
Abstract
Tauopathy is characterized by the fibrillar tau accumulation in neurons and glial cells. In order to advance our understanding of the causative mechanisms of tauopathy, neuroinflammation, which has been suggested to play important roles in disease progression, will require particular attention. Neuroinflammation is characterized predominantly by microglial activation. At present, it is still under debate whether microglial activation is a cause or a result of neurodegeneration. To search for a temporal relationship between neurodegeneration and neuroinflammation, our group demonstrated that in vivo imaging (e.g., tau-PET, TSPO-PET, and volumetric MRI) of tauopathy mice strongly supports the evidence of microglial activation along with both pathological tau accumulation and brain atrophy. Both in vivo imaging and histochemical analysis confirmed that microglial TSPO accumulation was the late event during the pathogenesis of tauopathy. On the other hand, it is known that purinergic receptor P2Y12 as a marker of homeostatic microglia cells was reduced at an early stage of disease progression. In this review, we will introduce a phenotypic change of microglia in a mouse model of tauopathy and propose novel approaches to the establishment of imaging biomarkers, thereby targeting the early diagnosis of tauopathy.
INTRODUCTION
Neurofibrillary tangles (NFTs) composed of hyperphosphorylated tau protein are a primary neuropathological feature of a number of neurodegenerative diseases, collectively termed tauopathies, including Alzheimer’s disease (AD), progressive supranuclear palsy, corticobasal degeneration, Pick’s disease, and familial frontotemporal lobar degeneration (FTLD) with underlying tau pathology (FTLD-tau) (reviewed in [1]). Although tau pathology was initially dismissed as a secondary event in AD that was not integral to the neurodegenerative process, the discovery of tau mutations associated with the tauopathy FTLD-tau demonstrated that tau dysfunction could directly result in neurodegeneration. However, we are still lacking understanding of the potential mechanisms underlying the cause of the diseases.
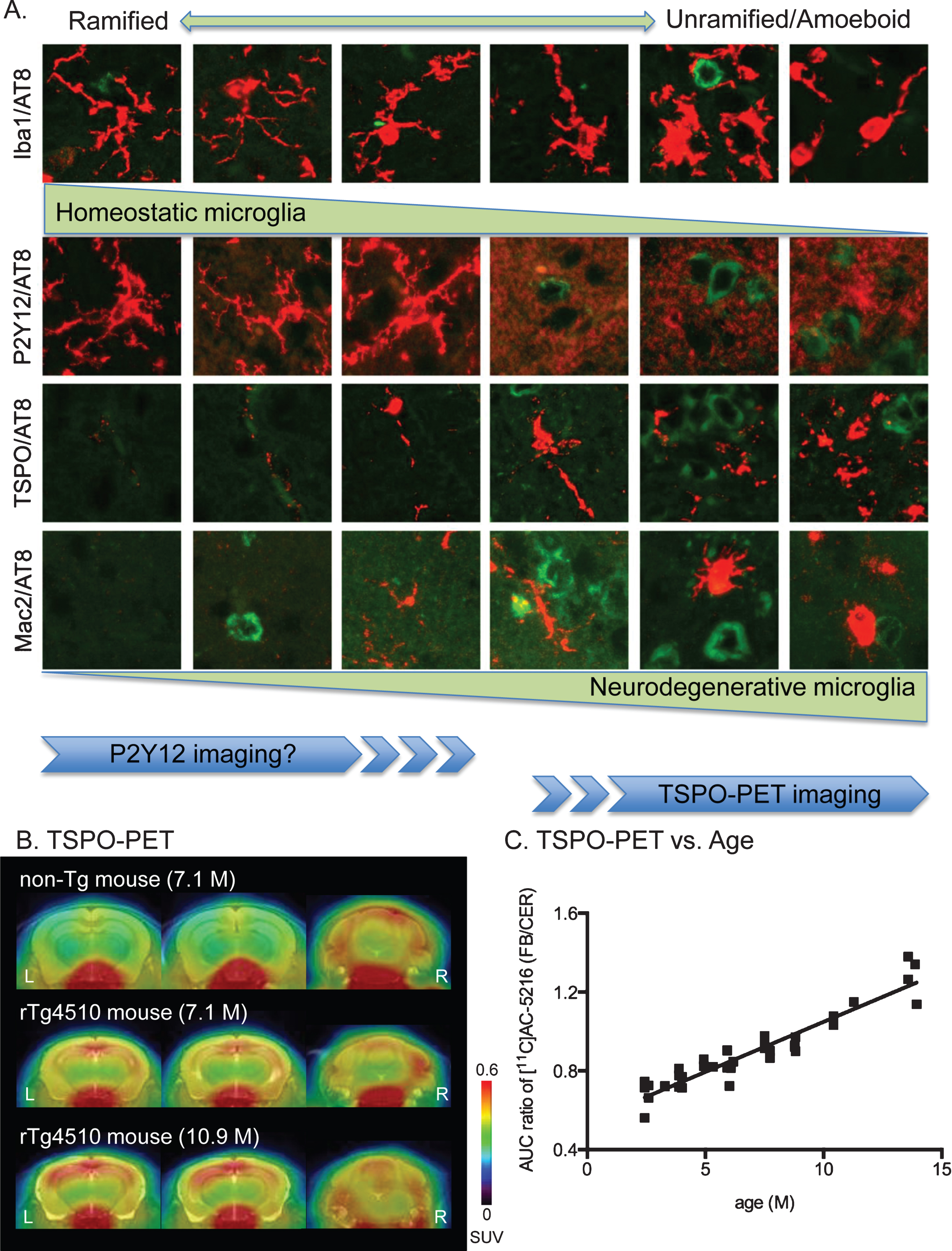
Morphological phenotypes and markers of microglia. A) Representative images of microglial morphologies were aligned from ramified shapes (left) to unramified/amoeboid shapes (right). Each morphology was labeled by the microglial markers Iba1 (rabbit polyclonal, Wako), P2Y12 (rabbit polyclonal), TSPO (rabbit monoclonal, Abcam), and Mac-2 (rat monoclonal, Cedariane) antibodies. Images were double-labeled with microglial marker (red) and AT8 (green). P2Y12 imaging and TSPO-PET imaging are anticipated to visualize distinct morphologies. B) Representative TSPO-PET imaging with [11C]AC-5216 radiotracer in mouse brains. Images of [11C]AC-5216 signals in brains of 7.1-month-old non-tg (top), 7.1-month-old rTg4510 (middle), and 10.9-month-old rTg4510 (bottom) mice were generated by averaged dynamic scan data. The images showed dorsal hippocampal level of coronal (left), ventral hippocampal level of coronal (middle) and cerebellum level of coronal (right) slices. SUV (standardized uptake value) was calculated by injected dose per tissue volume x body weight. C) Scatterplot of AUC (area under the curve of the time-activity curve) ratio (forebrain to cerebellum) for [11C]AC-5216 signals against age (months) of rTg4510 mice (n = 40).
For the purpose of modeling human tauopathy by showing prominent intracellular deposition of tau protein and associated neuronal loss, several transgenic mouse lines expressing FTLD-linked mutant tau have been developed [2]. Among them, the rTg4510 mouse line is one of the popular tauopathy models presenting features of age-dependent neuropathology as well as neurodegeneration both induced by the overexpression of P301L mutated human tau in cerebral cortex and hippocampus [3]. This mouse line is well studied not only for its application to cognitive impairment [3–5] but also for the advancement of tau positron emission tomography (PET) imaging [6, 7]. Especially, in vivo imaging studies have presented a new avenue for investigating causal mechanisms of neurodegenerative diseases via the monitoring of real-time brain functional changes.
Inflammation is considered a key factor in regulating both amyloid and tau pathologies, based on the fact that activated astrocytes and microglia are well associated with these pathological hallmarks (reviewed in [8]). Increasing numbers of studies have shown that microglia could become a chronic source of multiple neurotoxic factors (e.g., tumor necrosis factor-α, nitric oxide, interleukin-1β (IL-1β), and reactive oxygen species) [9]. Notably, it was suggested that microglial activation and resulting secretion of IL-1β triggered exacerbation of tau pathology [10–12]. A longitudinal cohort study (45–69 years old at the start of cognitive testing) showed that IL-6, a major proinflammatory cytokine [13], elevated in the sera of cognitively declined participants [14]. This also suggested that peripheral inflammation could contribute to neuronal damage. Furthermore, the immunosuppressive drug FK506 has been shown to attenuate the taupathology and increase the lifespan of the tau model PS19 mouse line, which expresses the P301S mutant human tau under control by the mouse prion promoter [15]. On the other hand, AD treatment trails using nonsteroidal anti-inflammatory drugs (NSAIDs) failed to demonstrate any clear clinical efficacy, suggesting that anti-inflammatory drugs may not be effective for the prevention of AD [16]. Nonetheless, the inflammatory mechanisms during the progression of neurodegenerative diseases are still largely obscure.
To determine whether microglial activation occurs prior to pathological tau accumulation, we have recently demonstrated longitudinal monitoring of both in vivo tau pathology and the mitochondrial 18-kDa translocator protein (TSPO), as a marker of microglial activation, in a tauopathy mouse model rTg4510 using small-animal PET imaging [7]. Our data showed age-dependent TSPO accumulation along with both pathological tau accumulation and brain atrophy. The rising phase of TSPO accumulation was relatively later than that of tau accumulation, suggesting that microglial activation might be downstream of the formation of pathological tau aggregates. However, the exact activated microglia state (e.g., change of cellular morphology, surface phenotype, secretory mediators, and proliferative responses (reviewed in [17]) has not been clearly understood. Here, based on our current studies of rTg4510 mice [7], we will introduce the temporal change of microglial phenotypes during the development of tauopathy. Furthermore, we will discuss the possible implications of in vivo imaging technologies for evaluating disease progression.
ACTIVATED MICROGLIA IN rTg4510 MOUSE BRAINS
The stages of microglial activation were defined based on morphological, molecular, and functional characteristics. To investigate microglial morphology, Iba1 (ionized calcium binding adaptor molecule 1) is the most reliable marker because it is expressed in microglia throughout various morphological states and is upregulated in activated microglia [18, 19]. As can be seen in the panels of microglial morphology from rTg4510 mouse brains, Iba1 immunostaining showed all types of microglial morphology (Fig. 1A). We recently demonstrated that Iba1 staining on rTg4510 sections showed age-dependent increases of unramified microglial cells in the cerebral cortex and hippocampus [7]. Double-labeling with Iba1 and TSPO antibodies further confirmed the increased activated microglia in 6-8-month-old rTg4510 mice [7]. This change was strongly associated with both pathological tau deposition and brain atrophy [7]. From the aspect of morphological phenotypes, microglia can transform into unramified and amoeboid shapes under pathological condition. Since Mac-2, a member of the galectin family of β-galactoside binding lectins, serves as a marker of a subtype of activated microglia [20], functional phenotypes of activated microglia can be examined by the immunoreactivity of Mac-2 antibody (Fig. 1A). In the aged rTg4510 mouse cerebral cortex, a number of roundish/oval microglia were labeled by Mac-2 antibody (Maeda et al. manuscript in preparation). In addition to the immunohistochemical observations, TSPO-PET imaging in live rTg4510 mice demonstrated age-dependent TSPO accumulation in the cerebral cortex of rTg4510 mice (Fig. 1B, C; PET imaging data and experimental procedures were also reported in [7]). Thus, the linkage between tau pathology and microglial activation was clearly confirmed by our multifaceted studies.
MICROGLIA STATUS IN EARLY STAGE OF TAUOPATHY
Microglial morphologies from ramified to unramified and amoeboid shapes were generally identified by Iba1 immunoreactivity (Fig. 1A). However, Iba1 immunolabeling may not be especially valuable for discriminating functional phenotypes, as this protein is expressed in most types of microglia as well as recruited monocytes [18]. As previously reported, the metabotropic purinergic receptor P2Y12 is known as a selective marker for the ramified phase of microglia (Fig. 1A) [21–23]. Microglia in P2Y12-/- mice showed dysfunction of directional branch extension toward sites of central nervous system (CNS) injury [21]. P2Y12 expression was dramatically reduced after microglial activation [21]. Extensive loss of P2Y12 immunoreactivity was observed in active cortical lesions of human multiple sclerosis (MS) [22, 24]. Similar to the result in MS, we observed that both AD and tauopathy mice had decreased P2Y12 receptor levels in brain regions with tau pathology (Maeda et al. manuscript in preparation). Notably, a reduction of P2Y12-positive microglia in the cerebral cortex and hippocampus regions occurred in young rTg4510 mice prior to pathological tau accumulation (Maeda et al. manuscript in preparation). P2Y12 and several other genes were recently identified as unique microglial genes showing homeostatic microglia phenotypes in mouse brain [25]. Therefore, these results indicate that microglia may lose their homeostatic molecular signature and functions before tangle formation. The age-dependent increase of Iba1 immunoreactivity observed in rTg4510 mice may also suggest the conversion from resting phase to active phase of microglial function at early stage of neurodegenerative disease [7]. Nevertheless, further investigations with identification of microglial molecular signatures (microglial gene and microRNA signatures in murine CNS-derived adult microglia were described in [25]) in rTg4510 mice during disease progression will be needed.

Characteristics of current tau-PET imaging. A) Process of pathological tau aggregations was determined by conformation, phosphorylation and filamentous structures. TOC1 (tau oligomer specific) antibody and AT8 antibody (mouse monoclonal, Thermo Fisher Scientific) can recognize pre-fibrillar oligomeric tau inclusions, whereas Thioflavin S and Gallyas silver label tangle-shaped tau inclusions. Current tau PET tracers principally recognize filamentous tau aggregates. B) Representative tau-PET imaging with [11C]PBB3 radiotracer in mouse brains. Images of [11C]PBB3 signals in brains of 2.3-month-old non-tg (top left), 7.1-month-old non-tg (bottom left), 2.3-month-old rTg4510 (top right), and 7.1-month-old rTg4510 (bottom right) mice were generated by averaged dynamic scan data. The images showed dorsal hippocampal level of coronal (left), ventral hippocampal level of coronal (middle), and cerebellum level of coronal (right) slices. SUV was calculated by injected dose per tissue volume x body weight. C) Scatterplot of AUC ratio (forebrain to cerebellum) for [11C]PBB3 signals against age (months) of rTg4510 mice (n = 40). D) Scatterplot of AUC ratio (forebrain to cerebellum) for [11C]PBB3 signals against AUC ratio (forebrain to cerebellum) for [11C]AC-5216 signals. Spearman’s correlation analysis showed significant correlation (p < 0.05).
P2Y12 RECEPTOR AS AN EARLY-STAGE MARKER FOR MICROGLIAL ACTIVATION
Ramified microglia in the healthy CNS were thought to be immobile due to the low expression of activation-associated molecules [26]. But, these ramified “resting” microglia actually surveyed the environment until any disturbance occurred (reviewed in [27]). Since the housekeeping activity of these surveillant microglia is largely unknown, searching for specific markers of surveillant microglia is essential for understanding the resting phase of microglia. Expression of P2Y12 receptor is remarkable in the resting state while it is significantly reduced after microglial activation [21]. As an early-stage marker, P2Y12 may be a feasible marker for identification of resting microglia. In vivo visualization of activated microglia is available with the use of PET tracers of TSPO, although several problems (e.g., non-specific binding, sensitivity for single nucleotide polymorphism) still need to be resolved [28]. Based on our findings, increase of TSPO signal in a tauopathy mouse model was a late event following pathological tau accumulation (Fig. 1C) [7]. Thus, the early phase of microglial activation is hardly detectable by current in vivo imaging techniques (Fig. 1A). The targeting of P2Y12 receptor for visualizing resting microglia is being anticipated. P2Y12 receptor is a potent marker for visualizing in vivo microglia functions by PET imaging, although positron labeled P2Y12 receptor antagonists have not yet been reported. As potential candidates, reversible competitive antagonists (e.g., cangrelor, ticagrelor) have been synthesized [29]. However, due to their lower lipophilicities and higher molecular weights, these molecules seem to have difficulty crossing the blood brain barrier. For future direction, researchers are looking for novel antagonists of P2Y12 receptor with higher lipophilicity and lower molecular weight.
LIMITATIONS OF CURRENT TAU PET IMAGING
In vivo visualization of tau pathology is one of the hot topics for diagnosing neurodegenerative diseases. Recent progress of tau PET imaging has led to successful clinical assessments in patients with tauopathy [30]. Although the off-target binding issue is still under investigation [30], it has become possible to assess the regional distribution and severity of tau pathology. Because current tau PET tracers are designed for targeting filamentous tau aggregates [31–33], these tracers theoretically do not bind with premature tau aggregates (e.g., tau oligomers, pretangles) (Fig. 2A). Our recent study confirmed that micro-PET imaging with [11C]PBB3 (11C-labeled phenyl/pyridinil-butadienyl-benzothiazoles/benzothiazolium 3 [31]) showed an age-dependent increase in [11C]PBB3 signals (Fig. 2B, C) and that [11C]PBB3 signals were positively correlated with TSPO tracer [11C]AC-5216 signals (Fig. 2D; detailed findings were presented in [7]). Since the increase in [11C]PBB3 signals reached a plateau at age 7 months (Fig. 2C), its significant correlation with [11C]AC-5216 disappeared after age 7 months. Notably, TSPO levels were a better indicator for the severity of disease progression at the late stage of tauopathy. On the other hand, our previous finding showed that intracellular tau accumulation labeled by TOC1 antibody (tau oligomer-specific antibody [34]) appeared as early as 1.5 months of age [35]. As the [11C]PBB3 signal increases at 6 months of age, this tracer may not be able to detect tau oligomers (Fig. 2A). Since an early diagnosis of tauopathy is essential, novel tools for detecting pre-fibrillar oligomeric tau inclusions need to be developed.
CONCLUSION
Current observations indicate that reduction of P2Y12-positive microglia appeared at early stage of human MS and tauopathy [22, 24] (Maeda et al. manuscript in preparation). There is accumulating evidence that microglia play important causative roles in neurodegenerative diseases. Imaging biomarkers for neuroinflammation have been developed, targeting specific proteins in activated microglia. As a most popular target for microglial activation, TSPO-PET imaging is now available in human study [28]. Since TSPO is not only overexpressed in activated microglia but also in reactive astrocytes, visualization of other targets for microglial phenotypes is greatly desired. Combinations of tau and microglia imaging will provide novel approaches for enabling an early differential diagnosis of tauopathy.
Footnotes
ACKNOWLEDGMENTS
This research was supported in part by grants from Grant-in-Aid for Science Research on Innovation Area (“Brain Protein Aging” 26117001 to N.S.) and Scientific Research (C) (15K06793 to N.S.) from the Ministry of Education, Culture, Sports, Science and Technology, Japan, and from the Strategic Research Program for Brain Sciences from Japan Agency for Medical Research and Development, AMED. M.H. holds a patent on compounds including PBB3 (JP 5422782/EP 12 844 742.3).