
Abstract
Alzheimer’s disease (AD) impairs memory and causes significant cognitive deficits. The disease course is prolonged, with a poor prognosis, and thus exacts an enormous economic and social burden. Over the past two decades, genetically engineered mouse models have proven indispensable for understanding AD pathogenesis, as well as for discovering new therapeutic targets. Here we highlight significant studies from our laboratory that have helped advance the AD field by elucidating key pathogenic processes operative in AD and exploring a variety of aspects of the disease which may yield novel therapeutic strategies for combatting this burdensome disease.
INTRODUCTION
Alzheimer’s disease (AD) is the leading cause of dementia among the elderly, and it is projected that, by 2050, 1 in 3 seniors will develop this insidious disease [1]. Despite immense efforts within academia and the pharmaceutical industry, as of today, there are no effective treatments available [2, 3]. Moreover, the course of the disease is prolonged and the prognostics are poor and a definitive diagnosis of AD is only established when the presence of amyloid plaques and neurofibrillary tangles are confirmed in the postmortem brain from the suspected patient [4].
Neuropathologically, AD is characterized by the abnormal accumulation of extracellular deposits composed primarily of the amyloid-β protein (Aβ), known as plaques, and intracellular aggregates consisting of hyperphosphorylated forms of the microtubule-associated protein, tau, known as neurofibrillary tangles [3]. Aβ is a heterogeneous mixture of peptides ranging from 37 to 43 amino acids in length produced through the sequential cleavage of a type-I membrane-spanning protein known as the amyloid-β protein precursor (AβPP), with 40- and 42-amino acid peptides being the predominant species. AβPP can be cleaved at three different sites, by proteolytic activities referred to as α-, β-, and γ-secretases. Aβ peptides are produced when AβPP is processed first by β-secretase, then by γ-secretase. Cleavage by β-secretase results in the secretion of the large amino (N)-terminal ectodomain of AβPP, known as sAβPPβ, into the extracellular space. The resulting carboxy (C)-terminal fragment is retained in the membrane and subsequently processed by γ-secretase. The vast majority of AβPP is, in fact, processed by an alternative pathway, being cleaved first by α-secretase, resulting in the secretion of an N-terminal ectodomain known as sAβPPα, followed by γ-secretase-mediated processing of the membrane-bound C-terminal fragment. Notably, α-secretase activity cuts at a site located within the Aβ sequence, thus precluding the creation of Aβ [5].
Once formed, Aβ peptides have a strong tendency to self-aggregate, something that is especially true for the longer species of Aβ like Aβ42. Aβ peptides coalesce to form a number of higher-order aggregates characterized by a beta-sheet conformation, including soluble, low-molecular-weight species, including dimers, trimers, and dodecamers, known collectively as oligomers. Aβ can also form a variety of high- molecular-weight aggregates, which are generally insoluble, including protofibrils, fibrils and, ultimately, plaques.
Tau is a microtubule-associated protein that has a role in stabilizing neuronal microtubules and, hence, in regulating axonal transport [6–10]. When released into the extracellular space, tau can modulate the signaling of synaptic receptors and, due to its interactions with scaffolding proteins, tau may also regulate receptors present in postsynaptic sites. Also, recent findings demonstrated that tau is involved in long-term depression in the hippocampus [11, 12]. Altogether, these mechanisms demonstrate a key role of tau in controlling the normal functioning of synapses, which can be severely affected in AD.
For the past few decades, genetically engineered mouse models have been the gold stars of basic AD research and have proven invaluable for understanding how AD pathology develops in the brain and to evaluate and discover new therapeutic targets and disease-modifying strategies. Our research group helped advance our collective understanding of the interrelationship between Aβ and tau pathology in AD by developing a mouse model that develops both amyloid plaques and neurofibrillary tangles. We accomplished this by generating a mouse model that harbors disease-causing mutations in three separate genes, AβPP, tau, and presenilin-1 [13]. Known as the triple-transgenic model of AD, or 3xTg-AD, this approach made it possible not only to investigate the two major pathological hallmarks within the same animal, but also to shed a light into the interaction between Aβ and tau.
In this chapter, we will focus on how our research group has ultimately changed and helped the AD field move forward through the understanding of key pathological mechanisms in AD such as neuroinflammatory processes, synaptic changes, comorbidities associated with AD and stem cell-related research.
NEUROINFLAMMATION: BUILDING UP TO THE STORM
Among the factors associated with aging that reduce the quality of life for the elderly are the alterations that affect the immune system. As we age, the innate immune system becomes dysregulated and is characterized by persistent inflammatory responses [14, 15]. Although inflammation is a fundamental protective response, age-related changes in the immune system can contribute to the increased susceptibility of the elderly to innumerous diseases including AD. More insight into the molecular pathogenesis of the disease is required to better translate basic biological discoveries into safe and effective clinical applications.
We have been particularly interested, over the past few years, in understanding how inflammation impacts Aβ and tau pathology (Fig. 1). Elderly individuals are susceptible to viral and bacterial infections, and these microbial agents could exacerbate the existing inflammatory condition in the brain, accelerating the cognitive decline. It is now well accepted that chronic inflammation mediated by inflammatory receptors such as IL-1R1, Toll-like receptor 4 (TLR4), and tumor necrosis receptor (TNFR) represents a key mechanism by which Aβ drives the development of tau pathology and cognitive decline in AD [16–18]. One important receptor implicated in AD, TLR4, is responsible for detecting microbial products and inducing innate and adaptive immunity [19]. Studies conducted by our group in the 3x-Tg-AD mouse model demonstrated that stimulation of TLR4 by Escherichia coli lipopolysaccharide (LPS) exacerbates tau pathology, via a glycogen synthase kinase-3β (GSK-3β)–dependent mechanism, with chronic inflammation leading to impairments in spatial memory [20]. The activation of TLR4 by pathogen-associated molecular patterns leads to the expression of proinflammatory cytokines, which will then start specific immune responses. Indeed, the brains of 3xTg-AD mice presented significant increased levels of interleukin-1β (IL-1β) after chronic LPS treatment.
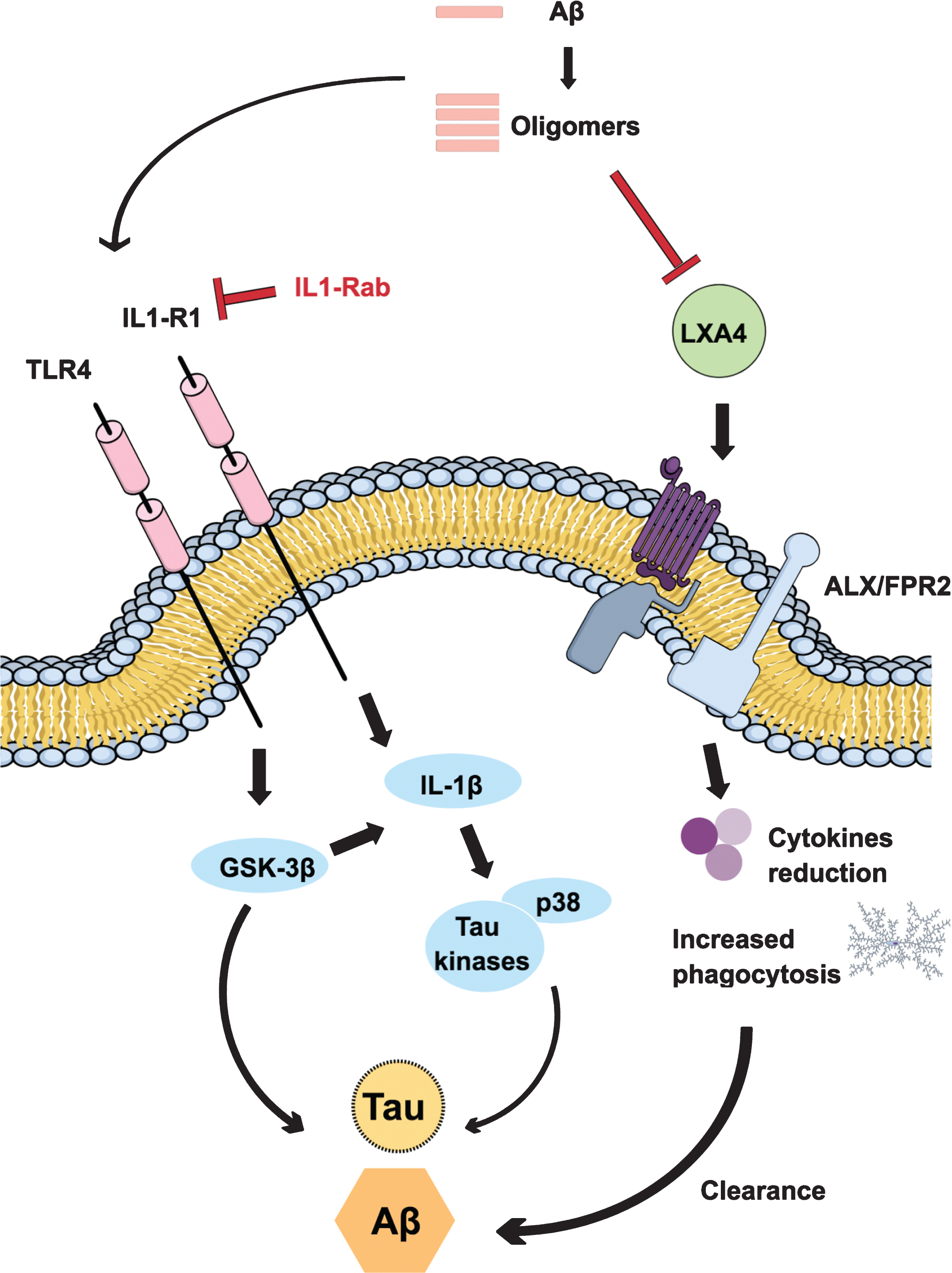
Inflammatory mechanisms linked to Alzheimer’s disease (AD). The activation of inflammatory receptors like IL1-R1 and TLR4 is a key mechanism by which Aβ leads to tau pathology and cognitive decline in AD. Stimulation of TLR4 leads to the expression of proinflammatory cytokines, via a glycogen synthase kinase-3β (GSK-3β)–dependent mechanism, converging on cognitive impairments and pathology progress. Inhibition of IL-1β signaling, by an IL-1R1 antibody, reduces the activation of tau kinases and p38, alleviating cognitive deficits and partly reducing some fibrillar and oligomeric forms of Aβ. During aging and in AD, there is a reduction of lipoxin A4 production, an endogenous pro-resolving mediator. Restoring its levels leads to an alternative activation of microglia, a reduction of overall inflammation, and the promotion of increased phagocytosis and Aβ clearance.
There is a growing body of evidence showing that IL-1β turns synaptic plasticity, learning and memory more susceptible to impairment, especially with age [21–23]. Aged animals present specific deficits for long-term potentiation (LTP) [24, 25] and hippocampal-dependent memory [26, 27] after a systemic immune activation, and all of these impairments are blocked by brain infusion of the IL-1 receptor antagonist, IL-1ra. In this regard, we demonstrated that inhibition of IL-1 signaling, by chronically treating 3xTg-AD mice with an IL-1R blocking antibody, reduced the activity of several tau kinases in the brain, including cdk5/p25, GSK-3β, and p38-MAPK, also reducing phosphorylated tau levels. Moreover, the treatment significantly altered brain inflammatory responses through the reduction of nuclear factor κB (NF-κB), alleviated cognitive deficits and partly reduced some fibrillar and oligomeric forms of Aβ [17]. Recently, it was demonstrated that IL-1β impairs LTP directly at the synapse and that sensitivity to IL-1β is augmented in aged hippocampal synapses, through an IL-1 receptor subunit reconfiguration [18]. Thus, ours and other studies provide evidence that modulation of IL-1β signaling may offer therapeutic benefit to AD patients, and it has been a constant target of investigation within our research group.
Immune responses need to be tightly regulated in terms of intensity, class, and duration to prevent molecular, cellular, and organ damage. Despite the fact that its neuropathological involvement and consequence in AD still remains to be elucidated, it has been suggested that inflammation plays a dichotomous role in the disease. In young individuals, inflammation is self-limited and resolves by means of an active termination program known as inflammatory resolution [28]. The discovery of this active and highly coordinated process controlled by endogenous pro-resolving mediators modified our understanding of diseases caused by chronic inflammation [29, 30]. In older subjects, however, disturbances in the immune system result in a state of low-grade chronic inflammation. In the brain, persistent and unresolved inflammation has been implicated, with a variable degree of importance, in almost all age-related neurodegenerative disorders. In AD, chronic inflammation, that is characterized by activation of microglia and astrocytes and excessive production of pro-inflammatory mediators, may lead to disease progression and neuronal loss [31–33]. Therefore, new approaches aimed to modulate the inflammatory response in AD might prove efficacious. To this end, we evaluated the role of an endogenous lipid mediator, lipoxin A4 (LXA4), generated during the resolution phase. Through agonistic actions at the G-protein coupled LXA4 receptor ALX/FPR2, lipoxins reduce neutrophil recruitment and activation, leukocyte migration, and cytokine production [34, 35]. In the central nervous system (CNS), LXA4 protects neurons against stroke, the development of neuropathic pain after spinal cord injury [36], and Aβ42 toxicity [37]. During aging and in Tg2567 mice, there is a significantly impairment of LXA4 production. Notably, restoration of this mediator signaling led to an alternative activation of microglia, with a reduction of overall inflammation, and the promotion of phagocytosis and Aβ clearance. All these effects were also accompanied by upregulation of synaptic proteins and cognitive improvement [38]. Additionally, aspirin-triggered lipoxin A4 (ATL) also reduced Aβ and phosphorylated tau enhancing the cognitive performance of 3xTg-AD mice [39]. Recently, it was demonstrated the reduction on the levels of LXA4 both in the cerebrospinal fluid (CSF) and hippocampus of AD patients, with a strong correlation with cognitive function [40]. Also, the ability to measure these important mediators in the CSF also provides incentive to explore their potential as diagnostic markers.
Altogether, these data suggest that the inflammatory resolution process is altered by AD, playing a role of great significance in brain homeostasis.
SYNAPTIC LOSS: THE BEGINNING OF THE END
AD is currently an important public health issue, leading to an increased effort over the past years to better understand the causes of it. Several epidemiological studies have demonstrated that synaptic loss has been strongly associated with the cognitive deficits observed in AD. Notably, these impairments are better correlated with the synaptic pathology than either plaques or tangles, therefore suggesting synaptic changes as a central factor for the disease process and progression [8, 13]. Several animal models and clinical studies utilizing familial forms of AD have widely documented the importance of Aβ and tau pathology in the progression of AD.
In this section, we will highlight research findings from our group on how Aβ and tau affects synaptic loss and cognitive deficits in animal models of AD. The idea of Aβ oligomers as toxins responsible for synapse dysfunction and cognitive deficits in AD has aided our understanding of the mechanisms of the disease [41]. However, new evidence has demonstrated that tau also regulates other important processes related to the synaptic function and it is also detected in the dendrites, as well as in pre- and postsynaptic components of normal healthy neurons [42, 43]. Nevertheless, in AD and several other neurodegenerative diseases, known as tauopathies, tau develops post-translational changes that will affect its affinity to microtubules. This process leads to neurofibrillary tangles, which may alter the axonal transport. Moreover, calcium signaling is essential for learning and memory processes; however, its dysregulation may be related to pathological tau changes [6, 7]. Our research group has previously demonstrated that calpain-active cdk5 and ERK1/2 kinases can phosphorylate tau and induce innumerable downstream tau-dependent and independent pathogenic effects, including impairments of synaptic plasticity and cognition [44].
The development of the 3xTg-AD mice by our research group have greatly advanced the AD field, as these mice together promote the development of Aβ and tau pathology and exhibit deficits in synaptic plasticity, including LTP that occurs before extracellular Aβ deposition and formation of tangles. Such finding demonstrates that synaptic transmission and LTP deficits precedes plaque and tangle formation in the 3xTg-AD mice and implies that synaptic dysfunction is an early manifestation of AD and that extracellular Aβ deposition is not the only factor underlying the synaptic dysfunction [13].
A question that still needs to be addressed in the AD field is how the molecular relationship between Aβ and tau affect the integrity of synaptic function and lead to profound and irreversible cognitive deficits. Bearing this in mind, there are multiple mechanisms by which Aβ and tau can impair synaptic function and lead to severe cognitive deficits (Fig. 2). It has been demonstrated that Aβ promotes tau and its misplaced localization in dendritic projections, and that overexpression of both toxic proteins accelerates synaptic and cognitive impairments [45–47]. Given that Aβ and tau coexist and interact directly between themselves within the synaptic compartment, both proteins may have a synergic role in affecting normal synaptic functions [8]. Our research group has demonstrated that 3xTg-AD mice highlight the importance of intraneuronal soluble Aβ as the initial mediator of tau pathology. Aβ induces tau pathology by altering the levels of the C terminus of heat shock protein 79-interacting protein (CHIP), a known tau ubiquitin ligase responsible for facilitating degradation of hyperphosphorylated tau and caspase-3-cleaved tau [48]. In addition, extracellular Aβ is also involved in the development of tau pathology. As it will be demonstrated in another section of this chapter, studies using induced neuronal-derived pluripotent stem cells (iPSCs) have shown that extracellularly generated Aβ increased tau levels in familial AD neurons and that extracellular Aβ has an important role in tau pathology mediated by inflammation.
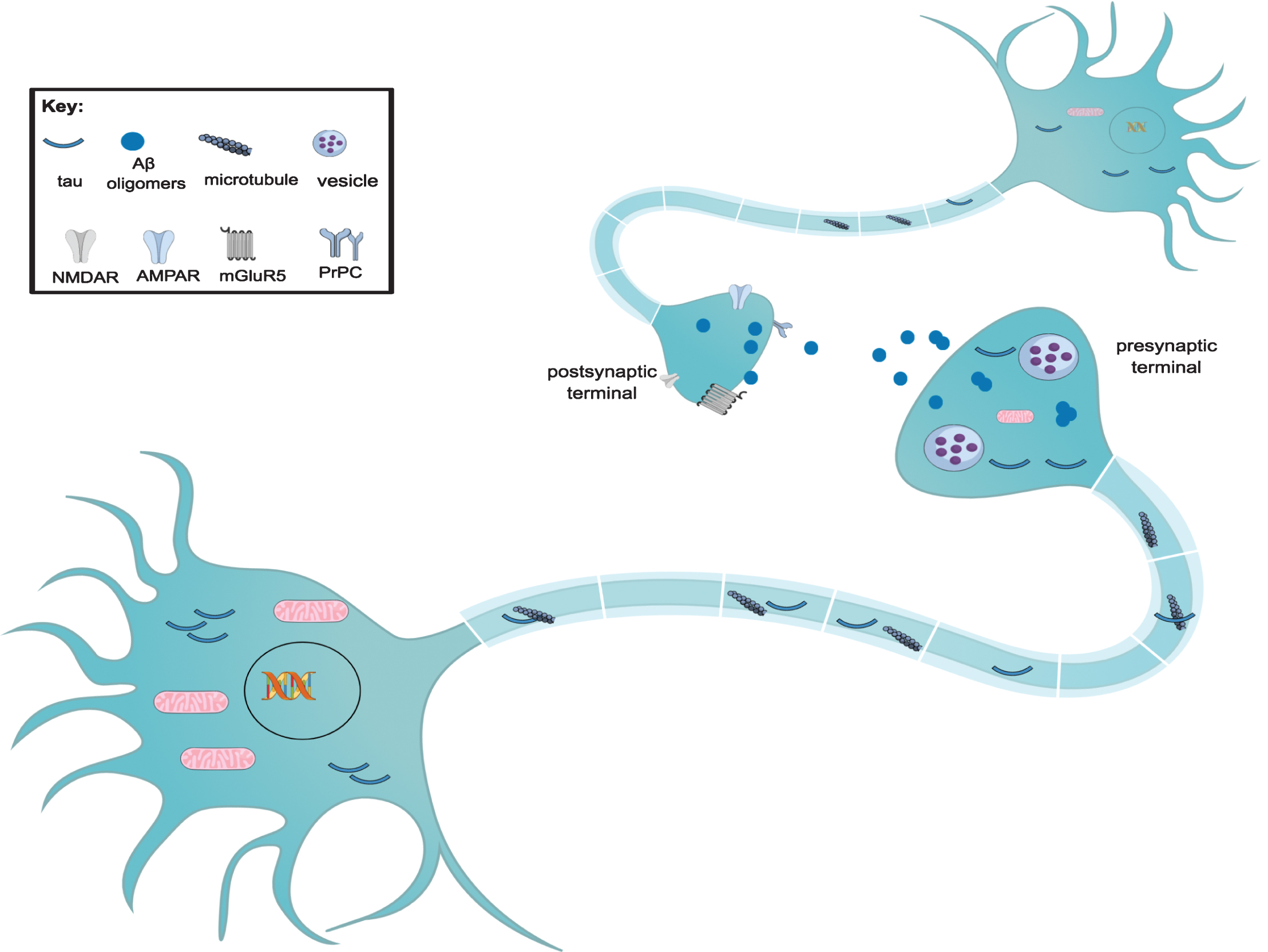
Formation and mechanisms of synaptic toxicity of tau and Aβ oligomers. During tauopathies, there is a reduction in the number of dendritic spines. Tau does not enter the nucleus of the neuron, resulting in DNA damage. There is a reduction in the number of mitochondria and also in the number of presynaptic vesicles, which leads to synaptic loss. Such loss is also due to the entrance of tau into dendrites and postsynaptic areas. Tau also aggregates extracellularly, enabling it to be captured by other neurons. Aβ oligomers may decrease the number of surface glutamate α-amino-3- hydroxy-5-methyl-4-isoxazolepropionic acid receptors (AMPARs); there is a decrease the synaptic strength via an NMDA-dependent pathway. The prion protein-containing oligomer receptor complex (PrPC) interacts with mGluR5, spreading the toxic effect of Aβ oligomers. Moreover, oligomers can interact with a variety of receptors on the pre- and postsynaptic membrane of neurons.
Further studies suggest that tau targets the tyrosine kinase Fyn, a member of the Src family, in the postsynaptic density and induces aberrant glutamatergic synaptic transmission via overactivation of NMDARs [49]. Moreover, the reduction in soluble Aβ oligomers is accompanied by a decrease in human tau pathology, including reduced association of tau with PSD-95, and a rescue of learning and memory deficits. Our data therefore indicate that soluble Aβ, particularly soluble Aβ fibrillar oligomers, facilitate wild-type tau pathology in vivo [47].
In summary, these findings highlight the complexity of Aβ and tau relationship and demonstrate how our research group has led to a better understanding of how tau impacts synaptic function and is related to the pathological role of Aβ in synapses.
DIABETES, STRESS, AND AD: THE CHICKEN AND EGG QUESTION
Despite intensive research efforts over the past few decades, the mechanisms underlying the etiology of sporadic AD (sAD), which represents the most common form of the disease, remains unknown. This is due, at least in part, to the fact that the majority of sAD patients are elder subjects that commonly suffer from a variety of co-morbidities (e.g., stroke, stress, diabetes, seizures, osteoporosis, and renal disease). On average, people living with dementia who are over 65 years old have four comorbidities (Fig. 3). These comorbidities add complexity to the pathogenesis of sAD, affecting its onset and progression [50, 51]. Over the past decade, multiple studies have been performed in animal models to understand the impact of these co-morbid medical conditions on AD pathogenesis [52, 53]. Here, we describe the most relevant studies in the last years and those in which our research group has been working on.

Comorbidities in Alzheimer’s disease (AD). Diabetes, osteoporosis, renal disease, obesity, hypertension and hypercholesterolemia/dyslipidemia, stroke, and seizure are the main comorbidities affecting the onset and progression of AD, adding intricacy to the pathogenesis of the disease. The mechanisms underlying this relationship are assorted and complexes and are highlighted in the blue square, including insulin resistance, inflammation, and oxidative stress. BBB, blood-brain barrier.
Among the variety of co-morbidities one of the most prevailing conditions is diabetes (Fig. 4). Interestingly, recent epidemiological studies indicate that diabetes significantly increases the risk of developing AD, suggesting that diabetes may play a causative role in the development of AD pathogenesis [54]. Moreover, AD and diabetes share several clinical and biochemical features, suggesting common molecular pathways underlying these two diseases [55–58]. The presence of insulin receptors (IRs) in the brain provides important evidence that the brain is a target organ for insulin. Specifically, IRs in the CNS are highly expressed in cognition-related regions, indicating that insulin signaling influence memory, neural plasticity, and cognition [59–66]. Recent evidence reveals that aberrant brain insulin signaling contributes to the pathogenesis of AD [67, 68], and brain insulin resistance is an early common feature of AD [62, 70]. Interestingly, data obtained from human [71] and animal models have shown that diabetes could induce Aβ pathology [72, 73] and promote aberrant tau modifications [74–76]. However, the underlying molecular mechanisms connecting these two disorders are still not well understood. Elucidating these mechanisms is crucial because the number of diabetic and AD patients is expected to increase exponentially in the next decades.
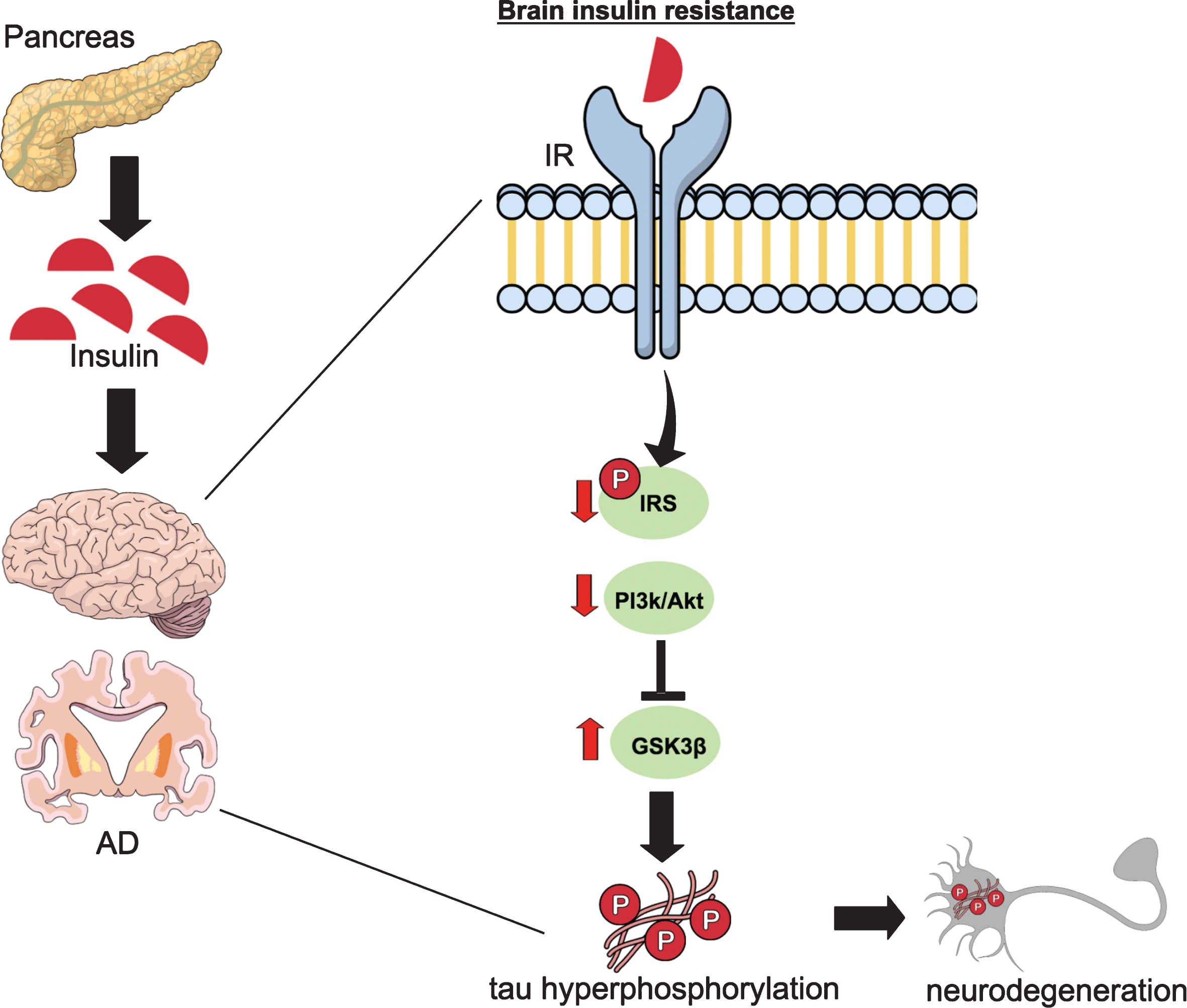
The role of impaired brain insulin signaling in tau pathology. Disturbance of brain insulin signaling has been suggested to be a key causative event underlying sporadic AD pathogenesis. In type 1 and type 2 diabetes, insulin deficiency and resistance, respectively, lead to an altered insulin signaling pathway in brain tissue. Impaired insulin/insulin receptor signaling leads to decreased insulin-mediated activation of PI3k/Akt signaling activity, resulting dephosphorylation (activation) of GSK-3β. Consequently, GSK-3β activation directly promotes tau hyperphosphorylation and formation of neurofibrillary tangles. Brain insulin signaling dysfunction culminates, then, in synaptic failure and memory decline. IR, insulin receptor; IRS, insulin-receptor substrate; PI3k/Akt, phosphatidylinositol 3 kinase/protein kinase B; GSK-3β, glycogen synthase kinase 3β.
Specifically, our group has focused on understanding how diabetes can alter tau pathology and affect the cognitive and synaptic function. Interestingly, several preclinical studies have shown that modeling type 1 (T1D) or type 2 (T2D) diabetes in rodents results in an increase in tau phosphorylation versus normal controls animals [52, 77]. Using streptozotocin (STZ) treatment, a glucosamine-nitrosourea compound that is toxic to the insulin-producing β-cells of the pancreas inducing hyperglycemia and insulin deficiency in mice, rendering them a valuable model to study T1D [78], we have demonstrated that depletion of endogenous tau mitigates behavioral and synaptic deficits induced in T1D-like mice [52]. In this sense, although induction of T1D in non- transgenic (Ntg) mice led to cellular and behavioral deficits, it did not do so in tau- knockout (tauKO) mice. We showed that STZ treatment causes hyperphosphorylation of tau in Ntg mice through activation of GSK-3β. These increments on hyperphosphorylated tau correlate with spatial cognitive deficits and changes in synaptic proteins. Notably, tauKO mice treated with STZ show no cognitive or synaptic deficits. Overall, our data indicate that T1D impairs cognition via tau-dependent mechanisms, and genetic deletion of endogenous tau gene prevents the synaptic degeneration and cognitive impairment. Hence, these data indicate that tau proteins are crucial downstream targets of the insulin pathway and mediators of cognitive deficits in a condition of insulin deficiency, representing a potential therapeutic target for patients with diabetes and AD. We are now investigating the role of tau mediating the cognitive/synaptic deficits in T2D, which represents the most common form of the disease.
Current epidemiological evidence indicates that life experiences, including chronic stress, are a risk for AD [79, 80]. In fact, hypothalamic-pituitary-adrenal axis dysfunction as well as elevated levels of cortisol in plasma and CSF are found in AD patients [81], and multiple key studies indicate that stress modulates synaptic plasticity and memory processes [82, 83]. Furthermore, recent studies in animal models have found that stress and stress hormones, including glucocorticoids and corticotrophin-releasing hormone, play a crucial role in AD pathogenesis by modulating Aβ production and degradation [84–86], and impairs tau pathology by modulating key kinases involved in tau phosphorylation or by mislocalizing tau protein to the somatodendritic compartment [85, 88]. Together, these findings suggest that stress and several stress mediators play key roles in modulating AD pathogenesis.
Our group has investigated the impact of short-term, multi-modal modern-life like stress, which often last for hours, on AD progression and its implication in synaptic plasticity and cognitive function. Several lines of evidence support the importance of stress duration and modalities on cognitive function [82, 89]. This matter is extremely important, because modern-life stress often involves multiple concurrent psychological, social, and physical stresses [90]. Therefore, it is fundamental to elucidate the effect of multiple concurrent stresses on the onset and progress of AD pathogenesis. We found that short-term multimodal stress, lasting for 5 hours, severely reduced the number of the spines in 3xTg-AD mice. In addition, this form of stress increased Aβ oligomers by modulation of AβPP processing via upregulation of beta-site amyloid precursor protein-cleaving enzyme 1 (BACE1) steady state levels without altering Aβ degradation. This increase of Aβ oligomers might impact the synaptic plasticity and induce robust synaptic loss in the 3xTg-AD mice [53]. Overall, our data suggest that short-term, complex (multimodal) stress, recapitulating salient features of modern-life conditions, is a key factor that triggers AD pathogenesis and severely affects memory and synaptic plasticity in 3xTg-AD mice.
In agreement with these results, we sought to evaluate if blocking the effects of glucocorticoids could help reduce pathology and cognitive decline in 3xTg-AD mice. With this purpose, we used the glucocorticoid receptor antagonist mifepristone (RU486). Mifepristone treatment leads to robust reductions in Aβ levels and plaques through the induction of a 17 kDa cleavage of AβPP, and reduces tau hyperphosphorylation via reduction in p25 levels [91]. Hence, our results show that compounds targeting the glucocorticoid system could be useful for the treatment of AD. However, further studies will be necessary to determine the long-lasting effect of this short-term multimodal stress event in AD pathogenesis.
Owing to the rapid growth in the number of both diabetic and AD patients, and the current impact of a stressful modern life, identifying the clinical associations between those disorders and elucidating the molecular mechanism that mediate their associations could provide protection from the profound medical and economic impact that AD will have over the ensuing decades.
STEM CELL THERAPY IN AD: BACK TO THE FUTURE
The timing for the development of therapeutic strategies that turn in real opportunities for AD patients is really critical, especially due to the lack of effective drugs to cure AD. Currently there are over 100 trials and about 80 drugs in the pipeline, and 99.6% of clinical trials have failed to translate into approved treatments [92, 93]. These disappointing results have encouraged an increased focus on the development of alternative novel and innovative methods. Over the past decade, the potential use of stem cells to treat neurodegenerative diseases, such as AD, Parkinson’s disease, and amyotrophic lateral sclerosis have received more attention because of its promising capacity as a regenerative and replacement therapy. With these lines, multiple different studies have shown that using murine neural stem cells have provided compelling evidence of their beneficial effects in motor and cognitive function after different models of brain injuries [94–96], thus the use of stem cell therapy may be a potential treatment for neurodegenerative diseases such as AD [97].
We have conducted pioneer preclinical studies in the 3xTg-AD mice, which develop amyloid plaques, tangles, and important synaptic and cognitive deficits [97], to determine whether neuronal stem cells (NSC) transplantation may offer symptomatic or disease-modifying effects in AD (Fig. 5). Our laboratory demonstrated for the first time that bilateral transplantation of mouse NSC in aged 3xTg-AD mice restored cognitive and synaptic deficits without modifying either plaques or tangle pathology. Among the possible molecular mechanisms underlying these benefits, we found that NSCs produces high levels of brain derived neurotrophic factor (BDNF) and a reduction of BDNF via shRNA-mediated mechanism prevent the cognitive benefit and reduces the effect in the synaptic density [98]. Similar findings were observed in a following study using a different AD transgenic model, the AβPP/PS. In this model, the restoration of both cognitive and synaptic deficits was associated with elevated levels of BDNF and its receptor TrkB. Interestingly, they also found that NSCs treatment did not affect Aβ pathology in APP/PS1 mice [99]. Therefore, these compelling preclinical findings suggest that this therapeutic approach may provide important benefits in patients with advanced existing pathology via improving multiple cognitive-related proteins.
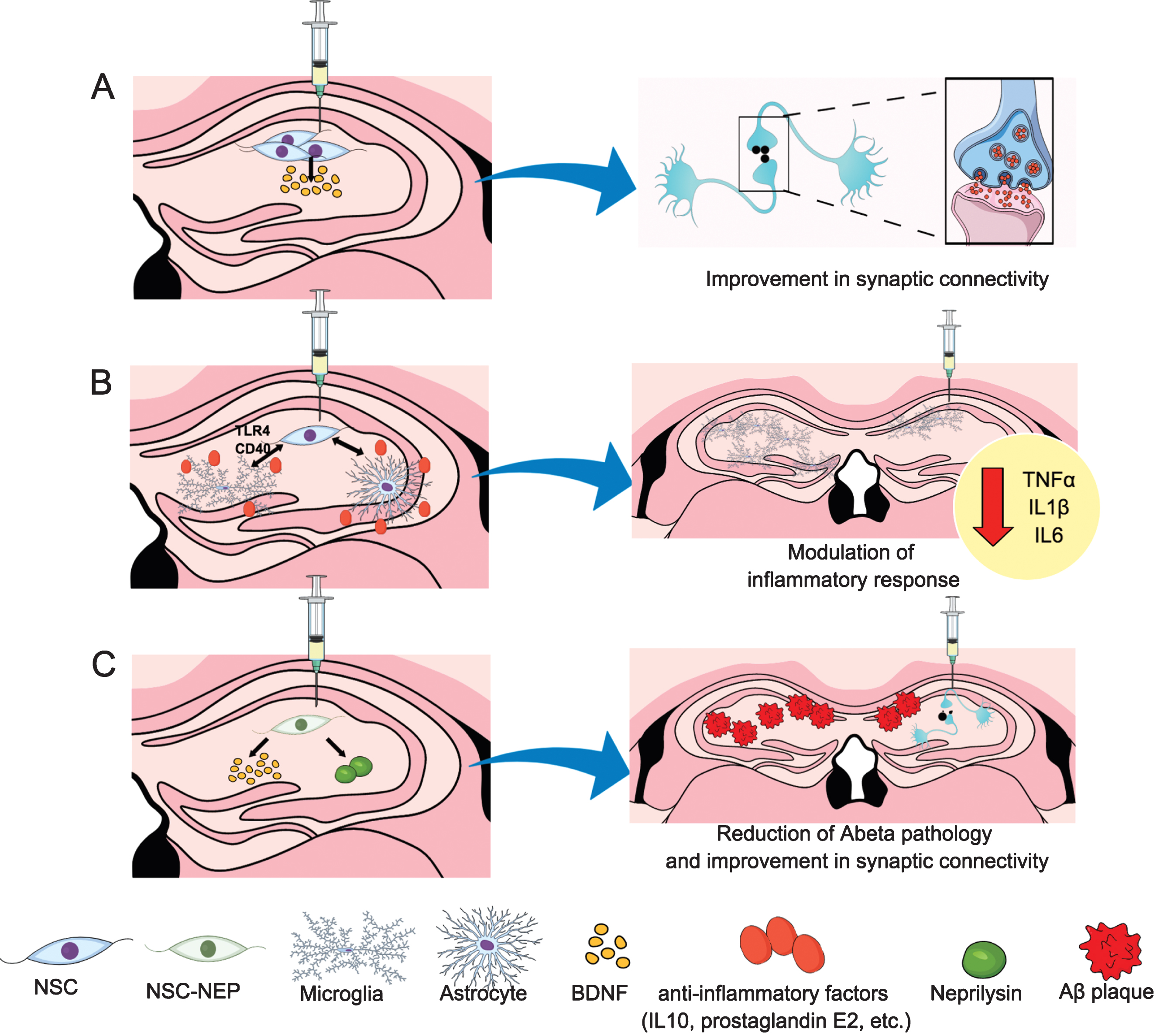
Underlying mechanisms to potential stem cells therapeutic effects. A) Hippocampal neural stem cells injection lead to an increase in BDNF production and a restoration of cognitive and synaptic deficits in 3xTg-AD mice. B) Stem cells can exhibit anti-inflammatory properties interacting with microglia and astrocytes. Among these effects, NSC might reduce microgliosis and the expression of proinflammatory cytokines such as TNFα, IL1β, or IL6, through CD40 or toll-like receptor 4 (TLR4) signaling pathways. C) The hippocampal injection of NSCs delivering neprilysin lead to a reduction in Aβ pathology in addition to the improvement in synaptic connectivity described in A. NSC, neural stem cell; BDNF, brain-derived neurotrophic factor; CD, cluster of differentiation; TNFα, tumor necrosis factor alpha.
However, for a successfully transition of stem cell-based approach into a clinical application, a suitable human stem cell line is necessary to be identified and tested in preclinical AD models in order to assess its efficacy and safety. Along with this idea we have used a human CNS stem cell line (HuCNS-SC) derived from fetal brain tissue to determine whether cognitive impairment could be restored in two relevant models of AD that exhibit either Aβ and tau pathology (3xTg-AD) and extensive neuronal loss (CaM/Tet-DTA). Our study demonstrated a robust therapeutic efficacy of clinically relevant human CNS stem cells in these two complementary models of AD [100]. Specifically, we observed that HuCNS-SC cells recover the cognitive function in both 3xTg-AD and CaM/Tet-DTA models via improving the synaptic connectivity as evidenced by an increase of synaptic levels and growth-associated proteins. Interestingly, our study also revealed that HuCNS-SC transplantation has no effect on Aβ and tau pathology suggesting that the mechanism of action occurs downstream from these pathologies and probably in a similar way to our previous study by using allogeneic murine NSCs, since HuCNS-SC also produces high levels of the neurotrophin BDNF [98]. Overall, our findings suggest that the mechanisms by which NSCs treatment improve AD cognitive symptoms is mediated via neuroprotection and trophic support rather than neuronal replacement, although we cannot discard that other possible mechanisms can take place. For example, certain stem cell population exhibits robust anti-inflammatory properties. In particular, several studies have shown important anti- inflammatory effect of mesenchymal stem cells through the production of anti- inflammatory mediators such as interleukin-10 and prostaglandin E2, or via stimulation of microglial phagocytosis or microglia production of the Aβ-degrading enzyme neprilysin and also by modulation of CD40 signaling [101–105]. Likewise, the effect of NSC in the immune system is currently under intensive research and new evidence suggests that NSCs could reduce microgliosis and the expression of proinflammatory cytokines such as tumor necrosis factor-α [106]. Another mechanism is via suppression of glial and TLR4 activation and its downstream signaling pathways [107]. Although these studies suggest an important role of stem cell in the modulation of the inflammatory response further studies remain necessary to determine the molecular mechanisms by which stem cell transplantation modulate inflammation in AD pathology. Moreover, another aspect to clarify is to determine if stem cell transplantation alters inflammation directly or simply as a result of tissue injury or xenotransplantation-associated artifacts.
Previously, we have indicated that NSCs can improve cognitive defects in an AD preclinical model through the improvement of synaptic connectivity, although they appear to have no effect on Aβ or tau pathology [98, 100]. Given the complex nature of this disease and the multiple pathways and regions affected, a single small molecule approach may not provide substantial benefit, and the NSC benefits may loss efficacy as pathology continues to develop. Therefore, a combinatory intervention may be a more realistic approach to treat AD patients. For example, supplementing NSC transplantation with Aβ and/or tau-targeting therapies could provide additional long-term benefits. In addition, NSCs could themselves be used to deliver therapeutic proteins due to its capacity to migrate throughout the brain and localize to areas of brain pathology [96, 108]. In this regard, we have tested whether NSCs that deliver disease-modifying proteins such as the Aβ-degrading enzyme, neprilysin (NEP) could provide more effective means. Our findings critically demonstrated that sNEP-expressing NSCs survive for a long period of time and secrete sNEP leading to a markedly reduction of Aβ pathology and enhancing the synaptic connectivity in two transgenic AD models (3xTgAD and Thy1-APP transgenic mice) [109]. Thus, sNEP-expressing NSCs represent a promising therapeutic approach that combines the neurotrophic-mediated benefits of stem cell transplantation with the widespread delivery of a disease-modifying protein and further studies will be needed to determine whether such approach can be translated to an eventual clinical application.
CONCLUDING REMARKS
We discussed in this chapter findings from our laboratory that illustrate critical factors to initiate AD pathology, co-morbidities that contribute to disease progression and cognitive decline, and potential cell-based treatments. The majority of our understanding on AD mechanisms has come from transgenic mice such as the 3xTg-AD model; however, improved models should be created, especially focusing on sporadic AD, in order to maximize the discovery and development of new therapies. Now more than ever, it is crucial to understand the exact pathological mechanisms of disease progression, with the broader purpose of sharply reducing the number of people suffering and dying from AD.
Footnotes
ACKNOWLEDGMENTS
The authors are supported by grants from Alzheimer’s Association NIRG-15-363477 (DBV) and #AARF-16-440760 (SF), The Larry Hillblom Foundation #2013-A-016-FEL (DBV) and 2016-A-016-FEL (ACM), the National Institute of Health (NIH) NIH/NIA AG027544, AG00538, and AG054884 (FML), and BrightFocus Foundation grant A2015535S (FML). The authors would like to acknowledge the use of Mind the Graph® for the design of the figures.