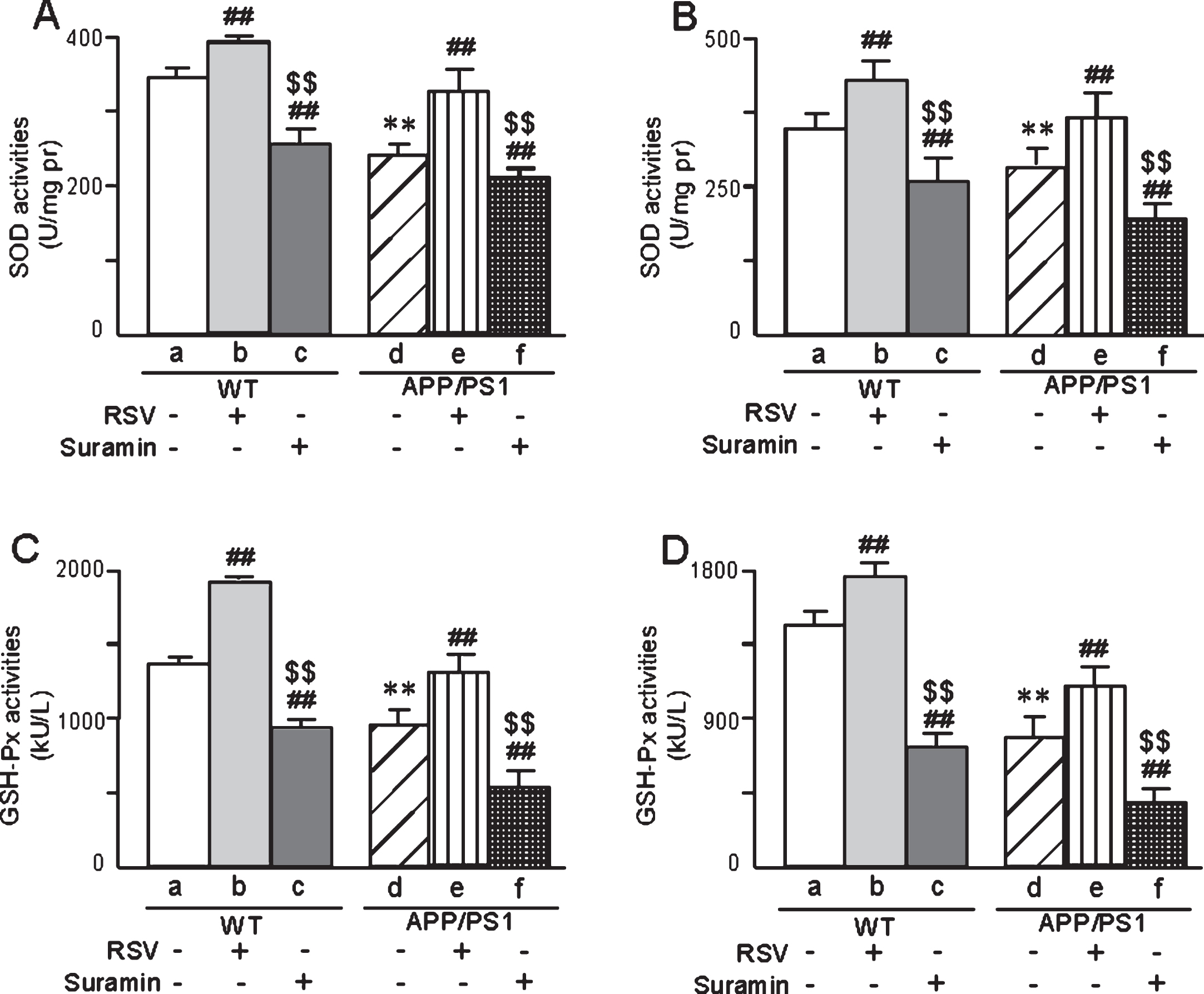
The activities of SOD and GSH-Px in the brains of APP/PS1 mice 6 (A, C) or 10 (B, D) months of age, respectively, and the effects of exposure to RSV (20 mg/kg) or suramin (20 mg/kg). The values shown are the means±SD of 8 samples. **p < 0.01 compared to WT, # #p < 0.01 in comparison to WT-a or APP/PS1-d and $$p < 0.01 in comparison to WT-b or APP/PS1-e, as determined by analysis of variance (ANOVA), followed by Student-Newman-Keul’s test.
Furthermore, the activities of SOD, SOD1, and SOD2 in brain mitochondria isolated from APP/PS1 mice exhibited similar changes (Fig. 8), with RSV again attenuating and suramin augmenting the decreases in these activities.
On the other hand, the levels of MDA and the contents of OH-, H2O2, and O2·- in the brains of APP/PS1 mice were higher than in WT animals (Fig. 9). Once again, RSV attenuated and suramin augmented these differences (Fig. 9).

The activities of SOD (A, B), SOD1 (C, D), and SOD2 (E, F) in brains mitochondria isolated from the brains of APP/PS1 mice at 6 or 10 months of age, respectively, and the effects of exposure to RSV (20 mg/kg) or suramin (20 mg/kg). The values shown are the means±SD of 8 samples. *p < 0.05 and **p < 0.01 compared to WT, #p < 0.05 and # #p < 0.01 in comparison to WT-a or APP/PS1-d, and $$p < 0.01 in comparison to WT-b or APP/PS1-e, as determined by analysis of variance (ANOVA), followed by Student-Newman-Keul’s test.
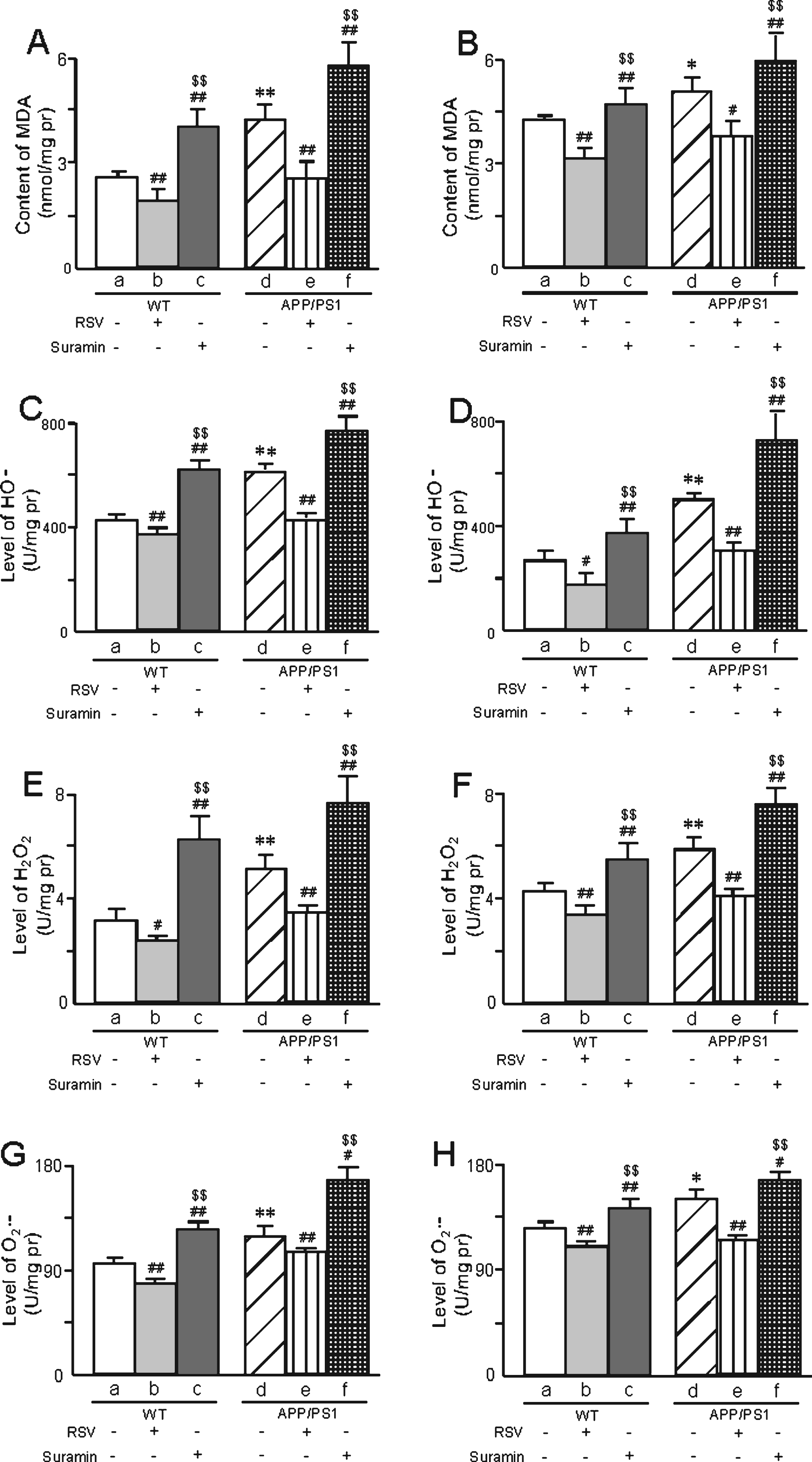
The levels of MDA (A, B) and ROS (OH- (C, D), H2O2 (E, F), O2·- (G, H)) in the brains of APP/PS1 mice at 6 or 10 months of age, respectively, and the effects of exposure to RSV (20 mg/kg) or suramin (20 mg/kg). The values shown are the means±SD of 8 samples. *p < 0.05 and **p < 0.01 compared to WT, #p < 0.05 and # #p < 0.01 in comparison to WT-a or APP/PS1-d, and $$p < 0.01 in comparison to WT-b or APP/PS1-e, as determined by analysis of variance (ANOVA), followed by Student-Newman-Keul’s test.
In the case of primary neurons exposed to AβOs, the activities of both SOD (SOD1 and SOD2) and GSH-Px were lower in both homogenates (Fig. 10) and in isolated mitochondria (Fig. 11) than in the corresponding control preparations, with the usual pattern of attenuation of these differences by RSV and augmentation by suramin (Figs. 10 and 11). In addition, as detected by both confocal laser scanning microscopy (Fig. 12A1-6) and flow cytometry (Fig. 12B1-6), the levels of ROS in primary neurons were elevated by exposure to AβOs, with the usual effects of RSV or suramin (quantified in Fig. 12C). The content of MDA in primary neurons exposed to AβOs was higher than in untreated cells (Fig. 12D). Furthermore, RSV attenuated and suramin augmented the elevation of MDA (Fig. 12D).
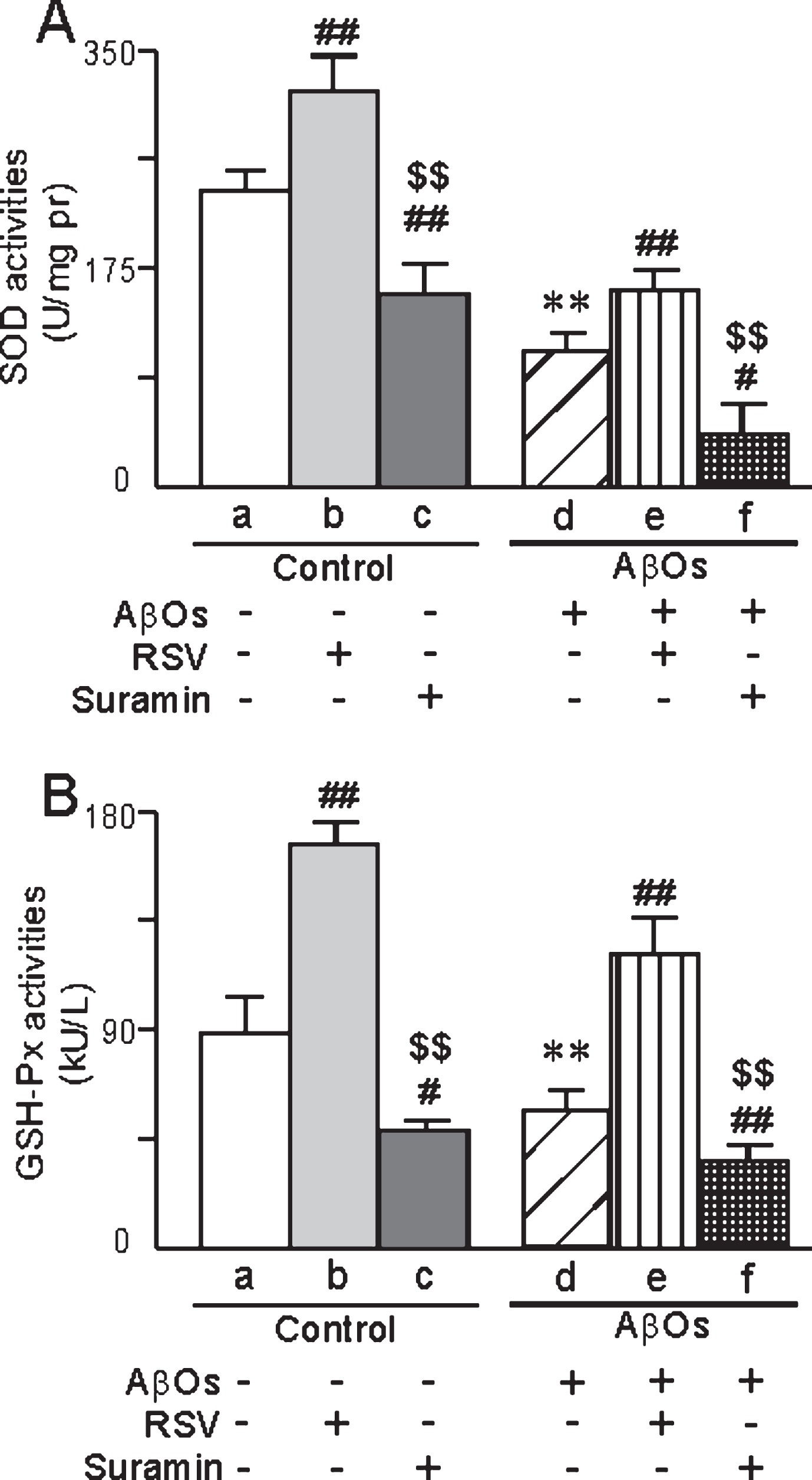
The activities of SOD (A) and GSH-Px (B) in primary neurons prepared from neonatal rats and exposed to AβOs (0.5 μM), RSV (20 μM) and/or suramin (300 mg/ml). The values shown are the mean±SD of 8 samples. **p < 0.01 compared to un-treated control, #p < 0.05 and # #p < 0.01 in comparison to control-a or AβOs-d, and $$p < 0.01 in comparison to control-b or AβOs-e, as determined by analysis of variance (ANOVA), followed by Student-Newman-Keul’s test.
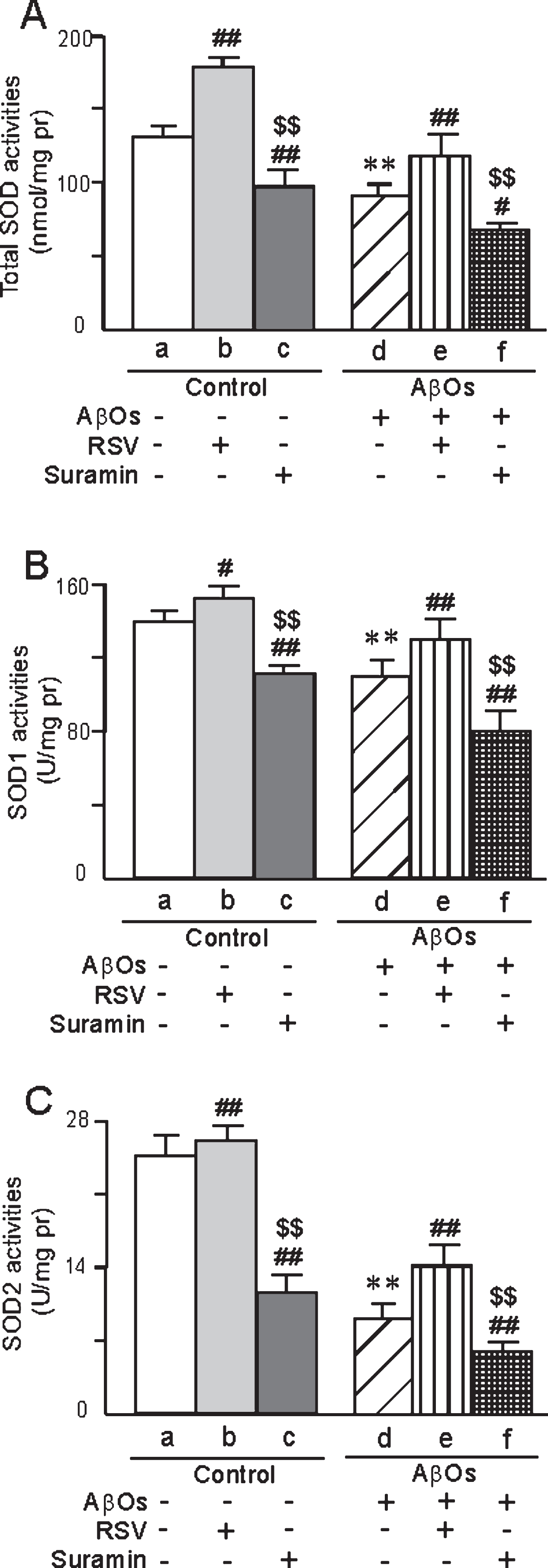
The activities of total SOD (A), SOD1 (B), and SOD2 (C) in mitochondria isolated from primary neurons prepared from neonatal rats and the effects of treatment with AβOs (0.5 μM), RSV (20 μM), and/or suramin (300 mg/ml). The values are shown as the mean±SD of 8 samples. **p < 0.01 compared to un-treated control, #p < 0.05 and # #p < 0.01 in comparison to control-a or AβOs-d, and $$p < 0.01 in comparison to control-b or AβOs-e, as determined by analysis of variance (ANOVA), followed by Student-Newman-Keul’s test.
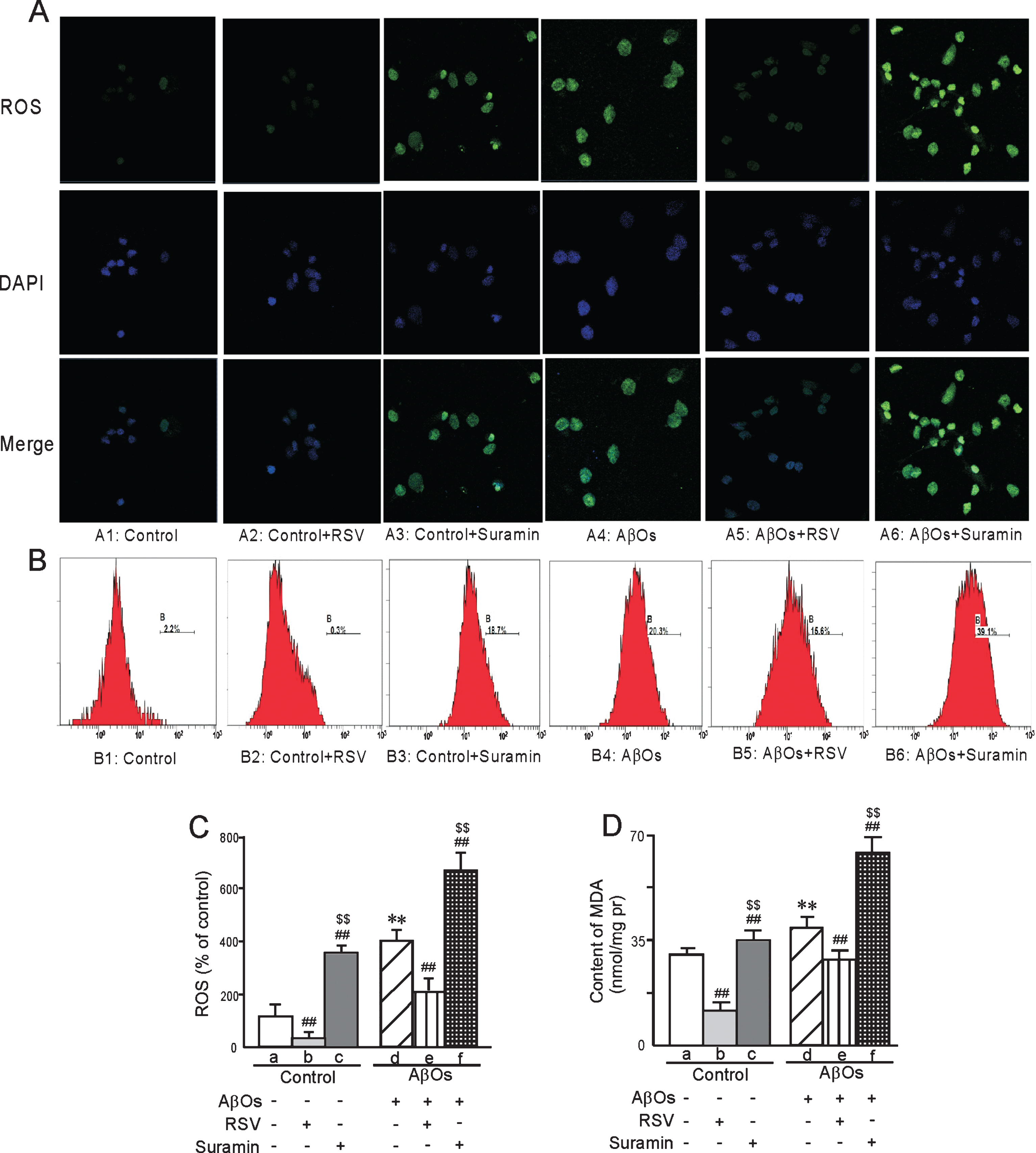
The levels of ROS and MDA in primary neurons prepared from neonatal rats and the effects of treatment with AβOs (0.5 μM), RSV (20 μM), and/or suramin (300 mg/ml). ROS (A) analyzed by confocal laser scanning microscopy and (B) flow cytometry, with the corresponding quantitation. In addition, the levels of ROS (C) and MDA (D) were quantified. The values shown are the means±SD of 8 samples. **p < 0.01 compared to un-treated control, # #p < 0.01 in comparison to control-a or AβOs-d, and $$p < 0.01 in comparison to control-b or AβOs-e, as determined by analysis of variance (ANOVA), followed by Student-Newman-Keul’s test.
DISCUSSION
Here, we employed APP/PS1 double-transgenic mice and primary neurons exposed to AβOs as models of AD neurotoxicity in which to examine the potential protective role of SIRT1, the most extensively studied member of the sirtuin family [29] that regulates numerous forms of neuroprotection [14]. The lowered expression of SIRT1 in both of our models is similar to previous findings [17, 31], as well as to observations on human brains and serum [32, 33]. SIRT1 is involved in several key pathways linked to the pathogenesis of AD, e.g., deacetylation of tau [34], promotion of the non-amyloidogenic AβPP processing pathway [31], protection against Aβ toxicity through inhibition of NF-κB (nuclear factor κ-light-chain-enhancer of activated B cells) signaling [35], and suppression of Aβ production by activating the ADAM10 α-secretase gene [30]. The decreased level of SIRT1 observed here indicates that its neuroprotective role may be lost during the progression of AD [32].
In the search for the mechanisms underlying AD, the amyloid cascade hypothesis, which has dominated this field for the past few decades, is now being challenged [36–38]. Interestingly, accumulating evidence suggests that oxidative stress plays a key role in late-onset sporadic forms of AD, which represent the majority of cases [39]. Oxidative stress, a potentially damaging pathophysiological imbalance between oxidants and antioxidants, has been detected in the blood, cerebrospinal fluid, and brains of patients with probable AD [40–44]. Accordingly, it has been proposed that AD develops as a direct consequence of oxidative stress in the brain, which may be caused, for example, by deposits of mercury, iron, and aluminum that promote the generation of free radicals through the Fenton and Haber-Weiss pathway and consequent lipid, protein, and DNA peroxidation or oxidation [45]. In addition, the activity of antioxidant enzymes such SOD or GSH-Px is lowered, thereby contributing to the accumulation of oxidative damage [46].
Our present findings confirm that the activities of SOD and GSH-Px in the brains of APP/PS1 mice at 6 or 10 months of age are lower than in WT mice. Furthermore, the activities of SOD1 (with a Cu/Zn site) and SOD2 (with a Mn site) in mitochondria isolated from these brains, as well as in primary neonatal rat neurons exposed to AβOs were significantly decreased, providing further evidence for the involvement of reduced levels of antioxidant enzymes in the pathophysiology of AD. In addition, in these same model systems the levels of MDA and ROS (such as OH-, H2O2, and O2·–) were elevated, which may explain the decreased cellular viability observed.
There is evidence that mitochondrial damage resulting in enhanced production of ROS contributes to the early stages of AD, prior to the onset of clinical symptoms and appearance of pathology [47]. Observations on both animal models and patients with AD indicate strongly that the phosphorylated tau protein may potentially connect these events and, therefore, therapy that both targets this protein and involves antioxidants might be of value [48]. For example, accumulation of both Aβ and phosphorylated tau may be required to initiate neurodegeneration in patients with AD [49–51]. Moreover, emerging data suggest that mitochondrial bioenergetics could independently influence both AβPP processing and Aβ production [52–58].
Interestingly, oxidative damage and expression of SIRT1 in the brains of APP/PS1 mice appear to be linked to their numbers of senile plaques [39, 45–47]. SIRT1 is attracting more and more attention for its role in resistance to oxidative stress, through mechanisms which may involve FOXOs (the mammalian forkhead transcription factors of the O class), NF-κB, NOX (nicotinamide adenine dinucleotide phosphate-oxidase), SOD, and endothelial nitric oxide synthase [18].
Binding of activated FOXO3 to the promoter region of the Mn-SOD (or SOD2) gene and subsequent enhanced expression of this enzyme protects quiescent cells from oxidative stress [59]. FOXO1, FOXO3a, and FOXO4 are indispensable for the ability of SIRT1 to promote cell survival in the presence of oxidative stress [60]. In response to oxidative stress, SIRT1 and FOXO3 form a complex, and under both in vivo and in vitro conditions, SIRT1 deacetylates FOXO3 to enhance its binding to DNA and promote resistance to oxidative stress [61].
In addition, NF-κB, a major inducer of inflammatory responses, was the first eukaryotic transcription factor shown to respond directly to oxidative stress induced by H2O2 [62]. Deacetylation and thereby inactivation of NF-κB by SIRT1 impairs its downstream signaling, attenuates the cellular load of ROS and promotes resolution of inflammation [63, 64].
Furthermore, a decrease in NAD+ content induced by ROS tends to impair SIRT1 activity [65]. At the same time, increased NOX activity may raise the NAD+ content and SIRT1 level to create an oxidized state in the cell [18]. SIRT1 activates eNOs through deacetylation of lysines 496 and 506, enhancing NO production and thereby resistance to oxidative stress [66, 67]. Finally, at least i n vitro, through its control of proliferator-activated receptor coactivator-1α (PGC-1α) activity, SIRT1 is able to regulate the expression of oxidative stress-related genes, including GSH-Px, CAT, and SOD2 [68].
RSV and suramin are well characterized as a specific activator and inhibitor of SIRT1, respectively. Here, expression of SIRT1 in the brains of WT mice and in primary neurons was elevated by exposure to RSV and reduced by suramin. Under all conditions, the level of SIRT1 mRNA was significantly correlated to the corresponding protein level. These findings are in agreement with previous reports [69, 70].
It has been proposed that loss of SIRT1 is associated with the accumulation of Aβ in the cerebral cortex of individuals with AD [71]. We show here for the first time that the number of senile plaques in the hippocampus and cortex of APP/PS1 mice was reduced significantly by feeding with RSV, but elevated by suramin. Simultaneously, the level of oxidative stress in mitochondria isolated from these brains rose.
A more recent report has shown that SIRT1 can directly activate transcription of the gene encoding the metallopeptidase domain 10 (ADAM10), an α-secretase, probably via deacetylation of the retinoic acid receptor β (RARβ) [30]. Overexpression of SIRT1 in murine neural N2α cells carrying the mutant APPswe gene significantly elevates the levels of both ADAM10 and the soluble amyloid-β protein precursor α (sAβPPα); while cilostazol, an inhibitor of type III phosphodiesterase, appears to suppress Aβ production in a manner dependent on SIRT1, RAR, and ADAM10 [72]. Protection of AD-susceptible neurons by SIRT1 may be preceded by enhanced α-secretase-mediated non-amyloidogenic AβPP processing, and a decline in the SIRT1 level in the aging brain might therefore predispose its neurons to amyloidogenic AβPP processing [73].
In addition, promotion of non-amyloidogenic AβPP processing by SIRT1 is corroborated by our finding that RSV treatment led to dose-dependent increases in the level of sAβPPα, an extracellular fragment generated through cleavage of AβPP by α-secretase [17]. Recently, it has been reported that α-secretase activity in SIRT1 transgenic mice is increased and that Aβ promotes loss of SIRT1 [31]. Importantly, since RSV increased the level of SIRT1 here, this level may play a key role in protecting against neurotoxicity [74].
Several lines of evidence indicate that elevation of the level of and/or activation of SIRT1 is beneficial in connection with AD and dementia [16, 75]. It has been proposed that in late-onset AD, ROS may activate cleavage of the AβPP molecule by β-secretase to generate Aβ peptides [76–78]. In aging and AβPP transgenic mice, chronic exposure to ROS results in oxidative damage to mitochondrial and other cellular proteins, lipids, and nucleic acids, shutting down mitochondrial energy production [79]. Recent studies on familial AD have revealed that Aβ enters mitochondria, where it interacts with an alcohol dehydrogenase (the expression of which it induces) to disrupt the electron transport chain, generate ROS and inhibit ATP production [80, 81]. Age-dependent interactions of Aβ with mitochondrial proteins may lead to mitochondrial dysfunction in association with AD [79, 82]. SIRT1 helps cells tolerate oxidative stress [83], perhaps by increasing catalase activity [84]. Overexpression of SIRT1 enhances the resistance of neuronal cells to free radical toxicity [85, 86]. In addition, SIRT1 can block p53-induced apoptosis through p53 deacetylation and induction of SOD2 [87, 88].
In our case, treatment of APP/PS1 mice or primary neurons exposed to AβOs with RSV reduces the levels of ROS (OH-, H2O2, and O2·–) and lipid peroxidation, while elevating the activities of antioxidant enzymes (such as total-SOD, SOD1, SOD2, and GSH-Px). On the other hand, inhibition of SIRT1 by suramin enhanced the level of oxidative stress. These results provide direct evidence that SIRT1 may play an important role in eliminating damage by ROS associated with the pathogenesis of AD. The decrease in the number of senile plaques in the brains of APP/PS1 mice achieved through activation of SIRT1 by RSV may mean that SIRT1 can reduce production of Aβ by acting on several secretases involved in AβPP cleavage [30, 73].
In conclusion, attenuated expression of SIRT1, elevated numbers of senile plaques and high levels of oxidative stress, as well as decreased cellular viability were clearly apparent in the brains of APP/PS1 mice or in primary neurons exposed to AβOs. Interestingly, the increased number of plaques and the elevated level of oxidative stress were attenuated when SIRT1 was stimulated by RSV and enhanced when this protein was inhibited by suramin, which indicates that SIRT1 may be neuroprotective in the pathophysiology of AD.
Footnotes
ACKNOWLEDGMENTS
This work was supported financially by grants from the Natural Science Foundation of P. R. China (81760571); the Foundation of the Ministry of Education of P. R. China (IRT13058); and the Scientific Foundations in Guizhou Province of China ([2014]06, [2014]4010 and [2014]6008).