
Abstract
Background:
Human tauopathy brain injections into the mouse brain induce the development of tau aggregates, which spread to functionally connected brain regions; however, the features of this neurotoxicity remain unclear. One reason may be short observational periods because previous studies mostly used mutated-tau transgenic mice and needed to complete the study before these mice developed neurofibrillary tangles.
Objective:
To examine whether long-term incubation of Alzheimer’s disease (AD) brain in the mouse brain cause functional decline.
Methods:
We herein used Tg601 mice, which overexpress wild-type human tau, and non-transgenic littermates (NTg) and injected an insoluble fraction of the AD brain into the unilateral hippocampus.
Results:
After a long-term (17–19 months) post-injection, mice exhibited learning deficits detected by the Barnes maze test. Aggregated tau pathology in the bilateral hippocampus was more prominent in Tg601 mice than in NTg mice. No significant changes were observed in the number of Neu-N positive cells or astrocytes in the hippocampus, whereas that of Iba-I-positive microglia increased after the AD brain injection.
Conclusion:
These results potentially implicate tau propagation in functional decline and indicate that long-term changes in non-mutated tau mice may reflect human pathological conditions.
INTRODUCTION
Alzheimer’s disease (AD) is a neurocognitive disorder that is typically characterized by impaired short-term memory at onset and involvement in other cognitive domains at later stages. In accordance with this symptomatic development, neurofibrillary tangles (NFTs) appear in a progressive and stereotypical manner. The distribution of NFTs is generally restricted to the transentorhinal and entorhinal regions (Braak stages I-II), but extend into the limbic (stages III-IV) and neocortices (stages V-VI) [1]. In AD, positron emission tomography targeting the tau protein confirmed the stereotypical progression patterns of neuropathological tau corresponding to Braak’s stage [2–4] and revealed a relationship with cognitive impairment in a region-specific manner [5]. Consistent with its progressive appearance in AD, deposited tau protein may exhibit prion-like self-propagation and spreading in experimental settings [6].
Tau aggregates injected into the unilateral hippocampus and the overlying cerebral cortex of transgenic (Tg) mice or non-transgenic (NTg) mouse brains induced tau pathology via connected networks in the contralateral hippocampus, fimbria, fornix, lateral septal nucleus, mammillary area, entorhinal cortex, and locus coeruleus [7–9]. However, only a few behavior analyses have been conducted to date and the findings obtained have been inconsistent. Extracts from the human AD brain 3 to 11 months post-injection into the hippocampus or cortex of NTg mice, tau Tg mice, or 5×FAD mice did not result in any obvious behavioral deficits in the object memory task, Y maze test, or rotarod test even though these mice showed an argyrophilic tau pathology [10–12]. K18 recombinant fibrils (residues Q244-E372 of the longest human tau isoform) with a P301L mutation 6 months post-injection into the bilateral hippocampus of PS19 mice induced a deficit in object recognition memory, whereas a unilateral injection did not induce any changes [13]. PHF tau extracted from AD brain injection 3 months post-injection into the unilateral hippocampus and overlying cortex of NTg mice showed behavioral deficits in the elevated zero maze and contextual fear conditioning; however, no significant changes were observed in the Y-maze test or open field test [14]. Furthermore, signs of neurodegeneration, such as neuronal loss and gliosis accompanied by tau propagation, were not reported in these mice. ALZ 17 mice overexpressing wild-type human tau injected with the P301S mouse brain 15 months post-injection into the unilateral hippocampus and overlying cortex did not show astrogliosis (GFAP), microgliosis (Iba-1), or neuronal loss [15]. Other mice seeded with the P301S transgenic mouse brain only showed a slight increase in Iba-1-positive microglial proliferation, even in the hippocampus with a robust tau pathology [7]. Although astrogliosis and microgliosis were observed in the area of Tg tau P301L mice injected with K18 fibrils with a P301L mutation, the reaction decreased with time [16]. These findings suggest that the molecular tau species responsible for propagation and neurotoxicity are not identical because mutated tau transgenic mice showed an obvious NFT pathology, neuronal loss and gliosis, or behavioral abnormalities [17, 18]. One contributing factor may be a short treatment period because the majority of studies performed an injection into young tau transgenic mice and completed the observation period before the development of the NFT pathology.
In the present study, we injected the insoluble tau fraction of the human AD brain into the unilateral hippocampus of NTg and Tg 601 mice, which overexpress human wild-type tau and do not develop NFTs at 24 months. After a long-term (17–19 months) incubation time, these mice exhibited obvious learning deficits, as detected by the Barnes maze test. A histochemical analysis showed silver-positive and phosphorylated tau-positive neurons and neuropil threads in the bilateral fimbria, hippocampus, and locus coeruleus that were more prominent in Tg mice than in NTg mice, showing Iba-1-positive microglial proliferation in the hippocampus.
MATERIALS AND METHODS
Mice
NTg and tau Tg (Tg601) mice expressing high levels of wild-type human tau (2N4R) under the control of the calcium/calmodulin-dependent protein kinase IIα (CAMKIIα) promoter were used in the present study [19]. These mice were littermates and had the same genetic background, C57BL/6. All mice used in this study were female. Mice were housed under a 12 h light/dark cycle, with ad libitum access to food and water. All experiments were performed in accordance with the Guidelines for Animal Experiments of Juntendo University (Permit number: 260199). Procedures involving animals and their care conformed to the international guidelines set out in the ‘Principles of Laboratory Animal Care’ (NIH publication no. 85–23, revised 1985).
Preparation of sarkosyl-insoluble tau
Frozen brain tissue samples from an autopsy-confirmed AD patient were obtained from the Juntendo University Hospital brain bank. All experimental procedures for brain autopsy and the use of human brain samples were approved by the Juntendo University School of Medicine Ethics Committee (Approval number: 2012068). Five hundred microliters of 20% sarkosyl solution was added (final 2%), incubated in a water bath at 37°C for 30 min, and sonicated for 15 s. Samples were centrifuged at 20,000×g for 10 min. Supernatants were collected and centrifuged again at 100,000×g at 25°C for 20 min. The resulting pellets were washed with 500μl of saline and centrifuged at 100,000×g for 5 min. Pellets were resuspended with 40–50μl of 30 mM Tris-HCl. For immunoblot analysis, samples were mixed with 5×SDS sample buffer and loaded on 5–20% gradient SDS-PAGE gels (WAKO) and electrophoresed with Tris-glycine buffer system. Proteins in the gels were transferred onto polyvinylidene difluoride membrane (Millipore) and blocked with 3% gelatin. The blots were incubated overnight with a monoclonal tau antibody (clone T46, recognizes 404–441 aa of tau, Thermo Fisher Scientific) in 10% calf serum at an appropriate dilution (1:1000) at room temperature. Membranes were washed with 1×PBS and incubated for 2 h with a biotin-labeled secondary antibody (Vector) at room temperature. Protein bands were visualized using an ABC staining kit (Vector).
Stereotaxic surgery
Mice ranging between 2 to 3 months of age were anesthetized with 10% pentobarbital sodium. Using a Hamilton syringe, the hippocampus (A/P, –2.5 mm from bregma; L, ±2.0 mm; D/V, –2.0 mm) was unilaterally infused with 2.5μl sarkosyl-insoluble tau at a speed of 0.25μl per minute. Two micrograms of total protein was injected into each mice. After the injection, the needle was kept in place for an additional 10 min before gentle withdrawal. The surgical area was cleaned with sterile saline and the incision was sutured using medical adhesive. Mice were monitored until recovery from anesthesia and checked weekly after surgery. Sarkosyl-insoluble tau was injected into 23 Tg601 mice (TgAD) and 23 non-transgenic mice (NTgAD). As a control, the same amount of protein-free phosphate-buffered saline (pH 7.4) was injected into 14 Tg601 mice (TgC) and 12 non-transgenic mice (NTgC).
Behavioral testing
The balance beam, elevated plus maze, Y-maze, and platform recognition tests were evaluated in mice 12 months post-injection. The methods described by Arendash et al. [20] were modified and used to perform these 4 tasks. The methods for the tests are detailed in the Supplementary Material. To assess cognitive deficits in learning and memory, the Barnes maze test was performed at 19 to 21 months of age [21, 22].
Barnes maze
At 17 to 19 months post-injection, the Barnes maze test was performed according to a shortened protocol [21]. This shortened Barnes maze protocol revealed memory deficits at 4 months of age in the triple-transgenic mouse model of AD. The maze was made from a circular, 13 mm-thick, white polyvinyl chloride slab with a diameter of 1220 mm. Twenty holes with a diameter of 50 mm were made on the perimeter at a distance of 25 mm from the edge. This circular platform was then mounted on top of a rotating stool, 1000 mm above the ground. The escape cage was made by using a mouse cage and assembling a platform and ramp 31 mm below the surface of the maze. The outside of the walls of the cage were covered with black to make the inside of the cage dark and, thus, attractive to mice. The maze was placed in the center of a room and 120 W lights were placed on the edges of the room facing towards the ceiling. Eight simple colored-paper shapes (squares, triangles, and circles) were mounted around the room as visual cues, in addition to the asymmetry of the room itself. After testing each mouse, the cleaning of the quadrant of the maze around the target hole was alternated with cleaning of the whole maze using 70% ethanol. The maze was rotated clockwise after every 3 mice to avoid intra-maze odor or visual cues. All sessions were recorded using a CCD camera.
Animals interacted with the Barnes maze in three phases: habituation (1 day), training (2 days, 5 sessions), and probe (1 day). Primary latency was defined as the time to identify the target hole for the first time because mice did not always enter the hole upon first identifying it. Hole search was defined as nose pokes and head deflections over any hole. Primary hole search was defined as the hole search before identifying the target hole for the first time. Parameters were assessed by a blinded observer. During the probe phase, measures of time spent per quadrant and hole search per quadrant were recorded. In these analyses, the maze was divided into quadrants consisting of 5 holes with the target hole in the center of the target quadrant. The other quadrants going clockwise from the target quadrant were labeled positive, opposite, and negative.
Histochemistry
After completing the Barnes maze test, mice were sacrificed at the age of 19 to 21 months. Mice were deeply anesthetized with pentobarbital (50 mg/kg) and then transcardially perfused with 20 ml cold PBS, followed by 20 ml of 4% paraformaldehyde in PBS. The brains of mice were promptly removed and immersion-fixed in the same fixative for 24 h. After paraffin embedding, 6 -μm-thick coronal sections were prepared. In tau and microglial immunolabeling, sections were permeabilized and endogenous peroxidase was quenched by treating paraffin sections with methanol containing 0.3% H2O2. Regarding antigen retrieval, sections were autoclaved in citrate buffer at 121°C for 10 min. Primary antibodies, such as AT8 (specific for tau phosphorylated at Ser202/Thr205, 1:200; Invitrogen) and anti-Iba-1 (specific for microglia, 1:500; Fujifilm Wako Chemicals Japan), were applied at 4°C overnight. Sections were immersed in simple stain MAX PO (Nichirei Biosciences Inc.) at room temperature for 1 hour and visualized by the DAB reaction. Regarding the immunostaining of astrocytes and neurons, sections were heat-treated in Cell Conditioning Solution (CC1) (Ventana Medical Systems) and then incubated with the anti-NeuN antibody (1:400, Millipore) or anti-mouse GFAP antibody (1:400, Thermo Fisher Scientific). As secondary antibodies, biotin-conjugated horse anti-mouse IgG (Vector Laboratories) or biotin-conjugated goat anti-rabbit IgG (Dako) were used. Sections were stained using the iVIEWTM DAB Detection Kit (Ventana) and Hematoxylin Counterstain II (Ventana) in an automated immunostainer (BenchMark™ Ventana).
To evaluate AD patient brain, tau immunostaining was performed using the above protocol. The methods for the Thioflavin S staining are described in the Supplementary Material.
Quantification analysis
The numbers of GFAP-positive astrocytes, Iba-1-positive microglia, and Neu-N-positive neurons were counted using Stereo Investigator software ver. 11.7 (MicroBrightField Japan, Inc., Chiba Japan). The counting frame was set to measure 5–10 cells inside. To measure GFAP-positive astrocytes in the hilus of the hippocampus, cells were counted in a 20×20μm counting frame inside a 100×100μm grid size using a×20 objective. A 30×30μm counting frame inside a 150×150μm grid size was used for Neu-N-positive neurons in the pyramidal cell layer of the hippocampus. Iba-1-positive microglia in the hippocampus were counted in 20×20μm counting frame inside a 300×300μm grid size. AT8-positive threads and silver-positive threads in the fimbria were counted in a 20×20μm counting frame inside a 100×100μm grid size. To quantify AT8-positive areas in the locus coeruleus and dorsal raphe nucleus, ImageJ software (NIH) was used. Three slides for each mice were used.
Statistical analysis
Statistical analyses were performed using JMP (SAS Institute Inc., NC, USA) software. Behavioral tasks, including the balance beam, elevated plus maze, Y-maze, and platform recognition tests, were analyzed using a one-way ANOVA followed by Tukey’s post hoc test. A two-factor factorial ANOVA was used for the Barnes test and histochemical quantification. Kruskal-Wallis test was used to assess microglial proliferation because the data for the NTg groups was not normally distributed. Data were expressed as the mean±standard error of the mean (SEM), and differences with p values less than 0.05 were considered to be significant.
RESULTS
Biochemical and neuropathological characteristics of AD brain
This case was an 84-year-old woman with a 4-year history of progressive mental deterioration [23]. The frontal cortex of this patient showed numerous phosphorylated tau and thioflavin s-positive pathologies (Supplementary Figure 1a, b). Braak NFT staging was stage V. The western blotting of the sarkosyl-insoluble fraction exhibited a typical AD band pattern (Supplementary Figure 1c). 2.5μl sarkosyl-insoluble tau extracted from the frontal cortex of this patient was injected into the unilateral hippocampus.
NTg and Tg mice showed learning deficits 17 to 19 months postinjection of the AD brain into the unilateral hippocampus
To examine whether the AD brain injection into the unilateral hippocampus affected behavioral dysfunction in Tg and NTg mice after injection at 2 to 4 months of age, several tasks were performed (Fig. 1a). Four tests were performed at 12 to 14 months of age (10 to 12 months incubation). Since a histochemical analysis were not performed at these ages, the relationship of the tests and tau pathology was not examined. In the balance beam test, which assesses motor function, the total time spent by the animal on the beam before falling was not significantly different between Tg and NTg mice or between AD brain-injected and PBS-injected control mice (Supplementary Figure 2a). The Y-maze test, evaluating working memory, showed that the AD brain injection did not affect the percent spontaneous alterations (Supplementary Figure 2b). To evaluate anxiety/emotionality, the elevated plus maze test was performed. The AD brain injection did not influence the time spent in the open arms, whereas the effect of the mice genotype was noted (Supplementary Figure 2c), which was consistent with our previous findings [19]. The platform recognition test was conducted to test for the ability to recognize/locate a variably placed platform, and no significant differences were observed between the 4 groups (Supplementary Figure 1d).
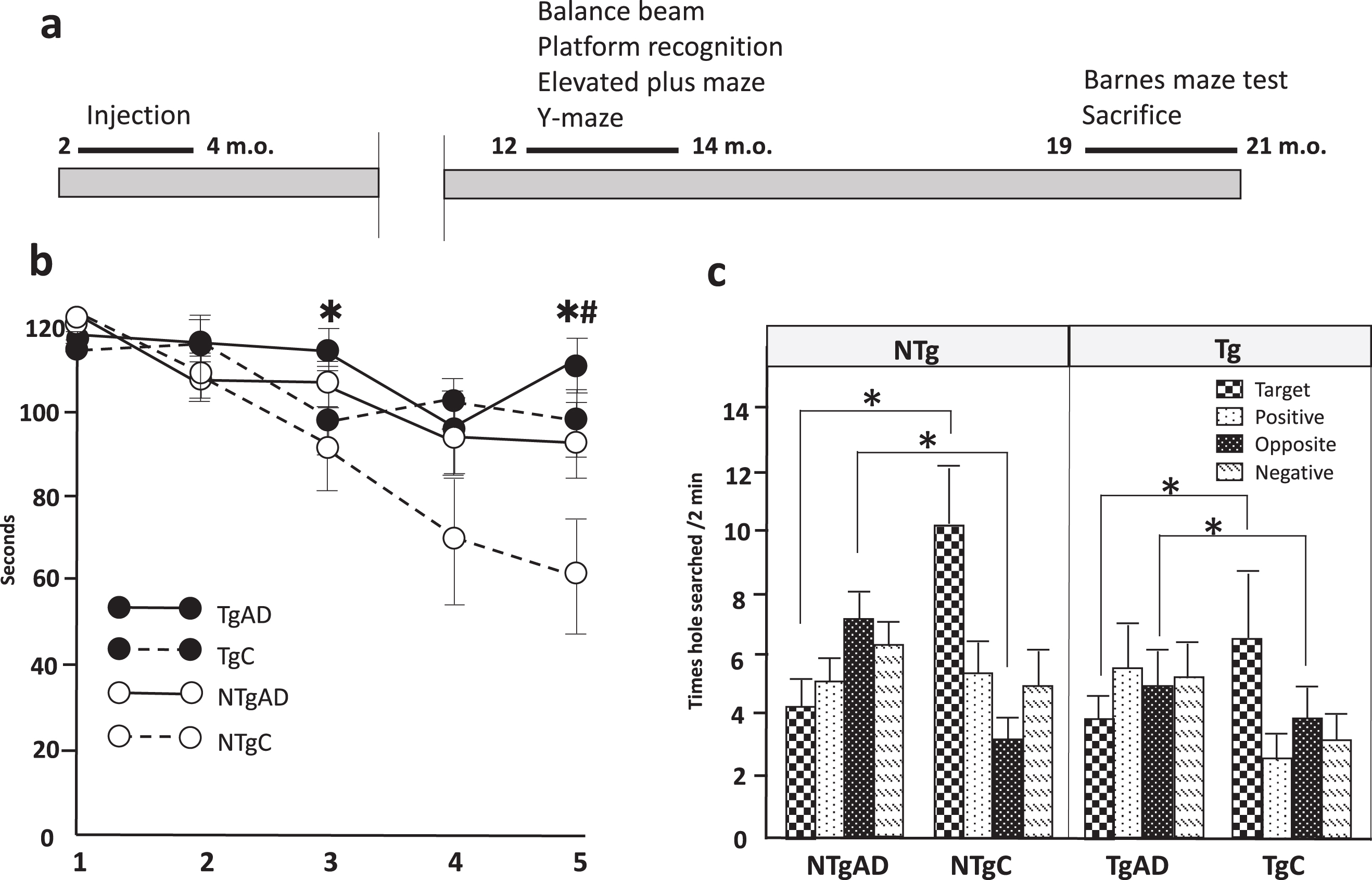
Learning deficits in NTg and Tg mice 17 months after an AD brain injection into the unilateral hippocampus. a) The order and timeline of behavioral tasks evaluated. b) Primary latency, out of 120 s, over 5 training trials for Non-Tg and Tg mice. Two-way factorial ANOVA noted the effects of the AD brain injection were detected at the 3rd and 5th sessions (*). Mouse genotype differences were only significant in the 5th session (#). c) Time holes searched in each of the four quadrants on probe day by NTg and Tg mice. The AD brain injection reduced hole search times in the target quadrant and increased them in the opposite quadrant. Twenty non-transgenic mice injected with the Alzheimer’s disease brain (NTgAD), 18 transgenic mice injected with the Alzheimer’s disease brain (TgAD), 13 transgenic mouse controls injected with phosphate-buffered saline (TgC), and 10 non-transgenic mouse controls injected with phosphate-buffered saline (NTgC) were evaluated. m.o: months old, A two-way factorial ANOVA was used. *, #p < 0.05. * and # indicate the effect of AD brain injection and mice genotype, respectively.
The Barnes maze test was conducted at 19 to 21 months of age (17 to 19 months incubation) to assess memory deficits in AD brain-injected mice (Fig. 2a). The Barnes maze was originally developed by Carol Barnes to overcome the stress induced by swimming in the Morris water maze (MWM) [24]. Similar to the MWM, the Barnes maze allows for the evaluation of spatial reference memory and learning without inducing the stress and anxiety that are commonly observed in the MWM related to floating. A two-way factorial ANOVA for each training session revealed the significant effect of the AD brain injection at the 3rd session (F (3, 61) = 4.1277, p = 0.0469) and 5th session (F [3, 61] = 5.6253, p = 0.021, Fig. 2b). Mouse genotype differences also significantly affected primary latency at the 5th session (F [3, 61] = 8.9882, p = 0.0040); however, the interaction between the mouse genotype and AD brain injection was not significant. These results may be related to those shown in Fig. 1b, namely, Tg mice did not learn during the 5 sessions, indicating that Tg mice at this age already showed learning deficits. In our previous study, 16-month-old Tg mice showed impaired place learning for 10 days in the MWM test [19]. Forty-eight hours after the last training day, the escape cage was removed and mice were given 2 min to explore the maze. During the probing phase, the number of hole search per quadrant were recorded (Fig. 2c). Hole search in the target quadrant significantly decreased after the AD brain injection (F [3, 57] = 9.2362, p = 0.0036), while the AD brain injection increased hole search times in the opposite quadrant (F[3, 58] = 4.9642, p = 0.0298). Mouse genotype differences did not affect hole search times in any quadrants. These results indicate that the AD brain injection into the unilateral hippocampus impaired learning ability in Tg and NTg mice.
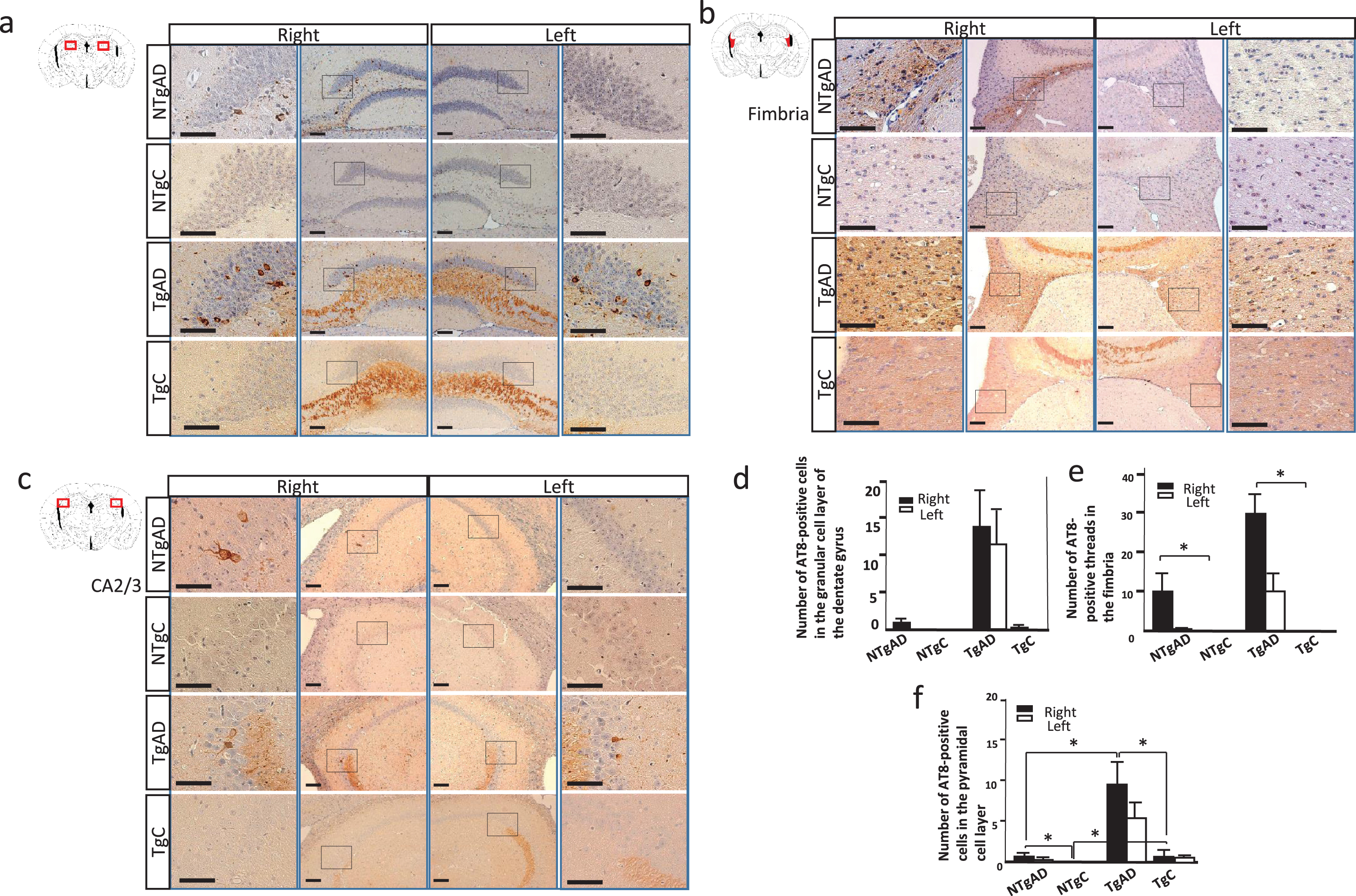
The AD brain-induced hippocampal tau pathology was more prominent in Tg than in NTg mice. a) AT8 immunostaining of the hilus. At 17 to 19 months postinjection (19 to 21 months of age), AT8 immunohistochemistry identified a neuronal pathology in the dentate gyrus in NTg and Tg mice after AD brain injection. The pathology was more prominent in the injected side (right). In Tg mice, mossy fiber was stained probably due to tau overexpression. PBS-treated NTg mice (NTgC) did not show an AT8-positive structure. b) AT8 immunostaining of the fimbria. In AD brain injected NTg and Tg mice, the AT8 antibody stained thread-like structures in the fimbria. c) AT8 immunostaining of the CA2 and CA3 areas. In NTg and Tg mice, AT8-positive neurons were observed in the pyramidal cell layer. Mossy fiber was stained in Tg mice. d) Quantification of AT8-positive cells in the granular cell layer of the dentate gyrus. A two-way factorial ANOVA did not show a statistical significance probably because of high standared deviation in TgAD mice. e) Quantification of AT8-positive threads in the fimbria. A two-way factorial ANOVA of the right fimbria showed significant effect of AD brain injection (F [3, 35] = 9.0027, p = 0.0052); however, there was no significant effect of mice genotype. f) Quantification of AT8-positive cells in the pyramidal cell layer. A two-way factorial ANOVA of the right hippocampus revealed the effects of the AD brain injection (F [3, 39] = 4.6283, p = 0.0382) and mouse genotype (F [3, 39] = 4.5484, p = 0.0398). The interaction between the mouse genotype and AD brain injection was not significant. An analysis of left pyramidal cells showed no significant differences. Scale bars: a–d, 100μm. *p < 0.05. a-c) The lateral panels indicate higher magnifications of the rectangle in the inner panels.
Tau pathology was more prominent in Tg mice than NTg mice 17 to 19 months post injection into the unilateral hippocampus
To assess the distribution of the AD brain injection, an immunohistochemical analysis was performed using the AT8 antibody. An examination conducted 17 to 19 months postinjection (19 to 21 months of age) at Bregma –2.12 revealed AT8-positive pathology in AD brain-injected NTg and Tg mice (Fig. 2). In the dentate gyrus of the molecular layer in the hippocampus, the AD brain injection induced AT8-positive neurons in Tg mice, but only a few positive neurons in NTg mice (Fig. 2a). The lack of a significant difference was attributed to variations in tau pathology among Tg mice (Fig. 2d). In the fimbria, AT8-positive threads were more prominent in Tg mice than in NTg mice (Fig. 2b, e). The number of AT8-positive cells in the pyramidal cell layer was higher in Tg mice than in NTg mice (Fig. 2c, f). The external capsule, and lacunosum moleculare, also showed AT8-positive threads (data not shown). Tau-positive lesions were always more prominent on the infused side (right), but also spread to the contralateral side. PBS-injected NTg mice (NTgC) did not show an AT8-positive pathology in the hippocampus (Fig. 2); however, in Tg mice (TgC), the AT8 antibody stained mossy fibers and a few neurons in the CA4 region of the pyramidal cell layer, probably due to the overexpression of human tau in Tg mice unrelated to the AD brain injection (Fig. 2a, c). Other than the hippocampus, the medial septum showed AT8-positve neurons and threads in one NTg mouse (Supplementary Figure 5); however, Tg mice did not show obvious AT8-positive staining. In the brain stem, the locus coeruleus, a major site of synthesis of noradrenaline, and the dorsal raphe nucleus, a serotonergic nucleus, have been reported to show hyperphosphorylated tau inclusions during AD precortical stages [25, 26]. The locus coeruleus showed AT8-positive tau pathology in NTg and Tg mice, whereas no significant difference was observed between Tg and NTg mice, partly because of the high standard deviation (Supplementary Figure 6a-c). The dorsal raphe nucleus also had AT8-positive cells after the AD brain injection into NTg and Tg mice (Supplementary Figure 6d-f). The olfactory bulb, caudate putamen (Supplementary Figure 3), amygdala, subiculum, entorhinal cortex, and cerebral cortex (Supplementary Figure 4) did not show obvious AT8-positive propagated lesions.
Gallyas silver staining methods were performed to stain neuropil threads and neurons in the fimbria, external capsule, lacunosum moleculare, pyramidal cell layer, and granular cell layer of the dominantly injected side of the dentate gyrus in both NTg and Tg mice (Supplementary Figure 7a-d). The number of silver-positive threads in the fimbria was higher with the AD seed injection than with the control in both NTg and Tg mice (Supplementary Figure 7e) however, no significant differences were observed in silver-positive cells in the granular cell layer of the dentate gyrus or in the pyramidal cell layer partly because of the high standard deviation (Supplementary Figure 8).
AD brain injection induced a microglial response
To investigate the involvement of brain inflammatory cells after AD brain seeding, we examined microglial proliferation using the microglial marker, Ca2 +-binding adaptor molecule-1 (Iba1). The AD brain injection increased the number of Iba-1-positive microglia in the bilateral hippocampus of both NTg and Tg mice (Fig. 3a–e). No significant difference was noted between Tg and NTg mice. GFAP-positive astrocytes were counted in the bilateral hilus of the hippocampus, and the results obtained showed no significant increase after the AD brain injection (Supplementary Figure 9). To assess neurodegeneration, NeuN-positive neurons were counted in the pyramidal cell layer of the bilateral hippocampus; however, the AD brain injection did not exert any significant effects (Supplementary Figure 10).

Microglial proliferation in the hippocampus after the AD brain injection. The Iba-1 antibody immunostained microglial cells and processes in the hippocampus in AD brain-treated NTg (a) and Tg (b) mice, and PBS-treated Non-Tg (c) and Tg (d) mice. Kruskal-Wallis tests showed the effects of the AD brain injection (p = 0.0059) on the injected side (right) and (p = 0.0476) contralateral side (left); however, there was no effect of genotype. *p < 0.05, Bars, SE Scale bar: a, b, d, e, 50μm.
DISCUSSION
We demonstrated learning deficits in NTg and Tg601 mice after injection of insoluble tau from AD brain 17–19 months postinjection (19 to 21 months of age). Although tau-positive structures were more prominent in Tg601 mice than in NTg mice, only a slight difference was observed in learning deficits between them. This is consistent with Braak’s NFT staging in that disease severity and progression basically depends on extent of NFTs [1]. In the hippocampus, Iba-1-positive microglial proliferation was noted in the AD brain-injected groups.
We demonstrated learning deficits and microglial proliferation in very old NTg mice (19–21 months of age) after a unilateral hippocampal injection of the AD brain despite only slight tau pathological changes where AT8-positive neurons and thread-like structures are mainly limited to the ipsilateral fimbria, and CA4 regions in the hippocampus (Fig. 2). In a previous study, Tg mice, such as Tau P301S transgenic mice, exhibited a robust tau pathology after a short-term (6 months) incubation; however, behavioral abnormality were not reported [13]. Young NTg mice (5 months old) and a short-term incubation (3 months) also failed to develop learning deficits [14]. Therefore, even in NTg mice, a long-term incubation may have contributed to the development of learning deficits in the present study.
The increase observed in the number of microglia, not astrocytes, in the hippocampus, may play a role in the development of behavioral abnormalities. In an in vitro system, tau aggregates directly activated microglia and neurons with tau filaments exposed phosphatidylserines, which act as an “eat-me” signal to microglia [27]. Aggregated tau seeds activated NLRP-3-ASC-inflammasome in primary microglia and ASC inflammasome influenced tau pathology in mice [28]. The adeno-associated virus vector delivery of tau protein to young (3-month-old) and aged (20-month-old) rats led to more severe microgliosis and a greater behavioral deficit in the aged groups [29]. A genome-wide analysis of mouse microglia from discrete brain regions revealed that hippocampal microglia maintained a more immune-alert state accompanied by the stronger expression of energy metabolism genes than the striatum and cortex; however, aging from 12 to 22 months reduced this distinction [30]. The senescence of hippocampal microglia may impair their physiological ability to maintain synaptic functions, thereby contributing to learning deficits in NTg mice in spite of the weak hippocampal tau pathology.
Sex differences in microglial function may also contribute to the development of learning deficits and microglial proliferation because we only used female mice in the present study. Soma size and cell density in the hippocampus were higher in male mice than in female mice [31]. Hippocampal male microglia showed the stronger expression of major histocompatibility complex (MHC)I and MHCII than female microglia, suggesting that male microglia are more responsive to stimuli. After an immune challenge with lipopolysaccharide, male microglia showed a stronger immune response [32]. Therefore, female microglia may be more vulnerable to oxidative stress via immune responses, leading to hippocampal dysfunction.
Tg601 mice overexpress wild-type human tau under the CAMK-II promoter and, thus, the amount of soluble human tau is 5.5-fold higher than endogenous mouse tau [19]. In Tg mice, AD seeds recruited higher amounts of soluble tau, leading to larger amounts of tau aggregates than in NTg mice (Fig. 2); however, the regional distribution of tau aggregates was the same. Only a slight difference was observed in the severity of learning deficits between Tg and NTg mice, and neurodegeneration, such as neuronal loss and astrogliosis, was not detected in either mouse. These results suggest that phosphorylated or aggregated tau consisting of wild-type tau is not a toxic substance because mutated tau transgenic mice demonstrated learning deficits with neuronal loss and gliosis [33, 34]. AD seed-injected Tg and NTg mice showed tau pathology in the locus coeruleus and median raphe, which were axon-connected to the hippocampus at a relatively long distance. These findings indicate that a disturbed axonal flow with tau protein transmission rather than cell toxicity contributes to learning deficits. Additionally, wild-type tau-recruited mice by AD seed injections may represent a closer model to the human pathological condition than transgenic tau models overexpressing mutant tau.
Tau aggregates in our NTg mice were mainly restricted to the hippocampus and brain stem at a very old age showing only memory impairment with the preservation of other cognitive domains. This mouse phenotype shares several features with a recently recognized common tauopathy, namely, primary age-related tauopathy (PART). PART is neuropathologically characterized by a mainly neurofibrillary pathology in the hippocampus and entorhinal cortex, basal forebrain and brain stem but not extending into the neocortex with a minimal to absent amyloid-β pathology [35, 36]. This tauopathy is commonly observed in 21.7% of the very elderly (85 years and older) and cognitive domains declined in PART patients are simpler than AD [37].
One potential limitation of this study is that only one patient brain was injected as seeding. Tau seeding activity measured by cell-based bioactivity assays in 32 patients with AD varied and correlated with the amount of spread in the mice and the aggressiveness of the clinical disease [38]. However, the other study of NTg mice using tau extracts obtained from 3 different AD cases resulted in a similar neuroanatomic distribution and a similar amount of tau pathology [8]. Our cell-based study also showed a similar amount of insoluble tau after 3 different AD brain seed additions (Supplementary Table 1, Supplementary Figure 11a, b) [39]. Another potential limitation of this study is that an injection of PBS was used for control groups. Thus far, the control experiments have been performed using brain extracts prepared from non-demented control cases [40] or by samples immunodepleted for tau [15]. In this respect, using 3 control brains, cell based assy was done (Supplementary Figure 11b).
NTg mice and Tg601 mice showed learning deficits and microglial proliferation in the hippocampus 17 to 19 months postinjection (19 to 21 months of age) of the AD seed. The amount of tau aggregates was higher in Tg601 mice than in NTg mice in the hippocampus. A long-term postinjection in non-mutated tau mice may recapitulate the human pathological condition more clearly rather than that in mutated tau mice.
Footnotes
ACKNOWLEDGMENTS
This study was supported by a grant from MEXT KAKENHI (Grant number 18K07456 to YM). We are grateful for the excellent immunohistochemical technique from Akiko Sumii (Department of Neurology, Juntendo University School of Medicine). We thank the members of the Laboratory of Morphology and Image Analysis, Research Support Center, Juntendo University Graduate School of Medicine for their technical assistance with microscopy.