
Abstract
The vascular hypothesis of Alzheimer’s disease (VHAD) was proposed 24 years ago from observations made in our laboratory using aging rats subjected to chronic brain hypoperfusion. In recent years, VHAD has become a mother-lode to numerous neuroimaging studies targeting cerebral hemodynamic changes, particularly brain hypoperfusion in elderly patients at risk of developing Alzheimer’s disease (AD). There is a growing consensus among neuroradiologists that brain hypoperfusion is likely involved in the pathogenesis of AD and that disturbed cerebral blood flow (CBF) can serve as a key biomarker for predicting conversion of mild cognitive impairment to AD. The use of cerebral hypoperfusion as a preclinical predictor of AD is becoming decisive in stratifying low and high risk patients that may develop cognitive decline and for assessing the effectiveness of therapeutic interventions. There is currently an international research drive from neuroimaging groups to seek new perspectives that can broaden our understanding of AD and improve lifestyle. Diverse neuroimaging methods are currently being used to monitor normal and dyscognitive brain activity. Some techniques are very powerful and can detect, diagnose, quantify, prognose, and predict cognitive decline before AD onset, even from a healthy cognitive state. Multimodal imaging offers new insights in the treatment and prevention of cognitive decline during advanced aging and better understanding of the functional and structural organization of the human brain. This review discusses the impact the VHAD and CBF are having on the neuroimaging technology that can usher practical strategies to help prevent AD.
Keywords
INTRODUCTION
It is a chicken or egg question. Which comes first, neurodegenerative changes prior to Alzheimer’s disease onset or chronic cerebral hypoperfusion?
The question is not trivial because the answer likely points to the pathogenesis of Alzheimer’s disease (AD). It is self-evident that knowledge of the cause of AD lays the groundwork for the construction of a plan to prevent this malady. There is therefore a need to sort out what has been done and should be done in the field of AD so previous failures are not repeated and new ideas can move forward. One of these ideas is neuroimaging the human brain to detect hemodynamic patterns that can predict AD.
It is now evident from the expanding literature that major advances in the field of neuroimaging are being reported almost daily and that a major target of this technology are patients with cognitive difficulties or mild cognitive impairment (MCI), a suspected precursor of AD. The main objective of many neuroimaging studies is focusing on regional cerebral hypoperfusion and how the consequential hemodynamic instability can affect cognitive function in the elderly.
This review is not meant to be exhaustive but rather an attempt to take a snapshot of a pathological problem involving declining cerebral blood flow (CBF), advanced aging, cognitive impairment, and cutting edge neuroimaging
Clinical failures that have plagued AD research for the past two decades are discussed in the context of the amyloid cascade hypothesis. Some first-class neuroimaging papers are cited to give the reader a taste of what needs to be done to decipher the why and how of cognitive dysfunction during advanced aging.
CONNECTING THE DOTS IN THE AD PUZZLE
It has been 24 years since we proposed that the neurodegenerative changes seen in AD, occur downstream from the insidious start of chronic brain hypoperfusion [1]. At the time, we believed that AD was a vascular disorder with neurodegenerative consequences, not the other way around, as was widely assumed. Chronic brain hypoperfusion can be described as an unstable hemodynamic activity that can progress in the arterial brain circulation to negatively impact brain cell metabolism and cognitive function.
This beginning of the vascular hypothesis of Alzheimer’s disease (VHAD), as it later became known, gathered some support several years following our 1993 [1] paper with further observations from our laboratory [2–5] and key epidemiological studies, which indicated that two common medical conditions, chronic hypertension and carotid atherosclerosis, appeared to be independently associated with AD [6, 7].
It did not take long to realize that hypertension and carotid atherosclerosis in elderly patients were risk factors to AD. To us, these two risk factors and others discovered soon after, represented the elusive precursors of chronic brain hypoperfusion that we believed initiated AD [4, 8]. Moreover, the correlation of risk factors to AD and their negative effect on cerebral perfusion seemed to reasonably explain the neurometabolic energy crisis that led to cognitive decline and cataclysmic neurodegeneration [4, 5]. But there was still the mystery of how brain hypoperfusion began. Could vascular risk factors play a role in altering cerebral hemodynamics, and if so, what was the mechanism?
After much thought, we concluded that perhaps the progressive decline of cerebral blood flow (CBF) in people at risk of AD, was worsened by vascular risk factors through a phenomenon we dubbed critically attained threshold of cerebral hypoperfusion (CATCH) [8–10]. CATCH can be likened to a slow but progressive decline in neuronal effectiveness that expands irreversibly towards the final stage of non-expansion, when most cognitive skills essentially disappear and the ability to survive vanishes. Mechanistically, CATCH is the beginning of CBF insufficiency that reaches a threshold where cerebral hemodynamic deterioration rises from an imbalance between CBF delivery and neuronal demand and supply dyshomeostasis. Protracted brain hemodynamic deterioration will damage endothelial cells that control CBF reactivity, vessel tone and vessel compliance as well as affect signaling pathways between astrocytes and neurons [11].
A simplified flow chart of how we theorized the cascading neuropathologic steps that begin with normal advanced aging to AD onset is summarized in Fig. 1.
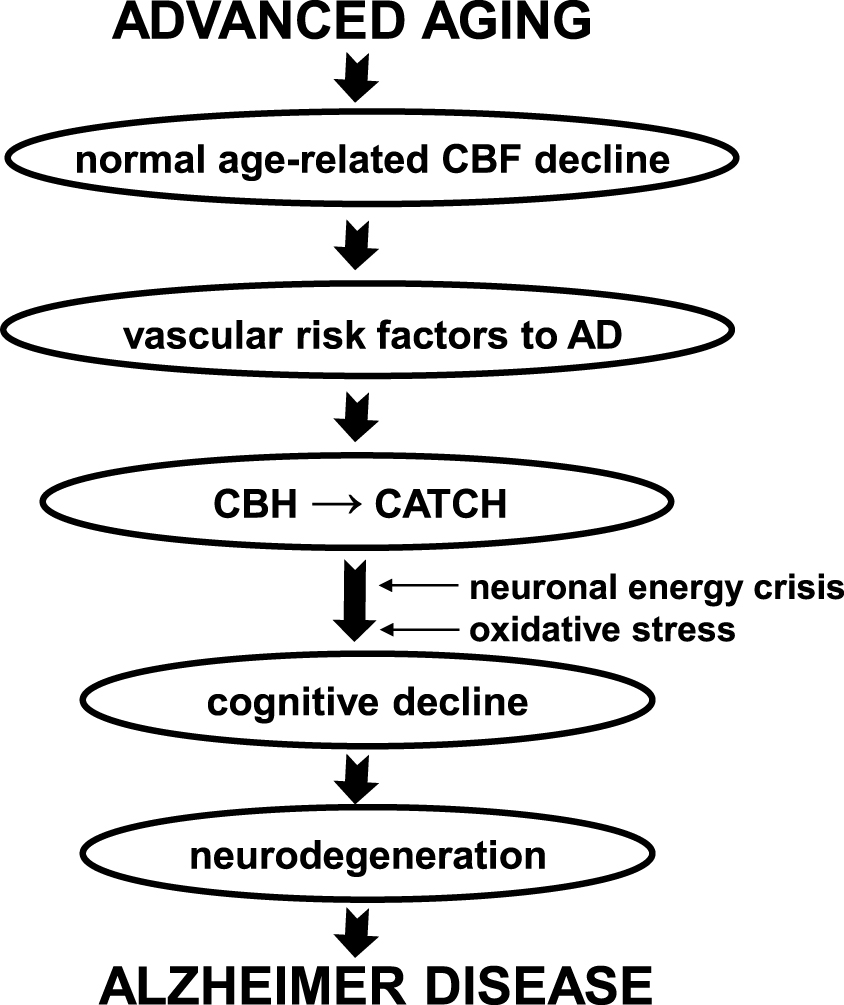
Neurodegeneration arising from chronic brain hypoperfusion leading to Alzheimer’s disease (AD) onset. Basic neuropathologic steps from normal advanced aging to AD onset based on the vascular hypothesis of AD (see review, [15]). Vascular risk factors to AD when acquired can burden normal age-related cerebral hypoperfusion to a point where chronic brain hypoperfusion (CBH) and critically attained threshold of cerebral hypoperfusion (CATCH) slowly develop. CATCH can lead to oxidative stress and a neuronal energy crisis, the precursors of cognitive decline. The process may take decades to develop until progressive neurodegeneration appears and in time, AD. CBF, cerebral blood flow.
The critical steps that turn normal aging into a cognitively dysfunctional vortex begins with the acquisition of vascular risk factors responsible for the persistent cerebral hypoperfusion that follows. An important side note is the finding that acquisition of multiple vascular risk factors to AD results in a greater rate of cognitive decline [12]. The reason may be due partly to the lowering of CBF and its delivery of high energy nutrients to the brain, heart, other organs, and periphery.
The way CATCH was conceived was based on the observation that from age 20 to 65, an age-related drop in CBF of about 15% (0.5 ml/y) normally occurs which is independent of brain atrophy or CNS damage [13, 14]. If an additional burden to the normal age-related CBF decline materializes, for example, from vascular risk factors, CATCH will likely develop [9–11]. Over the span of decades, depending on the patient’s age, state of health, lifestyle, gender, and genetics, CATCH can compromise neuron-astroglial metabolism by limiting the delivery of high energy nutrients to the brain, for example, oxygen and glucose and for introducing a mildly but sustained ischemic-hypoxic state at an early stage of cognitive impairment [9, 15].
A chronic, ischemic-hypoxic state also induces the formation of amyloid-β (Aβ) in the brain [16, 17]. Hypoxia-inducible factor 1-alpha (HIF-1a) is a transcriptional regulator that is overexpressed as an adaptive response to hypoxia [18]. This hypoxia regulator has been shown to activate both β (BACE 1) and γ (PS1)-secretases, eventually increasing the production of Aβ in the brain and cerebrovasculature [17, 20]. This finding would suggest that even mild oxygen deprivation in the vulnerable elderly brain is capable of stimulating neurodegenerative markers to AD that can contribute to neuronal loss.
Since virtually all vascular risk factors to AD can lower CBF to some extent [4, 7], the additional CBF burden to age-related CBF decline appears to result from acquiring any or several vascular risk factors to AD, such as diabetes 2, hypertension, dyslipidemia, smoking, obesity, atherosclerosis, or a vasculopathic condition affecting the heart or brain circulation [21–25].
There is now considerable evidence from longitudinal studies showing a significant association between vascular risk factors and cognitive dysfunction [26–29].
Apart from the intracranial and peripheral vascular risk factors to AD mentioned above that are known to lower CBF in the elderly, a set of structural and functional cardiac deficits are reported to be associated with brain hypoperfusion and cognitive impairment. The cardiac deficits include coronary heart disease, heart failure, valvular damage, aortic stiffening, left ventricular hypertrophy, and left ventricular systolic/diastolic dysfunction [30–33]. The cardiac conditions cited above can lower cardiac output and in time will also reduce regional CBF [34–36]. It has been determined that even a subtle but persistent drop in cardiac output can affect cerebral perfusion homeostasis and increase the risk of cognitive decline in the elderly [35, 37].
Peripheral artery disease is a common cause of hypoperfusion in the heart and brain by its association with aging, smoking, diabetes 2, arterial stiffness, hypertension, dyslipidemia, atherosclerosis, and obesity, conditions which are also risk factors to AD [38].
Another common condition of the elderly that can cause brain hypoperfusion is cerebral amyloid angiopathy (CAA). CAA consists of amyloid deposits outside and inside the vessel wall in mostly cortical and meningeal microvessels [39, 40]. The amyloid deposits within the vessels can effectively diminish blood flow delivery and focal oxygen supply to brain cells.
CAA is now the topic of intense investigation because of its clinical impact on cognition, stroke, and brain hemodynamics involving cerebrovascular reserve capacity, which determines how far brain perfusion can rise from baseline following a stimulus. Other brain hemodynamic changes affected by CAA include cerebral blood flow, cerebral blood volume, flow velocity, and mean transit time.
The effect of CAA on brain hypoperfusion occurs downstream to cognitive impairment since CAA is a comorbid condition that appears with Aβ formation [41]. Since CAA forms at the start of neurodegenerative changes, it is preceded by the advent of vascular risk factors and their effect on cognitive deterioration years before neurodegeneration is manifested. Paradoxically, CAA appears patchy in the brain and tends to localize in neocortical regions attacking the occipital and parietal cortex while sparing the hippocampus, an essential region of the brain for forming new memories and an initial site of neuronal loss at the earliest stages of AD [42].
Collectively, these findings highlight the importance of early detection and intervention when cardiovascular and cerebrovascular risk factors are first identified and much before dyscognitive symptoms appear at midlife or after [43].
THE VASCULAR HYPOTHESIS OF ALZHEIMER’S DISEASE: HOW CEREBRAL PERFUSION IMPACTS COGNITIVE BEHAVIOR
The VHAD was promptly dismissed and ignored for years by most researchers and funding agencies since it played no foreseeable role in the Aβ paradigm [1, 44]. Oddly, after many years of being cast out, the VHAD was embraced by several well-known researchers who “borrowed” most of our ideas that made up the concept behind VHAD but never cited its source or us.
Despite its humble beginnings, the VHAD did offer a compelling argument. It claimed that AD primarily develops from age-related chronic brain hypoperfusion which worsens relative to the presence of vascular risk factors during aging (Fig. 1). To our thinking, this happens because sustained, inadequate CBF in the elderly will reach a critical threshold where brain cells can no longer cope with the dwindling energy supply being delivered, thus creating a neuronal energy crisis. Moreover, the slow, sequential, hypoperfusion process slowing the energy supply to brain cells was to us the key development that initiates a pathway to progressive neuronal hypometabolism, cognitive decline, oxidative stress, neurodegeneration, AD, and death (Fig. 1) [1, 8–11].
The fulcrum of the VHAD consolidates two common interconnecting conditions: 1) normal age-related CBF decline and 2) the presence of vascular risk factors. Pernicious chronic brain hypoperfusion appears when these two events converge during advanced aging.
Aside from activating numerous biomolecular changes in the brain, chronic brain hypoperfusion methodically reduces the delivery of oxygen and glucose thereby slowly creating a neuronal energy crisis featuring oxidative stress and an ischemic-hypoxic state [9, 45–47]. From our previous laboratory studies on aged rats, we reasoned that mechanically reducing CBF in the aged experimental animals for 12 months, led to a chronologic set of cognitive deficits that began with mild memory loss, learning disability, neurometabolic slowdown, reduced protein synthesis, and later, absence of grooming behavior, neuronal loss in the hippocampus and posterior cingulate cortex, gliosis, severe memory loss, and extensive posterior parietal cortex atrophy [2, 49]. These findings were confirmed by others [50–53].
The neuropathologic consequences of chronic brain hypoperfusion in the aged rats thus appeared to produce a model that mimicked the progressive clinical course seen in AD [2, 54]. Curiously, this pathologic process occurred in the hypoperfused rats without the presence of neuritic plaques and the pathologic process could be fully reversed if CBF was restored after 2-3 weeks [2, 55].
The VHAD started to take traction when it was realized that over two dozen risk factors to AD were linked to poor blood flow to the brain [4, 56–58].
Some vascular risk factors appear to take a heavier toll on lowering resting CBF than other risk factors. The reason for this outcome remains unsolved. However, a cluster of vascular risk factors poses more peril to develop AD than a single risk factor [12]. Fortunately, nearly all these vascular risk factors are modifiable with the proper management or therapy if given before irreparable brain or organ damage occurs.
One of the strongest clues that AD is a vascular disorder is its similarity to vascular dementia (VaD), an undisputed vascular disorder [59, 60]. Not only do both dementias share nearly identical symptoms and signs, but both show comparable microstructural changes, negative impact on cognitive function and a link to more than two dozen vascular risk factors [60, 61]. This overlap between AD and VaD is often due to both disorders displaying neurodegenerative and cerebrovascular pathology (mixed dementia) but in some patients, each dementia can appear as relatively pure, suggesting a different pathomechanism underlying each condition [62].
THE Aβ FAILURES: A CURIOUS LEGACY
The anticipated value of the observations made about vascular risk factors to AD and the impact of CBF to cognitive dysfunction was dampened by the enthusiasm generated by Aβ peptide, the principal component of senile plaques in AD brain and cerebrovasculature. The amyloid cascade hypothesis was then at its apogee in AD research idolatry and clearly overshadowed most other related research findings. The basic tenet of the early amyloid cascade hypothesis was that toxic, fibrillar Aβ overload in the brain was the cause of AD [63, 64]. This scientific monolith sucked the air out of the dementia think tank and virtually excluded competitive views from its dominating bubble for the next two decades.
Eventually, it was discovered that a significant load of Aβ plaques were present in cognitively normal individuals and that neuronal death was widespread in brain regions devoid of plaques [65, 66]. As mounting evidence showed that the fibrillar Aβ cascade was “too good to be true” to serve as a causative agent of AD [67], a surrogate explanation was quickly spun-out with a view to resurrect the original Aβ paradigm. The newer version of the amyloid cascade hypothesis would now involve soluble “toxic” Aβ oligomers [68]. This version remains highly contentious since toxicity exerted by these polypeptides in cultured cells and mice does not reflect human pathogenicity [45]. Moreover, very high concentrations of Aβ monomers are needed to reconstitute toxic oligomers in vitro [69]. Such a non-physiological concentration of monomers is unlikely to occur in human brain [69]. Additionally, therapeutic studies targeting Aβ oligomers are of questionable value since they appear to be based on flawed biochemical assays that detected toxic oligomer species where none existed [70, 71]. Toxic Aβ oligomers in vitro thus present a weak and unproven argument relative to the AD cause.
Despite its complex physicochemical interactions with membranes, binding sites and regulatory subunits, Aβ, in our judgment remains a downstream pathological ‘product’ that is part of a neurodegenerative chain of events caused by vascular dysfunction initially beginning with brain hypoperfusion and hypometabolism.
It must be said in retrospect, that when we proposed cerebromicrovascular perfusion impairment as the primary cause of AD, the reaction by researchers and big pharma was to ignore and dismiss such heretical nonsense aimed at the untouchable amyloid cascade hypothesis [1, 3–5]. At the time, the role of Aβ in AD was considered by many workers as paradigmal gospel.
We believed, and still do, that Aβ is an important pathological product of neurodegeneration, especially its critical role in CAA, but it is not the cause of AD [4, 73]. This view has been costly. Our thinking and those of others who contradicted the role of Aβ in AD pathogenesis were (until recently) simply brushed aside by the ‘don’t rock the boat’ followers of Aβ. The eminence gained by the amyloid cascade hypothesis has allowed it to enjoy extensive jurisdiction over much of AD research, conference programs, and funding opportunities for over 20 years. For the few investigators who remained outside this expanding Aβ bubble, it became a challenge to obtain non-Aβ related grants, being able to present material outside the amyloid-centric microcosm at Alzheimer conferences and having articles unrelated to Aβ rejected by major scientific journals that were not relevant to Aβ research.
It is estimated that in the last 10 years, about 35,000 papers dealing with Aβ research have been published at a cost of billions of dollars to big pharma, government, special interest agencies, and taxpayers. Within that vast literature, evidence-based medicine into the amyloid cascade hypothesis has uncovered a parade of consistent flaws that seem incompatible with the claim that Aβ is the cause of sporadic AD [45, 74–80]. Despite the unsupportive research, which would have rung a death knell to any other hypothesis, big pharma and its paid academic consultants continue to plow this line of reasoning hoping they can turn a spent cartridge into a magic bullet.
Presently, dozens of clinical trials costing billions of dollars have ended in disaster and failed to prove the predicted clinical value of the amyloid cascade hypothesis [80–85]. Marketing hyperbole delivered to the media from big pharma consistently trumpet an imminent ‘breakthrough’ using their ‘newest’ anti-Aβ me-too drug. This soap-selling technique is cruel and deceptive in that it raises and dashes hopes of AD victims as it lines up new stock buyers.
As a result of these drug failures, other competitive views are slowly seeping into the dementia think tank, among them the VHAD. Meantime, a growing legion of scientists [45, 86–88] are questioning the logic of the amyloid cascade hypothesis, some even wondering why, after two decades of failing to treat, stop, or prevent AD using drugs that wipe out Aβ-containing plaques from the brain, the claims for this peptide remain exactly the same as they were 25 years ago [89, 90].
Not only has the treatment of AD with anti-Aβ therapy been proven ineffective but many patients were harmed from the treatments [91, 92]. It is self-evident that if the amyloid cascade hypothesis remains clinically indefensible, patient management and treatment will be suboptimal. Moreover, testing a treatment that consistently does not work and even causes harm to patients is an affront to good medical practice.
The amyloid cascade hypothesis has become a living zombie under the magic spell of big pharma money and its well-paid advisors. Editorializing the many clinical trial failures of the amyloid cascade hypothesis in The New England Journal of Medicine, the question in many people’s mind was asked: “when is a dead hypothesis really dead?” [93].
Since it is impossible to completely prove or disprove a hypothesis, it will be interesting to see whether the vast clinical data generated on Aβ will have a positive social effect someday [94]. One needs to remember, however, what the British philosopher Karl Popper thought about scientific proof when he said, “It must be possible for an empirical scientific system to be refuted by experience.”
Scientific proof in biology is generally difficult to demonstrate. One factor that strengthens a hypothesis is the amount of verifiable evidence available to support it. Even powerful evidence (i.e., strongly suggestive) is not proof that something is true.
CBF AS A PRECLINICAL BIOMARKER OF AD USING NEUROIMAGING
One of the most significant events in medical research in recent years is the emergence of a variety of neuroimaging methodologies used for direct and indirect monitoring of brain activity. This technological leap has given new insights into understanding the functional and structural organization of the human brain. The ability to predict abnormal cerebral changes associated with imminent dementia years before symptoms appear, could decidedly open the gates to more effective therapeutic interventions based on patient stratification of relative risk to cognitive decline. The strategy would result in a crucial health management initiative.
In a way, neuroimaging advancements to detect, diagnose, prognose, and predict cognitive decline in prodromal AD based on hemodynamic and CBF changes, can be described as “revolutionary” from what it was a decade ago. Presently, a number of neuroimaging techniques can predict conversion from MCI to AD based on CBF alterations and provide a differential diagnosis from other disorders causing dementia. Although still in its clinical infancy, neuroimaging is fast becoming the stethoscope of the brain. Its impact in medicine will soon be a turning point in our discovery of the role cerebral hemodynamics plays at each level of neurodegeneration and cognitive meltdown.
When one realizes that little over a decade ago there was ambiguous information on how AD was diagnosed using neuroimaging techniques, let alone how to predict or prevent it, it is reasonable to appreciate the remarkable progress made by this technology. This rapid growth of neuroimaging techniques now features non-invasiveness, high spatial and temporal resolution, consistent reproducibility, swift acquisition time, more robust or sensitive processing methods and ease of implementation. The neuroimaging techniques discussed in this review are based on brain hemodynamics and therefore only provide a slice of total brain activity, although a very important slice.
PREDICTING COGNITIVE DYSFUNCTION IN COGNITIVELY HEALTHY SUBJECTS
An intriguing study led by Sven Haller and colleagues from the University of Geneva could have important implications in predicting cognitive impairment and AD onset [95]. In that prospective study, 148 cognitively intact elderly participants and 65 MCI patients underwent baseline cerebral arterial spin labeling-MRI (ASL) and had repeated ASL and neurocognitive testing at follow-up 18 months after baseline. MCI is believed to represent a high risk state for developing dementia, including AD. Two groups were identified at the end of the observation period: stable cognitive function patients (sCON) and deteriorated cognitive function patients (dCON) whose CBF scans resembled those showing MCI scans at baseline (Fig. 2). The dCON group and MCI group revealed significantly reduced regional brain hypoperfusion, most notably in the posterior cingulate cortex when compared to sCON (Fig. 2).

Prediction of MCI in cognitively intact individuals. Two-dimensional pulsed ASL-MRI maps of CBF shows a global reduction in baseline cerebral perfusion notably in the posterior cingulate cortex (arrow) in cognitively intact elderly patients who went on to deteriorate cognitively (DCON) 18 months later as compared to cognitively stable (SCON) subjects. DCON images were similar to MCI subjects at baseline and are shown for comparison. See text for details. Courtesy of Xekardaki et al. [95].
This study demonstrates that ASL has the potential to predict MCI onset in cognitively normal individuals whose brains showed abnormal hemodynamic features. Furthermore, assuming more sensitive processing methods for neuroimaging acquisition are developed, detecting prodromal MCI should mature into a pivotal opportunity to evaluate novel interventions targeting brain hypoperfusion. The outcome could evolve as the tool of choice to explore brain activity and ways to prevent cognitive meltdown in the elderly.
PREDICTING MCI CONVERSION TO AD
A majority of neuroimaging methods are focusing on practical biomarkers in prognosing the potential course to AD. A preclinical biomarker, for example, regional CBF quantification with correlation from neurocognitive tests, can be applied to detect the proneness of people with cognitive impairment to convert to AD (see review [15]). The use of cerebral perfusion as a preclinical predictor of dementia is also suitable for measuring the rate of cognitive decline progression prior to AD onset and as stated above, for evaluating the effectiveness of therapeutic interventions.
Once a neuroimaging prognosis is made about the probability of MCI conversion to AD, patients can be stratified using a scoring system such as the Framingham Cardiovascular Risk Profile [96], allowing the clinician to determine which patients need more aggressive intervention and which can be followed-up for observation.
Some current neuroimaging techniques being used are portable and relatively safe. Many may become cost-effective if they help prevent or reduce AD onset. Several are non-invasive and highly accurate. These techniques can detect in real-time subtle brain changes associated with cognitive dysfunction and cerebral hemodynamic deficits. Rapid improvement of neuroimaging human brain could soon be able to routinely predict such changes in cognitively normal individuals and provide early information to begin appropriate interventions.
A few techniques are redefining how we view cognitive disturbances to better interpret these brain abnormalities and their pathological pathway to dementia. They include but are not restricted to: arterial spin labeling MRI (measures CBF non-invasively), functional MRI (measures functional connectivity, CBF and oxygenation) dynamic susceptibility contrast MRI (measures CBF and cerebral volume), [18F] fluorodeoxyglucose-positron emission tomography (measures uptake and local glucose metabolism in brain), single photon emission computed tomography (CBF assessment), transcranial Doppler (TCD) ultrasonography (measures flow velocity, pulsatile index), diffusion tensor imaging (evaluates white matter microstructure and fiber tract connectivity). These techniques have matured considerably in the last few years and are ready for clinical prime-time. All these techniques have their limitations which are generally discussed by the authors of most published research studies.
Several of these techniques are very powerful in providing information on cerebral hemodynamics as we discuss below. The methodologies are constantly being tweaked and modified to establish their validity and reliability and to improve their diagnostic, predictive, and prognostic value.
The evidence gathered so far from diverse neuroimaging studies indicate that CBF is likely the best marker to prognose cognitive decline and its conversion to AD and other indirect studies seem to support this conclusion [85, 97–102]. This is also the recommendation from the Alzheimer’s Disease Neuroimaging Initiative (ADNI) who have advised that imaging for brain hypoperfusion should be considered the primary predictive biomarker for AD [103].
PRECLINICAL PREDICTION OF AD USING MULTIMODAL NEUROIMAGING
If the concept of primary CBF insufficiency is the earliest pathologic event in the development of AD, then its preclinical detection in cognitively intact or in MCI patients provides a dramatic open-door for the prevention of cognitive decline and AD onset. This is the conclusion of Alan Evans’ and his group at the Montreal Neurological Institute [103]. These investigators amassed and analyzed 7,700 brain images in 1,171 healthy and diseased subjects at various stages of AD progression using multimodal PET and MRI scans to examine glucose metabolism, Aβ concentrations, and CBF in 78 gray matter regions. Then they determined each image or biospecimen as either pathologic or healthy. After a massive analysis, they concluded that vascular dysregulation appeared as the earliest and strongest brain pathologic factor associated with AD development [103]. This monumental study emphasizes the role brain hypoperfusion plays in the evolution of cognitive impairment, thus reaffirming a proliferating number of neuroimaging studies that target cerebral hemodynamics to determine the pathologic process involved in the development of dementia.
A follow-up study by the same authors took 561 individual baseline data from the ADNI and used structural MRI, FDG-PET, functional MRI, ASL, and Aβ PET to explore the predictability of cognitive deterioration in all patients [104]. The cohort was made up of normal, MCI and mild AD subjects. ADNI participants are 55–90 years and their baseline medical history and cognitive function has been determined by a consortium of universities and medical centers in the United States and Canada to test useful AD hypotheses based on clinical and biomarker data [105]. This follow-up study by Evans’ group confirmed their previous findings with the conclusion that cerebrovascular impairment is the primary pathologic event leading to AD [104].
ASL AND ASL-MRI NEUROIMAGING
It has been known for some time that CBF is much more reduced in AD patients compared to cognitively healthy subjects. Many investigators have attributed the CBF disparity on the effects of neurodegeneration and subsequent loss of neurons which no longer need CBF. This thinking was dispelled by some ASL findings that showed CBF was 20% lower in MCI patients as compared to CBF in healthy controls [102]. Although CBF is lower in MCI than in healthy controls, most MCI patients do not manifest the neuropathological features of AD at autopsy [107]. This observation indicates that Aβ accumulation and neurofibrillary tangles are downstream events from the initiation of brain hypoperfusion. Subsequent neuroimaging studies have essentially confirmed this conclusion.
Not surprisingly, there is a growing assumption among AD workers that cerebral hypoperfusion is involved in the pathogenesis of AD and that CBF can serve as a biomarker for predicting impending AD.
This thinking is now being studied by neuroimaging investigators. For example, Chao and his group [108] recruited 48 MCI patients and found 13 participants developed AD during a longitudinal period of observation lasting 2.5 years. Using ASL-MRI to measure baseline CBF, they were able to predict from the declining cerebral perfusion in the right precuneus and right frontal cortex, severe memory disturbance that led to dementia. This study was confirmed more recently when it was reported using ASL that regions of brain hypoperfusion in the right precuneus and right frontal cortex in MCI patients led to conversion of dementia [109, 110]. This finding indicates brain hypoperfusion is a significant predictor of AD conversion. Chao et al.’s [108] findings have been consistent with previous neuroimaging studies on MCI patients where reduced neurometabolism in precuneus and right temporoparietal cortex has been observed using FDG-PET and SPECT functional imaging [85, 111]. FDG-PET is a marker of synaptic activity while local glucose metabolism reflects neuronal function at a resting state, two key elements for analyzing memory impairment.
In a dual imaging technique of probable AD patients, ASL-MRI and FDG-PET were used to measure CBF and glucose metabolism respectively (Fig. 3), and Detre and colleagues [112] found a close association between abnormal neuroimaging patterns and working memory deficits obtained from neuropsychological tests. Since CBF and brain glucose metabolism are tightly coupled, an overlap of abnormal neuroimaging patterns correlating with the Boston Naming Test (BNT) and the Digit Symbol Substitution scores (DSS) were seen in the probable AD patients and in a small group of asymptomatic patients with subjective memory complaints (Fig. 3). Deficient BNT was associated with reduced CBF in the ventrolateral prefrontal cortex and inferior-middle temporal lobes (Fig. 3). By contrast, DSS low scores and abnormal neuroimaging patterns for glucose metabolism were found in the prefrontal cortex and the bilateral inferior parietal lobes (Fig. 3). Both CBF and glucose metabolism dwindled significantly in the posterior cingulate cortex, an early parenchymal focus of cognitive decline targeting learning and memory [112].
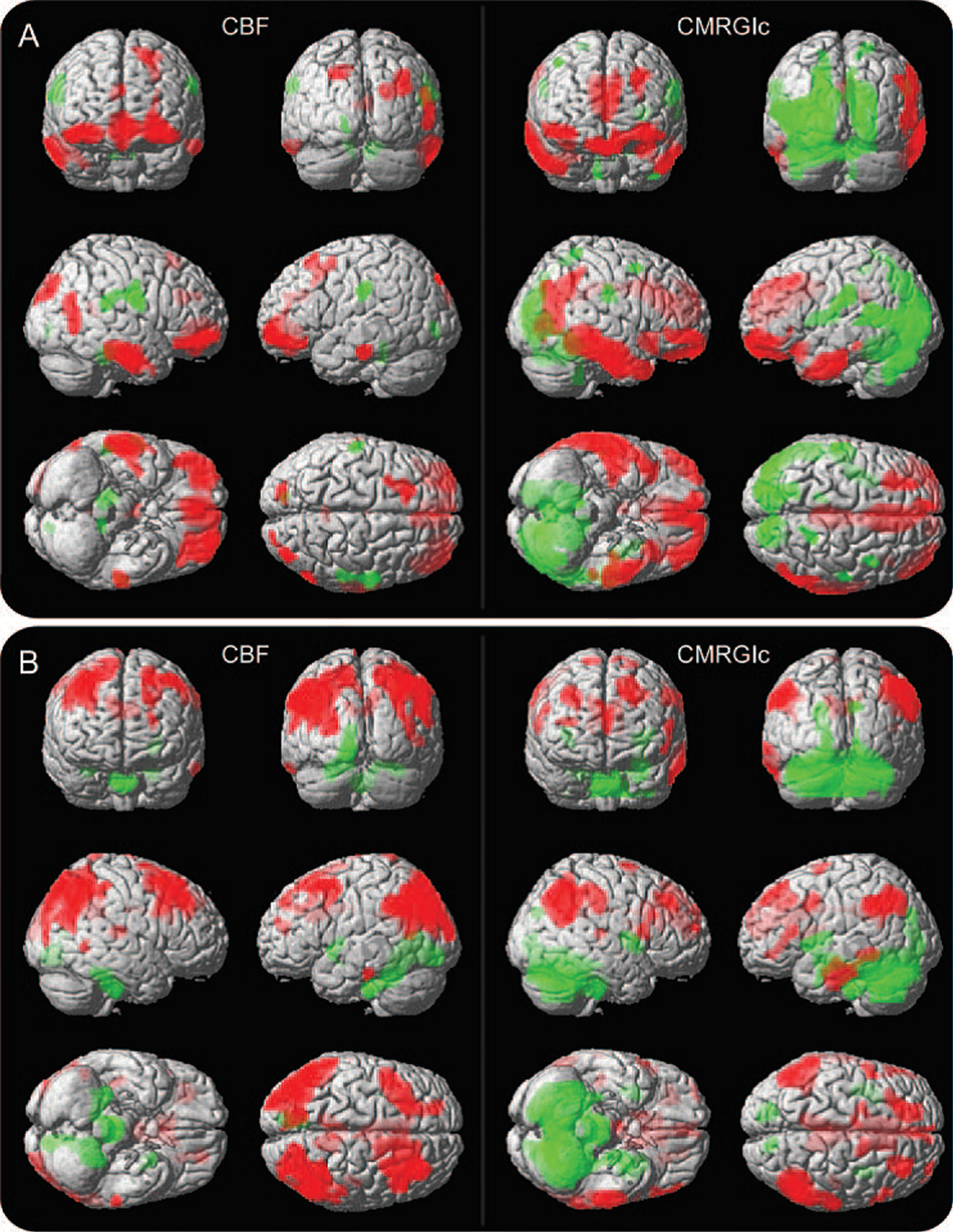
Correlation of task performance with CBF and CMRGlu in probable Alzheimer’s disease brain. A) Multimodal correlation between CBF (left side) using ASL and CMRGlu (right side) using FDG-PET with Boston Naming Test (BNT) scores. B) Correlation between CBF (left) and CMRGlu (right) with Digit Symbol Substitution (DSS) scores. Positive correlation between lowered CBF and CMRGlu neuroimages with deficient BNT performance are depicted by red color in the ventrolateral prefrontal cortex and inferior-middle temporal lobes (green color = negative correlation). Conversely, lowered CBF and CMRGlu is seen in the dorsolateral prefrontal cortex and bilateral inferior parietal lobes (red areas) correlating with deficient DSS scores. Red-color brain regions depicted are linked to diverse cognitive functions. See text for details. Image correlations were statistically significant (p≤0.005). CBF, cerebral blood flow; CMRGlu, cerebral metabolic rate of glucose; ASL, arterial spin labeling; FDG-PET, 18fluoro-deoxyglucose positron emission tomography. Courtesy of Chen et al. [112].
This important multimodal study identified patterns of brain hypoperfusion and neuronal hypometabolism associated with deficient memory task performance. As such, the technique could become useful in preclinical AD prognosis and in tracking disease severity following therapy [112].
The Framingham Offspring are the biological children of the original participants that made up the Framingham Cardiac Study (FCS). FCS was established in 1948 to study cardiovascular risk factors. A recent MRI investigation recruited 1,049 participants of these FCS offspring who received a battery of neuropsychological tests at baseline and were classified as MCI or normal cognition in a 6.5-year study [113]. MRI scanning was done on all participants to measure hippocampal volume and white matter hyperintensities (WMH). Both MCI and normal subjects who converted to MCI at follow-up showed smaller hippocampal volumes and WMH from baseline. These findings indicated a predictability of cognitively normal individuals to convert to MCI based on hippocampal and white matter abnormalities resulting from small vessel cerebrovascular disease [113]. The data also suggested that mild subclinical small vessel disease played a critical role in the expression and evolution of MCI and supported the authors contention that such changes predict prodromal dementia more reliably than measures of Aβ [113].
Although other MCI risk factors such as microhemorrhages or cortical thickness were not examined in this study, the authors point out that their findings confirm that traditional vascular risk factors, which are acknowledged risks for the development of brain pathology, should be a therapeutic target to prevent or delay the risk for MCI and AD [113]. A previous study by Bangen and her group [57], had also reported brain hypoperfusion in MCI patients adding that increased CBF in the medial temporal lobes correlated with better memory performance.
SPECT NEUROIMAGING
Many research groups have reported that Aβ containing plaques appear to cause severe brain disconnection and show up more frequently in the frontotemporal association cortex and limbic regions, but they are rarely found in white matter [114]. The finding that white matter lesions can be predictive of MCI conversion to AD, presents a curious paradox: if Aβ plaques cause no white matter damage, what does? The answer appears to point to vascular risk factors (hypertension, smoking, diabetes type 2, etc.), not a structural cause of damage.
Thirty-two elderly patients with subjective memory complaints were recruited to undergo SPECT imaging to determine their memory status using the Mini-Mental State Examination score in relation to regional CBF [115]. SPECT imaging indicated marked hypoperfusion in the left precuneus, left inferior frontal gyrus, bilateral temporal areas, bilateral anterior cingulate gyrus, bilateral parahippocampal gyrus, and right posterior cingulate gyrus. Memory dysfunction, particularly visual memory, showed a significant correlation with brain hypoperfusion [115]. A chronological study showing the pattern of the spreading hypoperfusion was not done by the authors of this study which could have indicated the initial brain regions affected by a declining CBF and the rate of such a decline in non-MCI subjects.
Functional brain imaging using SPECT was used on 42 amnestic MCI (aMCI) patients to determine conversion to AD in a 4-year follow-up [116]. SPECT data found differences between cognitively normal control patients and aMCI patients, the latter who showed lower brain perfusion in the posterior cingulate gyrus and the basal forebrain. The MCI individuals who converted to AD also had poorer performance on long-term visual memory when given neuropsychological tests suggesting a complementary interaction between pathological brain function and neuropsychological impairment. This study demonstrated signs of decreased blood flow prior to clinical expression of dementia in the MCI patients. These MCI patients went on to convert to AD within 4 years after baseline [116].
Reduced CBF has been consistently reported in a number of brain regions before and during cognitive dysfunction. The hypoperfused areas most notably affected are: medial temporal lobe, precuneus, posterior cingulate cortex and hippocampus [117–121]. It should be noted that brain regions where CBF is significantly reduced are also regions associated with MCI and are known to be the main parenchymal targets of neurodegenerative attack leading to AD.
DTI NEUROIMAGING
Diffusion tensor imaging (DTI) MRI was used for 3 years together with cognitive testing for 5 years to follow a group of patients diagnosed with small vessel disease (SVD) [122]. SVD was defined as symptomatic lacunar stroke with confluent WMH. All patients were monitored for possible progression to dementia. Cognitive impairment in SVD generally impairs executive function and processing speed but spares episodic memory. After the 5-year follow-up, 18 patients had converted to dementia. DTI predicted cognitive decline and dementia based on advancing white matter microvascular damage over a 3-year period. Findings from this study using DTI, indicate that cognitive decline and dementia can be predicted with accuracy during a time when cognitive dysfunction is not detectable by other means [122].
Although cerebral SVD implies the presence of brain hypoperfusion, the authors did not measure CBF to see if DTI measures correlated with perfusion changes in the brain regions analyzed.
DTI metrics have been used to assess cognitive function and predict cognitive decline in patients with MCI using specific cerebral structures for analysis [123]. Mean diffusivity of the left retrolenticular part of the internal capsule and left fornix were used as predictors in 66 elderly women with MCI. The authors reported that higher mean diffusivity values correlated with the Mini– Mental State Examination test for cognitive dysfunction [123].
FDG-PET NEUROIMAGING
PET studies play an important role in the early detection of MCI and recent work shows its usefulness may extend to prediction of MCI and conversion to AD.
Since virtually all energy activity in the brain is derived from CBF delivery of glucose to the CNS, neuronal and presynaptic activity as reflected by focal glucose metabolism and regional CBF can be accurately monitored by FDG-PET [124]. This capability endows FDG-PET as a versatile tool in the study of local brain function particularly in longitudinal studies.
For example, the value of 18F-FDG-PET for predicting MCI conversion to AD was examined in 114 elderly patients with MCI who were followed for 5 years [125]. Progression to AD was seen in 82 patients (72%) previously diagnosed with aMCI. Visual assessment of PET images predicted conversion to AD in hypometabolic MCI patients with an overall 82% diagnostic accuracy. Another observation from this study noted that conversion to AD from MCI was low in patients with no hypometabolism, suggesting that adequate CBF to maintain neurometabolic function during MCI may delay AD onset. Data from the Alzheimer’s Disease Neuroimaging Initiative shows that 18F-FDG-PET has a higher predictive power than structural MRI but only when patients are followed-up for less than 5 years [126, 127].
Previous studies using 18F-FDG-PET have been able to predict conversion of MCI to AD by examining images for changes in brain hypometabolism, a condition that reflects brain hypoperfusion, mainly in the precuneus, posterior cingulate and parietotemporal cortices [100, 128].
The sensitivity of resting glucose metabolism using FDG-PET was examined during 24 months of follow-up in order to determine the predictive value of cerebral glucose uptake in a group of elderly subjects with memory complaints [126]. The evidence showed that lower baseline FDG-PET could consistently predict longitudinal cognitive decline. FDG-PET consistently was seen associated with the Alzheimer’s Disease Assessment Scale-cognitive (ADAS-cog), a sensitive measure of cognitive function stressing analysis of memory, language and praxis [126].
In another study, MCI patients were evaluated using FDG-PET and followed on a yearly basis for 4 years [129]. This study examined 371 MCI elderly patients and combined functional brain imaging with cerebrospinal fluid biomarkers involving Aβ1 - 42, t-tau, and p-tau181p. However, the authors found FDG-PET combined with the Auditory Verbal Learning Test (AVLT) and Clock drawing more robust measures for predicting MCI conversion to AD than Aβ or tau markers [129]. These findings demonstrate the ability of cognitive tests to be robust predictors of MCI to AD conversion when aided by FDG-PET analysis detecting left middle temporal lobe thickness and left hippocampus volume [129].
Mental skill tests assess a combination of semantic knowledge, visual motor ability, and executive function, whereas the CSF biomarkers determine the presence of neurodegenerative changes which are not generally manifested at an early MCI stage. Moreover, the AVLT results are consistent with findings that have documented the association of the frontal lobes with confusion and confabulation of memory processes and which show the most consistent and pronounced degenerative changes over the years.
TCD DOPPLER ULTRASOUND NEUROIMAGING
One of the many advantages of transcranial Doppler ultrasound (TCD) is its ability to evaluate CBF hemodynamics in the intracranial arterial tree at the bedside or outpatient clinic. TCD uses a low-frequency (<2 MHz) transducer placed on the scalp that insonateshe basal cerebral arteries through thin cranial bones to measure CBF velocity [130]. TCD can provide continuous beat-to-beat measurement of the blood flow velocity. Apart from blood flow velocity, TCD can assess the pulsatility index, evaluate autoregulation, and analyze an assortment of physiological parameters which can be determined in outpatient clinic settings. TCD is also used to evaluate therapeutic interventions.
TCD is non-invasive, portable, inexpensive, repeatable and relatively easy to operate by a trained technician. These qualities make TCD an indispensable clinical tool in the detection of abnormal CBF. Because of its versatility, TCD is the most common neuroimaging method used to assess human cerebral hemodynamics [131]. This feature is especially useful in determining the role of cerebral hemodynamics at an early stage of cognitive decline [132].
It has been known for some time that blood flow velocity is diminished in AD patients. The question of whether this outcome results from neurodegeneration or from cerebral hypoperfusion was addressed in the Rotterdam Study by Ruitenberg and her colleagues [133]. TCD was used to examine 170 cognitively normal participants, some who later developed AD. It was found that subjects with higher CBF velocity showed less prevalence to develop cognitive decline and AD during a 6.5-year follow-up. MRI analysis also determined less hippocampal and amygdalar atrophy in the group with higher CBF velocity [133]. The authors concluded that these findings strongly suggested cerebral hypoperfusion preceded and was likely the cause of AD in the tested participants.
A more recent study by Roher and colleagues [134] used TCD to examine 16 arterial segments imaged from the circle of Willis. Mean flow velocity and pulsatility index in elderly participants was obtained in cognitively intact and presumptive AD patients. Findings from presumptive AD patients included increased pulsatility index, a sign of highly detectable cerebrovascular resistance coupled to reduced arterial mean blood flow velocity, reflecting low brain perfusion and possible risk of WMH.
Although Roher’s TCD study did not include MCI subjects, a recent report by Chung and colleagues did [135]. They reported testing 54 non-demented individuals with subjective memory decline and with MCI in a 6-year study [135]. The posterior cerebral and middle cerebral arteries were used to measure pulsatility index. High pulsatility in the left middle cerebral artery was found to independently predict MCI conversion to AD [135]. These findings confirm the versatility and practical advantage of TCD in evaluating the hemodynamics of the intracranial arterial vasculature.
CEREBRAL HYPOPERFUSION: IS IT PIVOTAL IN AD DEVELOPMENT?
The consensus that cerebral hypoperfusion is critical in the development of AD is not shared by all neuroradiologists. A Swedish group recently analyzed 11 patients with precerebral stenosis or occlusion that resulted in regional brain hypoperfusion [136]. Patients were scanned with PET using 18F-Flutemetamol uptake in the brain to determine long term accumulation of Aβ or tau aggregates in the hypoperfused regions. These investigators reported that no evidence was found to suggest chronic brain hypoperfusion increased Aβ deposition or tau aggregates. They concluded that these findings “strongly indicated” that brain hypoperfusion is not a pivotal event in the pathogenesis of MCI or AD [136]. This report did not cite contrary evidence nor the fact that brain hypoperfusion in combination with vascular risk factors to AD may lead to cognitive impairment and eventual AD independent of Aβ density in the brain [137]. Moreover, a number of studies have shown that AD can exist showing a significant CBF reduction but nearly absent Aβ brain deposition [69, 138–143].
WHERE DO WE GO FROM HERE?
The bulk of evidence presented in this brief review should be put into context. One can question the value of dozens of neuroimaging studies that link brain hypoperfusion and cognitive decline as either irrelevant or coincidental and that a proof of concept relationship has not been demonstrated.
Or, one can perceive from the findings thus far, and this review cites only a fraction of this evidence, that a growing neuroimaging data bank could soon provide a strategy to stop AD before it starts. This could be accomplished by focusing on vasculopathic imaging changes that may herald impending cognitive dysfunction at midlife and during advanced aging. Time will decide. There is always a chance of being wrong and the clinical history of AD for the last two decades have shown us what that can lead to.
For now, there is a choice as to where the funding opportunities should be aimed at and what topics AD conferences need to highlight. There is a cynical pecuniary shadow that hangs over the field of dementia that dramatically shrouds and impersonally impedes the progress that needs to move forward and serve the patient’s best interests.
The neuroimaging connection with brain hypoperfusion offers a realistic chance to correct or significantly delay abnormal brain function before irreversible structural damage occurs. This is only one approach of many that should be considered in future studies.
Twenty-five years from now, this AD story will be viewed by medical pundits as a clinical step forward or as a sad continuation of trackless clinical research prompted by money and profit. The time for diplomacy should now be replaced by first, open discussions of the dismal failure AD clinical research has generated in the last two decades, and second, how to best serve the interests of those at risk of AD.
How do we remedy the mind-set that promoted and keeps promoting big pharma-organized clinical failures? Time is running out for future AD victims. Researchers and clinicians should start thinking about AD in terms of flipping around the Hippocratic oath, “first do no harm”, to “first do some good”.
DISCLOSURE STATEMENT
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/18-0004r1).