
Abstract
The endocannabinoid system, which modulates emotional learning and memory through CB1 receptors, has been found to be deregulated in Alzheimer’s disease (AD). AD is characterized by a progressive decline in memory associated with selective impairment of cholinergic neurotransmission. The functional interplay of endocannabinoid and muscarinic signaling was analyzed in seven-month-old 3xTg-AD mice following the evaluation of learning and memory of an aversive stimulus. Neurochemical correlates were simultaneously studied with both receptor and functional autoradiography for CB1 and muscarinic receptors, and regulations at the cellular level were depicted by immunofluorescence. 3xTg-AD mice exhibited increased acquisition latencies and impaired memory retention compared to age-matched non-transgenic mice. Neurochemical analyses showed changes in CB1 receptor density and functional coupling of CB1 and muscarinic receptors to Gi/o proteins in several brain areas, highlighting that observed in the basolateral amygdala. The subchronic (seven days) stimulation of the endocannabinoid system following repeated WIN55,212-2 (1 mg/kg) or JZL184 (8 mg/kg) administration induced a CB1 receptor downregulation and CB1-mediated signaling desensitization, normalizing acquisition latencies to control levels. However, the observed modulation of cholinergic neurotransmission in limbic areas did not modify learning and memory outcomes. A CB1 receptor-mediated decrease of GABAergic tone in the basolateral amygdala may be controlling the limbic component of learning and memory in 3xTg-AD mice. CB1 receptor desensitization may be a plausible strategy to improve behavior alterations associated with genetic risk factors for developing AD.
Keywords
INTRODUCTION
Alzheimer’s disease (AD), the most common cause of dementia in the elderly, is characterized by a progressive impairment of memory and thinking skills, usually associated with agitation, psychosis, depression, apathy, disinhibition, or anxiety. Most of these symptoms are dependent on the cholinergic deficit described in AD [1, 2]. The cholinergic neurotransmission that controls learning and memory is specifically vulnerable in AD [3–8]. Impaired functionality of muscarinic receptors (mAChR) is found in areas that control cognitive processes, such as the amygdala and the hippocampus [8]. Different neuromodulators of the cholinergic system, including neurolipids, e.g., endocannabinoids (eCB), contribute to the alteration of cognitive and emotional processes [9]. Thus, a reduction of type-1 of cannabinoid receptors (CB1) in different layers of the hippocampus is described at advanced Braak stages of the disease [10], while an increase in CB1 receptor activity and density is found at early and moderated stages of AD [11]. Endogenous and exogenous cannabinoids seem to elicit modulatory effects in multiple AD-related processes, although the biochemical mechanisms need to be further investigated. Therefore, eCB system is foreseen as a novel potential therapeutic target to counteract the disease [12]. The triple transgenic mouse model of AD (3xTg-AD) shows impaired mAChR-mediated signaling in young animals (2–4 months). The cholinergic impairment is more evident at middle-aged mice (13–15 months), with a decrease in the activity of choline acetyltransferase. At 18–20 months the basal forebrain cholinergic neurons are affected together with hippocampal and cortical cholinergic neuritic dystrophy, in parallel with the progression of amyloid-β (Aβ) plaque formation [13, 14]. Altered CB1 receptor expression and functionality has also been described in the 3xTg-AD mice at different development stages (beginning at 4–6 months) [14, 15]. These mice harbor APPSwe, PS1M146V, and tauP301L transgenes and mimic several hallmarks of familial AD [16]. While Aβ and tau neuropathologies develop in middle age (12 months), deficits in synaptic plasticity and cognition have earlier onsets, when intraneuronal accumulation of oligomeric-Aβ, is clearly established at 6 months of age [17]. So far, learning and memory deficits are apparent in several cognitive paradigms such as the passive avoidance, the novel-object recognition and the Morris water maze [18, 19]. Fear and anxiety-like behaviors have also been shown in the open-field, the elevated plus maze, and the dark/light box [20–23]. Interestingly, in 6-month-old 3xTg-AD mice, the intraneuronal Aβ accumulation in the basolateral amygdala (BLA) has been shown to enhance innate and conditioned fear as assessed in fear conditioning paradigms [21].
The functional interplay of cannabinoid and muscarinic signaling was analyzed in relation to learning and memory of an aversive stimulus (step-through passive avoidance test). We examined the expression, neuroanatomical distribution, and functional coupling of CB1 receptor and mAChR to Gi/o proteins in seven-month-old 3xTg-AD mice and in age-matched non-transgenic (Non-Tg) counterparts, as well as following the subchronic eCB system activation.
MATERIALS AND METHODS
Animals
Seven-month-old male 3xTg-AD mice (n = 40) and age-matched Non-Tg mice (n = 18) were obtained from Universitat Autonòma de Barcelona, in collaboration with Dr. Lydia Giménez Llort. The 3xTg-AD mice harboring PS1M146V, APPSwe, and TauP301L transgenes were genetically engineered as previously described [16]. Also, nine-week-old male CB1−/− (n = 2) and CB1+/+ (n = 2) mice were used, provided by C. Ledent of the University of Brussels.
All the animals were housed (4–5 animals per cage) and maintained under standard laboratory conditions of 12 h light-dark cycle with light from 8 am to 8 pm and availability of food/water ad libitum. All procedures were performed in accordance with European Directive 2010/63/EU and the Spanish National Guidelines for Animal Experimentation and the Use of Genetically Modified Organisms (Real Decreto 1205/2005) and 178/2004; Ley 32/2007 and 9/2003). Experimental protocols were approved by the local Committee for Animal Research at the University of the Basque Country (CEIAB/21/2010/Rodríguez-Puertas).
Drugs and treatments
R-(+)-[2,3-Dihydro-5-methyl-3-(morpholinyl) methyl]pyrrolo[1,2,3-de]-1,4-benzoxazinyl]-(1-nap- hthalenyl)methanone mesylate (WIN55,212-2), (-)- cis-3-[2-Hydroxy-4-(1,1-dimethylheptyl)phenyl]-trans-4-(3-hydroxypropyl) cyclohexanol (CP55,940) and (2-Carbamoyloxyethyl) trimethylammonium chloride (carbachol) were acquired from Sigma-Aldrich (St Louis, MO, USA). 4-Nitrophenyl4- (dibenzo[d] [1, 3] dioxol-5-yl(hydroxy)methyl) piperidine-1-carboxylate (JZL184) and 5-(4-chloro-3-methylphenyl)-1-[(4-methylphenyl)methyl]-N- [(1S,2S,4R)-1,3,3-trimethylbicyclo[2.2.1]hept-2-yl] 1pyrazole3-carboxamide (SR144528) were acquired from Cayman-Chemicals (MI, USA), and (piperidin-1-yl)-5-(4-chlorophenyl)-1-(2,4-dichlorophenyl)-4-methyl-1H-pyrazole-3-carboxamide hydrochloride (SR141716A) from Tocris (Bristol, UK). WIN55, 212-2 and CP55,940 are potent synthetic cannabinoid agonists with similar affinity for CB1 and CB2 receptors. JZL184 is a potent, specific and irreversible inhibitor of monoacylglycerol lipase (MAGL) which increases the endogenous levels of 2-AG, an endocannabinoid with similar affinities for both cannabinoid receptors. SR141716A and SR144528 are specific antagonists of CB1 and CB2 receptors, respectively. Carbachol is a cholinergic agonist that activates muscarinic and nicotinic receptors.
WIN55,212-2 and JZL184 were administered intraperitoneally, once daily, in a volume of 5 ml/kg for seven consecutive days at the same time (between 8:00 and 9:00 a.m.). Both cannabinoid compounds were dissolved in pure DMSO and diluted with Kolliphor EL (Sigma-Aldrich) and 0.9% saline to a final proportion of (1:1:18) respectively, as vehicle. Mice were randomly assigned to one of the following seven groups: 1) Non-Tg-vehicle (n = 10), 2) 3xTg-AD-vehicle (n = 10), 3) 3xTg-AD-WIN55,212-2 (0.1 mg/kg) (n = 10), 4) 3xTg-AD-WIN55,212-2 (1 mg/kg) (n = 10), 5) 3xTg-AD-JZL184 (8 mg/kg) (n = 10), 6) Non-Tg-WIN55,212-2 (1 mg/kg) (n = 4), and 7) Non-Tg-JZL184 (8 mg/kg) (n = 4) (Fig. 1).
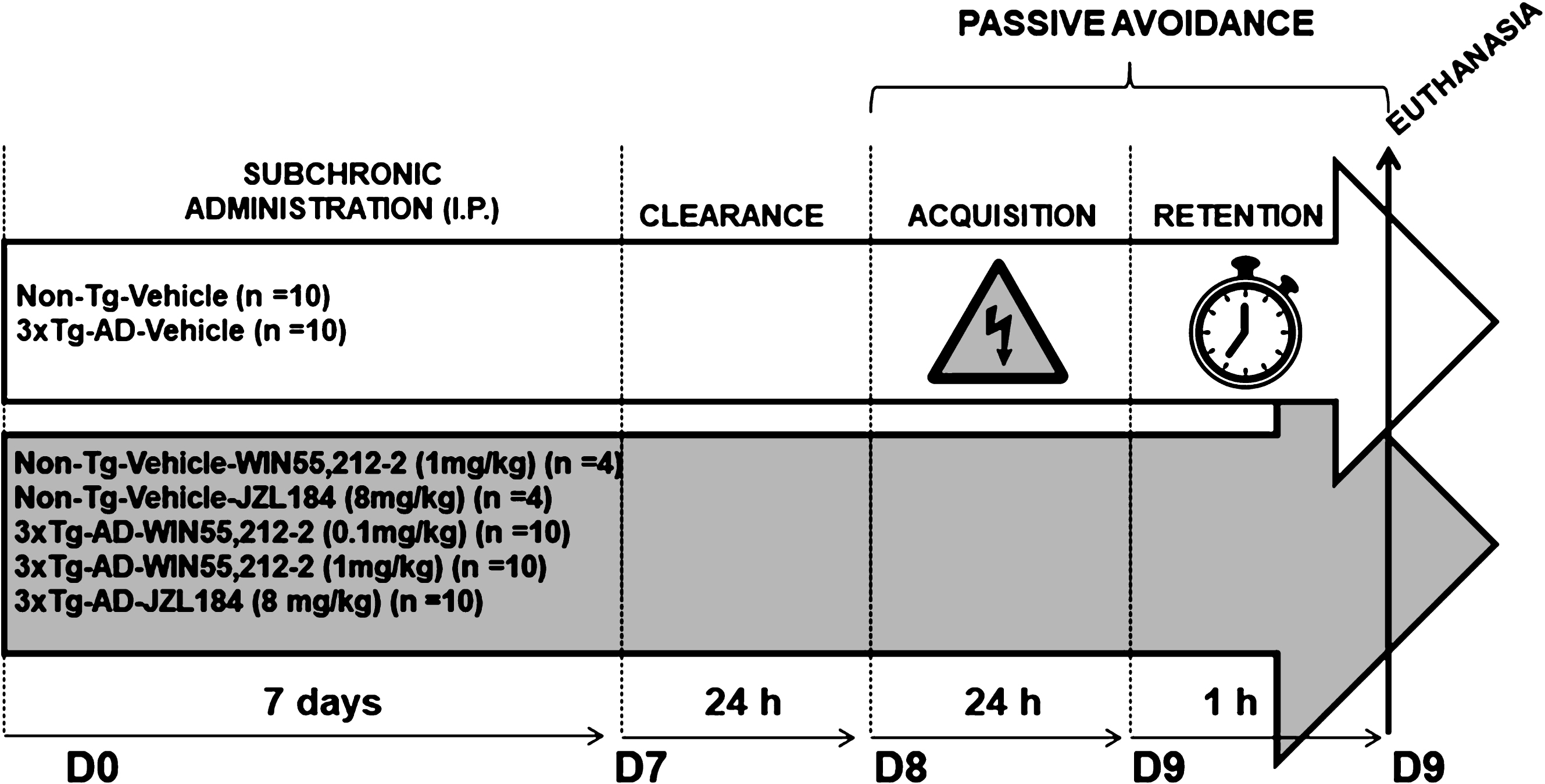
Synopsis of the experimental design including treatment schedule and behavioral assessment.
Behavioral test
On the two days following the last dose, the behavioral effects of cannabinoid administration in the step-through passive avoidance were studied (PanLab, passive avoidance box LE872). During the acquisition session, the animals were placed in the open and illuminated compartment with heads facing the door, and then allowed to explore for 30 s. Then, the door was opened, allowing the mice to enter the dark compartment. The acquisition latency, with a cut-off time of 60 s, was recorded. When the animals crossed the door, it was closed and a foot-shock (0.4 mA/2 s) was delivered. Twenty-four hours later, during the retention session, the animals were placed again into the illuminated chamber and allowed to explore for 30 s. Then, the door was opened and the step-through latency before entering the dark chamber, with a maximum cut-off time of 300 s, was recorded. No foot-shock was delivered in the retention session. Both sessions started at the same time (8:00 a.m.).
Tissue preparation
For immunohistochemical studies, 3 mice from groups 1 to 5 (see the section of “Drugs and treatments”) were anesthetized with ketamine:xylazine (90:10 mg/kg), and transcardially perfused via the ascending aorta with 50 ml warm (37°C), calcium-free Tyrode’s solution (0.15 M NaCl, 5 mM KCl, 1.5 mM MgCl2, 1 mM MgSO4, 1.5 mM NaH2PO4, 5.5 mM glucose, 25 mM NaHCO3; pH 7.4), 0.5% heparinized, followed by 4% paraformaldehyde and 3% picric acid in 0.1M PB (4°C) (100 ml/100 g b.w.). Brains were removed, post-fixed for 90 min at 4°C, and cryoprotected in 20% phosphate buffer-sucrose solution overnight. The tissue was immersed in isopentane (– 80°C). In order to get an appropriate penetration of the antibodies and acceptable signal to noise ratio, 10μm coronal sections were cryostat cut, mounted onto gelatin-coated slides, and stored at – 25°C.
For radioligand binding experiments, the remaining brain samples from groups 1 to 5 (n = 7 mice per group) and those from CB1−/− (n = 2) and CB1+/+ (n = 2) mice (including spleen samples) were removed, fresh frozen to preserve the receptor functionality, and cut into 20μm sections because the [3H] and [14C] microscales are calibrated for this thickness. Then, the sections were mounted onto gelatin-coated slides and stored at – 25°C.
Fixed and fresh frozen brain sections were obtained from five different stereotaxic coordinates in the coronal plane according to Paxinos and Watson (2001) [24]: Bregma 4.28 mm; Bregma 0.86 mm; Bregma – 0.82 mm; Bregma – 2.06 mm; Bregma – 3.28.
Radioligands and chemical reagents
[35S]GTPγS (1250 Ci/mmol) and [3H]CP55,940 (131.8 Ci/mmol) were purchased from PerkinElmer (Boston MA, USA). The [14C] and [3H]-microscales used as standards were purchased from American Radiolabelled Chemicals (St. Louis, MO, USA). The β-radiation sensitive films were purchased from Carestream. Bovine serum albumine (BSA), DL-dithiothreitol (DTT), adenosine deaminase (ADD), guanosine-5’-diphosphate (GDP), and guanosine-5’-O-3-thiotriphosphate (GTPγS) were acquired from Sigma-Aldrich. Finally, all the compounds necessary for the preparation of the different buffers were of the highest quality commercially available.
Cannabinoid receptor autoradiography
Two additional new consecutively cut sections from 3xTg-AD and Non-Tg mice (n = 7 mice per group) and from CB1−/− and CB1+/+ mice (including brain and spleen) were dried and submerged in 50 mM Tris-HCl buffer containing 1% of BSA (pH 7.4) for 30 min at room temperature, followed by incubation in the same buffer in the presence of the CB1/CB2 radioligand, [3H]CP55,940 (3 nM) for 2 h at 37°C. Nonspecific binding was measured by competition with non-labelled CP55,940 (10μM) in another consecutive slice. The CB1 receptor antagonist, SR141716A (0.1μM) and the CB2 receptor antagonist, SR144528 (0.1μM), were used together with [3H]CP55,940 in two consecutive slices to check the CB1 or CB2 receptor binding specificity. Then, sections were washed in ice-cold (4°C) 50 mM Tris-HCl buffer supplemented with 1% BSA (pH 7.4) to stop the binding, followed by dipping in distilled ice-cold water and drying (4°C). Autoradiograms were generated by exposure of the tissues for 21 days at 4°C to β-radiation sensitive film together with [3H]microscales used to specifically calibrate the optical densities to fmol/mg tissue equivalent (fmol/mg t.e.).
Labeling of activated Gi/o proteins by [35S]GTPγS binding assay
Additional newly cut consecutive sections (n = 7 mice per group) were dried, followed by two consecutive incubations in HEPES-based buffer (50 mM HEPES, 100 mM NaCl, 3 mM MgCl2, 0.2 mM EGTA and 0.5% BSA, pH 7.4) for 30 min at 30°C. Briefly, sections were incubated for 2 h at 30°C in the same buffer supplemented with 2 mM GDP, 1 mM DTT, ADD (3 Units/l), and 0.04 nM [35S]GTPγS. The [35S]GTPγS basal binding was determined in two consecutive sections in the absence of agonist. The agonist-stimulated binding was determined in another consecutive section in the same reaction buffer in the presence of the CB1/CB2 receptor agonist, WIN55,212-2 (10μM). Nonspecific binding was defined by competition with GTPγS (10μM) in another section. Then, sections were washed twice in cold (4°C) 50 mM HEPES buffer (pH 7.4), dried, and exposed to β-radiation sensitive film with a set of [14C] standards to specifically calibrate the optical densities to nCi/g tissue equivalent (nCi/g t.e.).
A similar procedure was followed for mAChR in the presence of the cholinergic agonist carbachol (100μM) (4 newly cut consecutive brain sections).
After 48 h, the films were developed, scanned, and quantified by transforming optical densities into nCi/g tissue equivalence units using a calibration curve defined by the known values of the [14C] standards (NIH-IMAGE, Bethesda, MD, USA). Nonspecific binding values were subtracted from both agonist-stimulated and basal conditions. The percentages of agonist-evoked stimulation were calculated from both the net basal and net agonist-stimulated [35S]GTPγS binding densities according to the following formula: ([35S]GTPγS agonist-stimulated binding×100/[35S]GTPγS basal binding)-100.
Immunofluorescence
Fixed 10μm coronal sections from Non-Tg and 3xTg-AD mice were air dried for 20 min and washed by immersion in PBS for 15 min at room temperature. Then, the sections were blocked with 5% normal goat serum in PBS buffer for 2 h at room temperature before being incubated with the primary antibody overnight at 4°C.
To label CB1 receptors, the primary rabbit antiserum against the CB1 receptor, PA1-743 (Affinity BioReagents, CO, USA), was diluted [1:500] in TBS (0.1 M Tris, 0.15 M NaCl, pH 7.4) containing 0.5% milk powder. The tyramide signal amplification method was used to amplify the signal associated with the CB1 receptor antiserum. Briefly, sections were washed for 30 min in TNT buffer (0.05% Tween 20 in TBS, pH 7.4) and blocked in TNB solution (10 ml TNT buffer, and 0.05 g blocking reagent, DuPont) for 1 h at room temperature. Later, the sections were incubated with horseradish peroxidase-conjugated goat anti-rabbit secondary antibody (Perkin Elmer) for 1 h followed by tyramide fluorescein-based amplification (Perkin Elmer) process in complete darkness for 10 min at room temperature. Sections were extensively rinsed in TBS.
To label the subtype-2 muscarinic receptor (M2 mAChR), rabbit anti-M2 mAChR (EMD Millipore, CA, USA) was diluted in PBS (0.1 M, pH 7.4) to a final concentration of 1:400. Then, the primary antibody was revealed by incubation (30 min at 30°C) with donkey anti-rabbit CY3. To study the cellular localization of CB1 receptor and M2 mAChR on glutamatergic or GABAergic neurons, tissue slices were incubated with primary guinea pig anti-vesicular glutamate transporter 3 (VGLUT3) and mouse anti-glutamic acid decarboxylase isoform 65kDa (GAD65) (EMD Millipore) diluted in PBS (0.1 M, pH 7.4) to a final concentration of 1:750 in both cases, and revealed by incubation (30 min at 30°C) with secondary Alexa488 or Alexa555 [1:250] donkey anti-guinea pig and FITC [1:80] or Alexa 555 donkey anti-mouse. Then, sections were incubated with Hoechst [1:106] for 15 min, washed, and mounted with p-phenylendiamine-glycerol.
Sections were screened with an Axioskop microscope (Zeiss). 630-fold magnification images for colocalization were acquired on an Axioskop Observer A1 inverted microscope (Zeiss) by optical sectioning (0.24μm/X-Y-Z-resolution) using structured illumination (ApoTome-Zeiss). Images were created with ZEN2014 software (Zeiss) and defined as signal being present without physical separation.
Statistical analyses
A two-tailed unpaired Student’s t-test was used to determine differences between genotypes (Non-Tg versus 3xTg-AD; groups 1 and 2) and one-way analysis of variance (ANOVA) for comparisons between all the groups of mice, including the vehicle-treated Non-Tg group (1 to 5), followed by Bonferroni’s post hoc test. The step-through latencies were represented as Kaplan-Meier survival curves, and for comparisons the nonparametric Log-rank/Mantel-Cox test was used which is appropriate when censored data must be analyzed, as explained in [25]. The existence of animals that reached the cut-off time of 300 s was the reason to choose this rigorous statistical analysis. Behavioral correlations with neurochemical data were analyzed with Pearson’s correlation. Statistical significance was set at p < 0.05.
RESULTS
Behavioral impairment in 3xTg-AD mice is restored following the subchronic cannabinoid administration
3xTg-AD and Non-Tg mice differed in acquisition and retention latencies during the passive avoidance test. 3xTg-AD mice took significantly longer to enter the dark compartment than Non-Tg mice (p = 0.0002, Student’s t-test; p < 0.01, one-way ANOVA followed by Bonferroni’s post hoc test for multiple comparisons) (Fig. 2A). Moreover, 40% failed to remember the foot-shock as compared to the positive response shown in 100% of Non-Tg mice (p = 0.029, Log-Rank/Mantel-Cox test) (Fig. 2B).
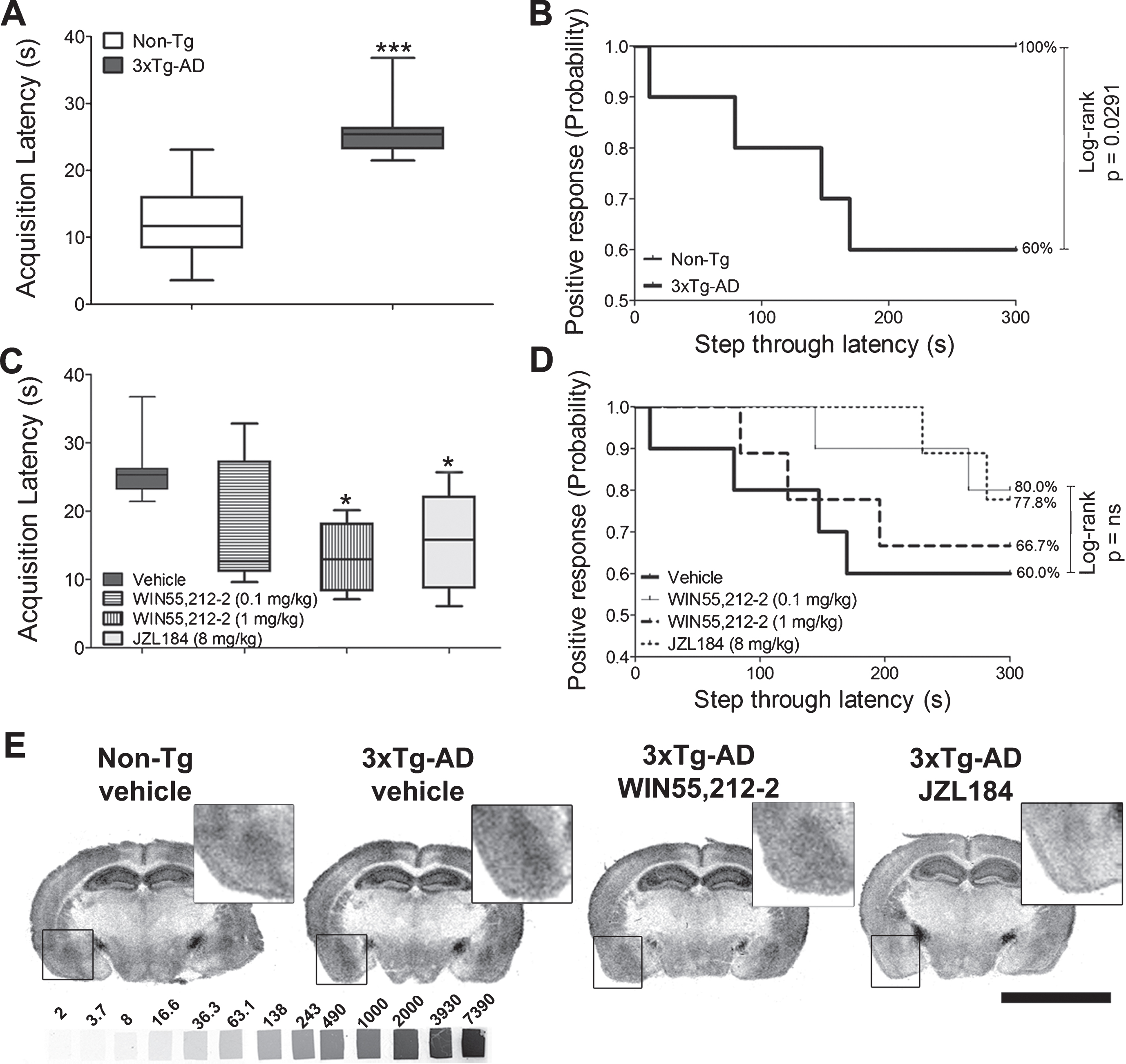
Passive avoidance test and CB1 receptor binding sites. A) Acquisition latency times during the learning trial in both genotypes in the absence of treatment; **p < 0.01 versus Non-Tg. B) Step-through latency times in both genotypes represented as Kaplan-Meier survival curves. C) 3xTg-AD mice treated with different cannabinoid agonists. The subchronic administration of WIN55,212-2 (1 mg/kg) and JZL184 (8 mg/kg) for seven consecutive days triggered a statistically significant decrease in the acquisition latency compared to that obtained in the Non-Tg group; *p < 0.05 versus 3xTg-AD mice treated with vehicle. D) Step-through latency times in 3xTg-AD mice represented as Kaplan-Meier survival curves. The probability is plotted over the step-through latency in 3xTg-AD mice after different cannabinoid-based treatments. Acquisition latencies were analyzed by a one-way ANOVA, followed by Bonferroni’s post hoc test for multiple comparisons. The step-through latencies were represented as Kaplan-Meier survival curves, and for comparisons the nonparametric Log-rank/Mantel-Cox test was used (n = 9-10 mice/group). E) [3H]CP55,940 binding autoradiography in representative brain coronal sections from both genotypes treated with vehicle and from 3xTg-AD treated with either WIN55,212-2 (1 mg/kg) or JZL184 (8 mg/kg). Note that both pharmacological treatments decreased the density of receptors in the whole grey matter including the basolateral amygdala (BLA) (boxed area). [3H]-microscales used as standards in μCi/g t.e. Scale bar: 5 mm.
No differences in the passive avoidance test were observed in Non-Tg mice, following the subchronic administration of the full agonist WIN55,212-2 (1 mg/kg). A similar effect was observed by the subchronic treatment with JZL184 (8 mg/kg), a potent inhibitor of monoacylglycerol lipase (MAGL) that increases the 2-AG endogenous levels (Supplementary Figure 1). However, the cannabinoid treatments in 3xTg-AD mice reduced the increase in acquisition latencies observed in the vehicle-treated 3xTg-AD group (one-way ANOVA followed by Bonferroni’s post hoc test for multiple comparisons), restoring the latencies to Non-Tg levels (Fig. 2C). Thus, WIN55,212-2 elicited a behavioral dose-response with slight (0.1 mg/kg, reduction 35%) or marked (1 mg/kg, reduction 50%, p < 0.05) reductions in acquisition latency as compared with those observed in the vehicle-treated 3xTg-AD animals. JZL184 (8 mg/kg), a MAGL inhibitor, induced a similar effect (reduction 42%, p < 0.05) (Fig. 2C). The treatments elicited subtle variations in memory (step-through latency) in 3xTg-AD mice, as shown in the Kaplan-Meier representation, although they did not reach statistical significance (Fig. 2D).
[3H]CP55,940 binding in different brain areas of seven-month-old Non-Tg and 3xTg-AD mice expressed in fmol/mg t.e. of CB1 receptors
Data are expressed as mean±SEM (n = 7 per group) and analyzed by one-way ANOVA, followed by Bonferroni’s post hoc test for multiple comparisons. *p < 0.05; ***p < 0.001 versus Non-Tg (vehicle). # p < 0.05; # # p < 0.01; # # # p < 0.001 versus Non-Tg (vehicle) (a); 3xTg-AD (vehicle) (b); 3xTg-AD (0.1 mg/kg WIN55,212-2) (c); 3xTg-AD (1 mg/kg WIN55,212-2) (d). $p < 0.05 versus 3xTg-AD (vehicle).
Endocannabinoid signaling in 3xTg-AD is restored following CB1 receptor desensitization
[3H]CP55,940 radioligand, that shows a similar affinity for CB1 and CB2 receptors, was used to analyze the cannabinoid receptor density. Quantitative densitometry showed increased density of cannabinoid receptors (specific [3H]CP55,940 binding sites) in the BLA (p = 0.0008, Student’s t-test; p < 0.001, one-way ANOVA followed by Bonferroni’s post hoc test for multiple comparisons) and the lateral olfactory tract nucleus (p = 0.0274, Student’s t-test; p < 0.05, one-way ANOVA followed by Bonferroni’s post hoc test for multiple comparisons), but a decrease in the glomerular olfactory bulb (p < 0.0001, Student’s t-test; p < 0.001, one-way ANOVA followed by Bonferroni’s post hoc test for multiple comparisons) of vehicle-treated 3xTg-AD mice (Figs. 2E and 5A) (Table 1 and Supplementary Table 1). [3H]CP55,940 autoradiography further revealed specific modifications in cerebral cannabinoid receptor density following the subchronic eCB system activation. The low dose of WIN55,212-2 (0.1 mg/kg) did not modify cannabinoid receptor density, but a higher dose of WIN55,212-2 (1 mg/kg) induced a significant decrease in BLA (22%; p < 0.05) which shows a dose-dependent effect in eCB signaling. Furthermore, the administration of JZL184 dramatically reduced the cannabinoid receptor density, including cortical (p < 0.01), hippocampal (p < 0.001), and amygdaloid regions (p < 0.001) (one-way ANOVA followed by Bonferroni’s post hoc test for multiple comparisons) (Figs. 2E and 5B) (Table 1 and Supplementary Table 1).
To determine the specific subtype of the cannabinoid receptor involved in the observed changes, SR141716A and SR144528, well-known selective antagonists for CB1 and CB2 receptors respectively, were used in combination with brain and spleen samples from CB1+/+ and CB1−/− mice. SR141716A blocked [3H]CP55,940 binding in brain slices from 3xTg-AD and CB1+/+ mice, but failed in spleen, as was expected in a tissue that exclusively expresses the CB2 receptor subtype. SR144528 completely blocked [3H]CP55,940 binding in spleen slices but failed in brain samples. CB1−/− mice showed an almost complete absence of [3H]CP55,940 binding in the brain, whereas they displayed similar binding levels in the spleen to those obtained in CB1+/+ mice, which demonstrates the selectivity of both antagonists and the specific deregulation of the CB1 subtype in 3xTg-AD mice (Fig. 3). The contribution of each cannabinoid receptor subtype to the observed changes was demonstrated by measuring [3H]CP55,940 binding in the presence of SR141716A, which specifically blocks the radioligand binding to CB1 receptors or, in the presence of SR144528, which specifically blocks the radioligand binding to CB2 receptors. Almost all the binding in the brain of 3xTg-AD mice and Non-Tg was blocked in the presence of SR141716A (i.e., the optical density was comparable to non-specific binding values). On the other hand, the optical density in the presence of SR144528 was comparable to that obtained in the total binding, i.e., in the absence of any antagonist (data not shown). These results demonstrate both the absence of detectable changes in CB2 receptor density in seven-month-old male 3xTg-AD mice and the CB1 receptor-mediated effects on the behavioral outcomes.
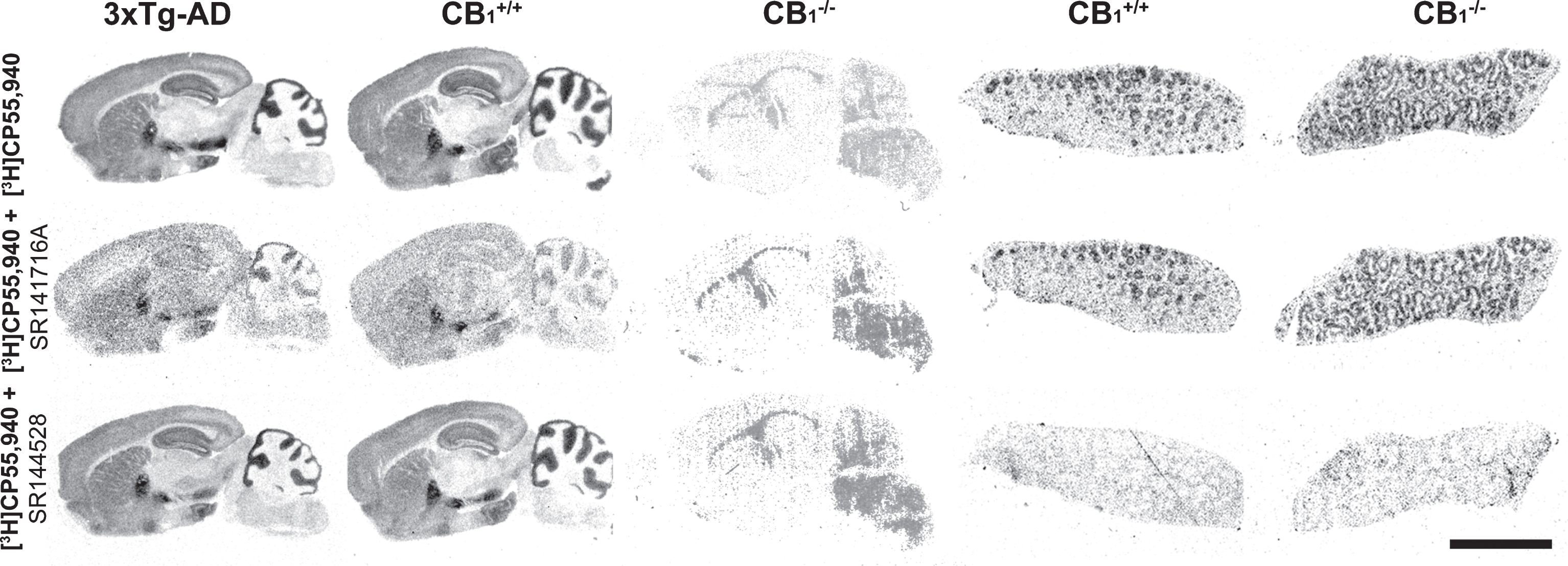
CP55,940 binding autoradiography in brain and spleen. The image shows the cannabinoid receptor distribution in brain and spleen samples from 3xTg-AD, CB1 receptor knockout (CB1−/−) and wild type (CB1+/+) mice. The total binding is shown in the top row, displaying the characteristic and well-described distribution of cannabinoid receptors in the brain, and surrounding the lymphatic nodules (white pulp) in the spleen. In the presence of 0.1μM of SR141716A, a CB1 receptor specific antagonist, binding is almost completely blocked in the brain but not in the spleen (middle row) while 0.1μM of SR144528, a CB2 receptor specific antagonist, completely displaced the [3H]CP55,940 binding in the spleen without affecting the binding in the brain (bottom row). Note the absence of binding in the brain from CB1−/− and the identical distribution in the spleen from both Non-Tg and knockout mice, revealing the preponderance of CB1 receptors in the brain and CB2 receptors in spleen tissue, and the specificity of the cannabinoid antagonists. Scale bar = 5 mm.
[35S]GTPγS binding in different brain areas of seven-month-old Non-Tg and 3xTg-AD mice evoked by WIN55,212-2 (10μM) expressed as percentage of stimulation over the basal binding
Data are expressed as mean±SEM (n = 7 per group) and analyzed by one-way ANOVA, followed by Bonferroni’s post hoc test for multiple comparisons. *p < 0.05 versus Non-Tg-vehicle; **p < 0.01 versus Non-Tg (vehicle). # p < 0.05 versus 3xTg-AD (vehicle).
Functional coupling of CB1 receptors in 3xTg-AD mice
GTPγS autoradiography allows anatomical localizing and quantification of receptor-dependent Gi/o protein activity directly in tissue. Basal activity was similar in the two genotypes. The activity of CB1 receptors evoked by WIN55,212-2 (10μM), was higher in the BLA of 3xTg-AD (p = 0.0303, Student’s t-test; p < 0.05, one-way ANOVA followed by Bonferroni’s post hoc test for multiple comparisons) (Figs. 4B and 5E) but lower in several areas such as the striatum (p = 0.0285, Student’s t-test; p < 0.05, one-way ANOVA followed by Bonferroni’s post hoc test for multiple comparisons), the glomerular olfactory bulb (p = 0.0043, Student’s t-test; p < 0.01, one-way ANOVA followed by Bonferroni’s post hoc test for multiple comparisons), and the molecular layer of hippocampal dentate gyrus (p = 0.0040, Student’s t-test; p < 0.05, one-way ANOVA followed by Bonferroni’s post hoc test for multiple comparisons) (Table 2 and Supplementary Table 2).

[35S]GTPγS autoradiography. [35S]GTPγS binding evoked by both WIN55,212-2 (10μM) for cannabinoid receptors (A-D) and carbachol (100μM) for M2/M4 muscarinic acetylcholine receptors (mAChR) (E-H), in representative coronal brain sections from Non-Tg and 3xTg-AD mice treated with vehicle and cannabinoid agonists. The highest CB1 receptor stimulation was found in the hippocampus, the most caudal portion of the globus pallidus, the deeper layers of the cortex, and the amygdaloid complex. Thus, in the amygdala, the latero-basolateral region (boxed area) (A-D) seems to be the most activated, displaying a hyperactivation in 3xTg-AD (B) mice, which is attenuated with both cannabinoids (C-D). Moreover, deregulation of mAChR functionality in 3xTg-AD mice was found. Note the decrease in the latero-basolateral region and in the pyramidal layer of the hippocampal CA1 region (boxed areas) (F) and the potentiation of muscarinic signaling in the amygdala following the subchronic administration of 1 mg/kg of WIN55,212-2 (G). [14C]-microscales used as standards in μCi/g t.e. Scale bar = 5 mm.
Functional [35S]GTPγS autoradiography of CB1 receptor activation showed a non-significant decrease of 24% and 32% in the BLA following treatment with WIN55,212-2 (1 mg/kg) and JZL184 (8 mg/kg), respectively (Figs. 4C-D and 5F) (Table 2 and Supplementary Table 2). The basal binding of [35S]GTPγS was not modified by the different cannabinoid compounds, which probably indicates the absence of changes in the constitutive activity of G protein-coupled receptors (GPCR). (Data analyzed with a one-way ANOVA followed by Bonferroni’s post hoc test for multiple comparisons).
Regression analyses
The regression analyses showed that 50% and 33% of the variation in the acquisition latencies recorded in 3xTg-AD mice were related to changes in CB1 receptor density ([3HCP55,940 binding) in the BLA (r2 = 0.5096, p = 0.0091) and/or to changes in CB1 receptor activity (evoked by WIN55,212-2) (r2 = 0.3299, p = 0.0508), respectively (Fig. 5C and G). No statistically significant correlations were found when other brain areas were compared such as the lateral olfactory tract nucleus and glomerular olfactory bulb (p = ns). Both treatments, JZL184 (8 mg/kg) and WIN55,212-2 (1 mg/kg), decreased the acquisition latencies of 3xTg-AD mice to Non-Tg mice control values due to the pharmacological desensitization of CB1 receptors to levels even lower than those observed in Non-Tg mice. When the groups were compared all together, a positive and statistically significant correlation between CB1 receptor density in the BLA and acquisition latencies was found, showing that 25% of the acquisition latency variation may be explained by changes in CB1 receptor density (r2 = 0.2499, p = 0.013), but not by changes in CB1 receptor activity (Fig. 5D and H).
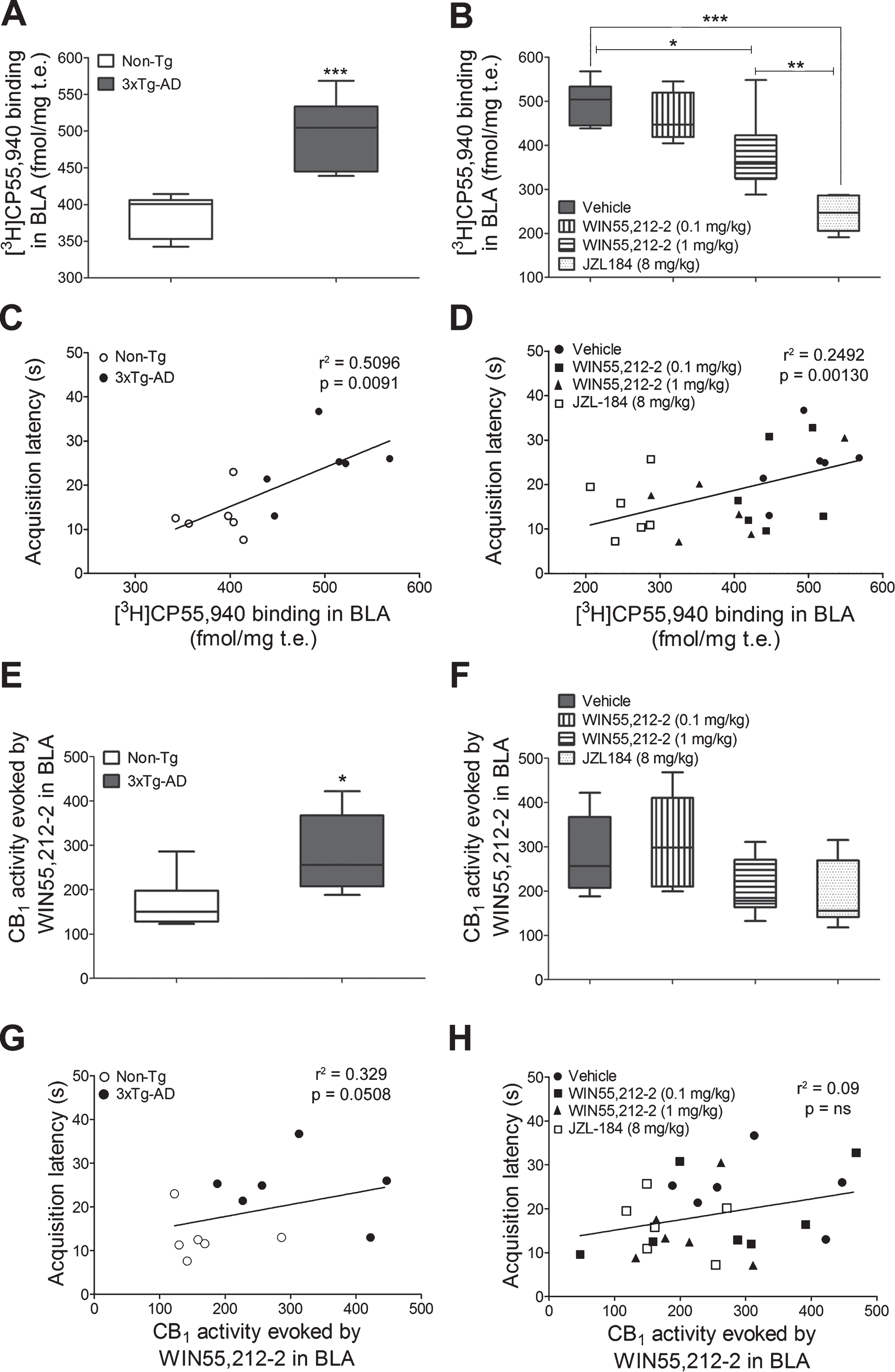
CB1 receptor-mediated signaling and behavior. [3H]CP55,940 binding in the BLA in both genotypes treated with vehicle (A) and in 3xTg-AD mice treated with WIN55,212-2 (0.1 mg/kg or 1 mg/kg) or JZL184 (8 mg/kg) (B). Correlation analyses between the CB1 receptor density in the BLA and the acquisition latency times of both genotypes (C) and of 3xTg-AD mice after cannabinoid treatments (D). Note that data are grouped according to both genotype and treatment. Quantification of CB1 receptor stimulation (% over basal activity) evoked by WIN55,212-2 (10μM) in the BLA of both genotypes (E) and of 3xTg-AD mice treated with WIN55,212-2 (0.1 mg/kg or 1 mg/kg) or JZL184 (8 mg/kg) (F). Correlation analyses between the endocannabinoid signaling in the BLA and the acquisition latency times of both genotypes (G) and of 3xTg-AD mice after cannabinoid treatments (H). Note that data are grouped according to genotype and not to treatment). Data are expressed as mean±SEM (n = 7 per group), and analyzed by a one-way ANOVA, followed by Bonferroni’s post hoc test for multiple comparisons. *p < 0.05; **p < 0.01 and ***p < 0.001. Behavioral correlations with neurochemical data were analyzed with Pearson’s correlation.
Decreased M2/M4 mAChR-mediated activity in 3xTg-AD is modulated by cannabinoid administration
We analyzed the functional coupling of mAChR to Gi/o proteins evoked by carbachol (100μM) in both genotypes and in cannabinoid-based treated 3xTg-AD mice. Transgenic mice showed decreased functional coupling in the BLA (p = 0.0258, Student’s t-test; p < 0.05 one-way ANOVA followed by Bonferroni’s post hoc test for multiple comparisons), in the lateral amygdala (p = 0.0303, Student’s t-test; p < 0.05 one-way ANOVA followed by Bonferroni’s post hoc test for multiple comparisons) and hippocampal pyramidal CA1 (p = 0.0227, Student’s t-test; p < 0.05 one-way ANOVA followed by Bonferroni’s post hoc test for multiple comparisons). Moreover, increased M2/M4 mAChR receptor activity was found in the glomerular olfactory bulb (p = 0.0095, Student’s t-test; p < 0.01 one-way ANOVA followed by Bonferroni’s post hoc test for multiple comparisons) of 3xTg-AD mice (Fig. 4F and Table 3). The administration of 1 mg/kg of WIN55,212-2 was able to increase the M2/M4 mAChR-mediated activity to similar values of Non-Tg mice; up to 60% in the BLA (p < 0.05 versus 3xTg-AD vehicle, one-way ANOVA followed by Bonferroni’s post hoc test for multiple comparisons) and up to 100% in the lateral amygdala (p < 0.01 versus 3xTg-AD-vehicle one-way ANOVA followed by Bonferroni’s post hoc test for multiple comparisons) (Fig. 4G). No modulation of M2/M4 mAChR-mediated activity was observed in other brain areas (Table 3 and Supplementary Table 3).
[35S]GTPγS binding in different brain areas of seven-month-old Non-Tg and 3xTg-AD mice evoked by carbachol (100μM) expressed as percentage of stimulation over the basal binding
Data are expressed as mean±SEM (n = 7 per group) and analyzed by one-way ANOVA, followed by Bonferroni’s post hoc test for multiple comparisons. *p < 0.05 versus Non-Tg (vehicle). # p < 0.05; # # p < 0.01 versus 3xTg-AD (vehicle).
CB1 receptors in BLA and M2 mAChR in hippocampus colocalize with GABAergic terminals in 3xTg-AD mice
To further understand the physiological cellular mechanisms of the observed changes in cannabinoid and muscarinic signaling, the immunofluorescence studies were carried out in BLA and in the ventral hippocampus (Bregma – 2.06 mm; Bregma – 3.28 respectively, according to Paxinos and Watson [24]). The different nuclei of the amygdala exhibited distinct CB1 receptor immunostaining patterns and were clearly defined. The dense CB1 receptor immunoreactive puncta observed at the BLA suggested a presynaptic localization of CB1 receptor. Immunofluorescence assays for VGLUT3 and GAD65 and the subsequent colocalization studies suggested the inhibitory nature of CB1 receptor containing presynaptic boutons (Fig. 6).

Localization of CB1 receptors in the BLA. Double labeling of tissue sections including the amygdaloid complex from seven-month-old 3xTg-AD mice processed for CB1 receptor (in green) and vesicular glutamate transporter type 3 (VGLUT3) (A2 and C2 in red) as a glutamatergic marker, and glutamic acid decarboxylase isoform 65kDa (GAD65) (B2 and D2 in red) as a GABAergic presynaptic marker. The different amygdaloid nuclei exhibited specific CB1 receptor-immunostaining patterns. VGLUT3 was distributed presumably in postsynaptic somatodendritic compartment (A2 and C2) while GAD65 immunostaining clearly delineated presynaptic inhibitory boutons (B2 and D2). In low magnification images, note the distribution of CB1 receptors surrounding positive glutamatergic neurons (A3) and sharing localization with GAD65 (B3); scale bar = 150 μm. High magnification images showed the intracellular localization of VGLUT3 (C2) closely surrounding the nuclei stained with Hoechst (C3 in blue) revealing the almost complete lack of colocalization with CB1 receptors (C4). Conversely, CB1 receptors were located on GAD65-positive terminals (D4), revealing its presynaptic localization on inhibitory synaptic boutons. Scale bar = 10μm. Bregma – 1.82 mm. CeL: central amygdaloid nucleus, lateral division; La: lateral amygdaloid nucleus; BLA: basolateral amygdaloid nucleus, anterior part; BLP: basolateral amygdaloid nucleus, posterior part; BMP: basomedial amygdaloid nucleus, posterior part.
M2 mAChR immunoreactivity was differentially localized along the hippocampal formation. The pyramidal neurons of CA1-CA3 displayed a dense network of fibers delineating the perikarya in basket-like formations. VGLUT3 displayed a somato-dendritic immunostaining, but GAD65 immunoreactivity was present as a dense plexus of fibers around pyramidal neurons with a similar distribution to M2 mAChR. Colocalization studies confirmed the presence of M2 mAChR in presynaptic GABAergic terminals with a high degree of co-immunolabeling with GAD65, but an almost total absence of expression on VGLUT3 positive cells (Fig. 7). However, in the amygdala, the M2 mAChR are not distributed in presynaptic GABAergic terminals or in VGLUT3 positive compartments (images not shown).

Localization of M2 mAChR in the hippocampus. Double labeling of tissue sections including the CA1 field of the hippocampus from a representative seven-month-old 3xTg-AD mouse processed for M2 mAChR (in red) and VGLUT3 (A2 and C1 in green) as a glutamatergic marker, and GAD65 (B2 and D1 in green) as a GABAergic presynaptic marker. The different hippocampal subfields exhibited specific M2 mAChR-immunostaining patterns delineating the perikarya of the large pyramidal neurons in basket-like formations. VGLUT3 was distributed near the nucleus (A2 and C1), presumably in the somatodendritic compartment of pyramidal neurons, while GAD65 immunostaining (B2 and D1) clearly delineated presynaptic inhibitory boutons. In low magnification images, note the complementary distribution of M2 mAChR-immunoreactivity to VGLUT3, surrounding the pyramidal neurons (A3), and the localization in GAD65-positive presynaptic terminals (B3); scale bar: 150μm. High magnification images revealed the intracellular localization of VGLUT3 (C1) closely surrounding the nuclei stained with Hoechst (C3 in blue) and the almost complete lack of colocalization with M2 mAChR (C4). Conversely, M2 mAChR were distributed in GAD65-positive terminals, revealing the presynaptic localization on inhibitory synaptic boutons (D4). Scale bar = 10μm. Bregma – 3.08 mm. Alv: alveus of the hippocampus; Or: oriens layer of the hippocampus; Py: pyramidal cell layer of the hippocampus; Rad: radiatum layer of the hippocampus; LMol: lacunosum molecular layer of the hippocampus.
DISCUSSION
The eCB system has emerged as a promising target for the treatment of several neurodegenerative disorders including AD. Here, we provide evidence of neuroanatomical and neurochemical modifications related to the eCB neuromodulatory system and muscarinic cholinergic signaling in the 3xTg-AD mouse model and their behavioral outputs at 7 months of age, once the cognitive impairment is clearly established and is concurrent with limbic system-mediated symptoms [17]. The results point to the eCB system as a key modulator of neuronal homeostasis involved in learning or acquisition processes.
The present study examines, for the first time, the neurochemical effects of cannabinoid agonism in 3xTg-AD mice and their behavioral correlates in a learning and memory task under fear conditions, which can be relevant in relation to clinical interventions at the onset of the disease.
CB1 receptor desensitization in BLA and decrease of acquisition latency in 3xTg-AD mice to Non-Tg levels
The results provide evidence that repeated cannabinoid administration was able to decrease the acquisition latency in 3xTg-AD mice to Non-Tg levels, which is related to the CB1 receptor desensitization recorded in the BLA. Interestingly, we observed both, a downregulation of CB1 receptors and an attenuation of their functional coupling to Gi/o proteins induced by the subchronic administration of JZL184, comparable to the results obtained in a previous study but at different doses [26]. Moreover, the administration of WIN55,212-2 (1 mg/kg) decreases the acquisition latency and slightly also the CB1 receptor functionality in the BLA.
Our results confirm previous findings reported for JZL184, which selectively increased brain 2-AG and pointed to the inhibition of the MAGL as a promising target to indirectly potentiate the activation of CB1 receptors [9, 27]. In this sense, pharmacological blockade or genetic deletion of MAGL dramatically raises brain 2-AG levels, downregulates CB1 receptors, and modulates synaptic plasticity, learning, memory and anxiety-like behavior [28]. A recent study shows that the intra-BLA administration of both AEA and 2-AG hydrolysis inhibitors is able to attenuate anxiety-like responses, which are dependent on deregulated levels of eCB in the amygdala [29]. Conversely, chronic CB1 receptor blockade induced an upregulation of CB1 receptor expression and modified anxiety-like behaviors [30]. The contribution of the eCB levels or the observed CB1 receptor signaling regulation in the BLA of 3xTg-AD mice, to the reported differences in learning or acquisition latencies, should be clarified in further studies.
The results of the passive avoidance test, used to evaluate learning and memory of an aversive electrical stimulus under stressful conditions, could indicate fear and/or diminished motivation to explore as shown in a lesioned rat model of AD [31]. 3xTg-AD mice displayed higher acquisition latencies as compared to controls. Fear or anxiety-like responses have been shown in the contextual fear-conditioning, in the open field, dark-light box and in the passive avoidance tests in this mouse model of AD at 6 months of age [18, 21]. Stover et al. [32] observed that 6-month-old 3xTg-AD mice showed enhanced motor performance on the rotarod, but there was no difference in voluntary motor activity between genotypes. We observed that the subchronic administration of cannabinoids to Non-Tg mice did not modify the behavior in the passive avoidance test, suggesting that the treatments do not cause changes in voluntary movement. A specific battery of motor behavior test in 3xTg-AD mice treated with cannabinoids at different ages is necessary since their effects depend on the used test, the age and rodent strain [9, 33].
Regarding the possible involvement of cannabinoid signaling in these behavior modulations, we report specific changes in density and activity of CB1 receptors, indicative that cannabinoid signaling is potentiated in the BLA and attenuated in the olfactory bulb and hippocampal dentate gyrus of transgenic mice. Our results support the studies which report a significant increase in CB1 receptor density in the BLA when only intracellular Aβ accumulation can be detected and may be related to the symptoms of fear shown by these mice [15, 21]. The specificity for CB1 receptor was demonstrated by the anatomical pattern of distribution of [3H]CP55,940 binding sites in brain compared to that of spleen. Therefore, tangentially to the objective of this work, an absence of significant CB2 receptor-mediated detectable signal in the CNS of seven-month-old 3xTg-AD mice was found. Although, upregulation of CB2 receptors has been previously associated to neuroinflammation in AD patients, these results show the lack of oligomeric-Aβ-associated neuroinflammatory response related to CB2 receptor signaling, coinciding with the onset of earlier markers of disease in 3xTg-AD mice [34–37].
Depending on the specific location of CB1 receptors, on inhibitory or excitatory neurons, the functional and physiological outcomes of deregulated endocannabinoid signaling may be useful to understand the present results. Previous studies had reported that stressing factors result in a modulation of the endocannabinoid levels in the amygdala, and also induce a subsequent CB1 receptor-mediated suppression of GABA release specifically in the BLA [34, 38–40]. The immunochemical results in the BLA showed that the localization of CB1 receptors is more frequent in GABAergic than in glutamatergic compartments, even though CB1 receptors have been previously detected in both of them [41–43]. The detected CB1 receptors in BLA were in the proximity of GAD65 (the enzyme glutamate decarboxylase; GAD, associated with inhibitory nerve termini) [44]. In addition, the detection of VGLUT3 was used to identify both excitatory presynaptic boutons and glutamatergic somatodendritic compartments [45, 46]. Although CB1 receptors are present in both GABAergic and glutamatergic cellular compartments in areas such as the hippocampus, their activity seems to be lower in the inhibitory terminals [47]. However, in BLA, CB1 receptors are highly expressed in axon terminals of GABAergic neurons modulating GABA release via a presynaptic mechanism [48]. Some authors have related long-lasting increase of anxiety-like behaviors with a hyperactivity of BLA as consequence of a decrease in the inhibitory synaptic transmission [49, 50]. Thus, eCB-mediated suppression of inhibitory inputs to BLA neurons is involved in the cellular mechanism for the stress-induced increases in anxiety-like behavior [51]. Different studies suggest that drugs targeting the endocannabinoid system (e.g., endocannabinoid degrading enzymes inhibitors) could be used as a potential treatment strategy for anxiety and mood disorders [38, 51–53]. Globally, the present findings suggest an upregulation of the eCB tone in 3xTg-AD mice in areas such as the BLA, which should alter the local excitatory– inhibitory balance, as a possible underlying mechanism that may be involved in the observed differences in the acquisition phase of the test. Furthermore, a reversion of the acquisition latencies to those of Non-Tg mice was recorded after the eCB signaling attenuation mediated by a pharmacological desensitization of CB1 receptors, suggesting that suppression of inhibition induced by increase of CB1 signaling in the BLA of 3xTg-AD mice, would result in an enhanced excitatory input.
This decrease in GABAergic neurotransmission would act as an important component of the neurobiological mechanisms controlling fear-related behavioral responses probably contributing to the observed differences in acquisition latency, which should be further confirmed using additional behavioral studies.
Moreover, the administration of WIN55,212-2 (1 mg/kg), but not JZL184, was able to induce a significant increase in the activity mediated by mAChR in the latero-BLA complex and hippocampus but not in the cortex or in the glomerular olfactory bulb. This possible crosstalk between both systems in limbic areas, suggests a selective effect dependent on the cannabinoid treatment and on the brain region. This specific CB1 receptor-driven modulation of cholinergic neurotransmission in the amygdala could also be involved in the behavioral outcomes recorded with the passive avoidance test. In addition, the results support previous studies describing the role of BLA cholinergic system, via mAChR, in memory retrieval in fear-induced learning [54, 55].
On the other hand, M2 mAChR, which are not localized in CB1-GABAergic terminals, could be responsible of the crosstalk between both systems in latero-BLA. Further anatomical and behavioral studies are necessary to understand the meaning of CB1 receptor-induced modulation of the muscarinic control on acquisition latency, as a possible indicator of states involving fear, attention, agitation or confusion.
The subchronic administration of WIN55,212-2 or JZL184 failed to induce significant modifications in step-through latency in either Non-Tg or 3xTg-AD mice
Regarding the memory process, step-through latency clearly distinguished the cognitively impaired AD-phenotype of 3xTg-AD mice, in accordance to previous studies [19, 56]. However, under the present experimental conditions, we cannot rule out the possibility that the differences found in the acquisition, or even in the consolidation, may also contribute to the performance of step-through latency. The desensitization of CB1 receptors by means of subchronic administration of WIN55,212-2 or JZL184 failed to induce significant modifications in step-through latency in either Non-Tg or 3xTg-AD mice. However, previous studies analyzing the effects of other CB1 agonists in a different transgenic mice model of AD have reported a reduction in cognitive impairment [57].
On the other hand, the analgesic effects of acute CB1 receptor activation are well known [58], and one may speculate that the administration of cannabinoids may contribute to alter the pain perception leading to increase the pain threshold of the foot-shock [59]. In this sense, the repeated administration of both high (40 mg/kg) or low (8 mg/kg) doses of JZL184, the latter being that used in the present study, were able to induce a loss of the CB1 receptor-mediated analgesic activity, probably as a consequence of the CB1 receptor downregulation [9, 61]. This has been extensively reviewed in a recent paper [62]. Therefore, the subtle variations in step-through latencies recorded after the cannabinoid administration should not be biased by a possible increase in analgesia. Moreover, the limbic system involving the cholinergic neurotransmission may be controlling specifically the consolidation and extinction of aversive or traumatic memories [63]. Further behavioral analyses by means of non-aversive stimulus-based learning and memory tests will contribute to clarify this issue since 3xTg-AD mice do not seem to differ from Non-Tg in pain thresholds [19, 64]. Interestingly, muscarinic activation, through the M2 mAChR subtype, modulates hippocampal neuronal plasticity, and the lack of these receptors leads to cognitive impairment in the passive avoidance test [65, 66]. The present immunofluorescence studies revealed the presynaptic localization of M2 mAChR in GABAergic terminals, presumably making contact with postsynaptic VGLUT3 immunoreactive pyramidal neurons in CA1-CA3. These results are consistent with those reported in rat brain, suggesting that ACh via M2 mAChR reduces GABA release from presynaptic inhibitory terminals. The final effect could be an increase of the activity in the dendritic region of pyramidal neurons, as previously described [67, 68]. The significant reduction in choline acetyltransferase activity described in the hippocampus from middle-aged 3xTg-AD mice, not associated with the loss of cholinergic neurons, may be related to the observed decrease in mAChR functionality leading to enhance the inhibitory tone of the pyramidal neurons from CA1 [13]. We suggest that intraneuronal accumulation of Aβ, beginning at 4 months of age, may trigger an early deregulation of the hippocampal muscarinic neurotransmission, as observed in seven-month-old 3xTg-AD mice, thereby contributing to the cognitive impairment observed in this model [17]. Moreover, an excitatory/inhibitory imbalance mediated by a deregulated presynaptic muscarinic neurotransmission in the hippocampus may underlie the impaired synaptic plasticity, i.e., the neurobiological substrate for creating and maintaining new memories.
Conclusions
We provide evidence that both endocannabinoid and muscarinic signaling are altered in seven-month-old male 3xTg-AD mice, when earlier pathological markers of disease are clearly established. CB1 receptor-mediated hyperactivity in BLA may have behavioral correlates that correspond with the restoration to control levels after pharmacological desensitization of CB1 receptors.
WIN55,212-2 administration restores muscarinic neurotransmission in vulnerable limbic areas to Non-Tg levels demonstrating a crosstalk between both systems.
CB1 receptor desensitization could be a plausible strategy to palliate specific behavior impairments associated with genetic variants of AD.
Footnotes
ACKNOWLEDGMENTS
This work was supported by the Departments of Economic Development (Elkartek KK-2016/00045) and Education (IT975-16) of the Basque Government. Technical and human support provided by General Research Services SGIker from University of the Basque Country (UPV/EHU), co-financed by Ministry of Economy and Competitiveness (MINECO) of the Spanish Government, European Regional Development Fund (ERDF), and European Social Fund (ESF). Dr. Frank M LaFerla, University of California Irvine, Irvine, CA, (USA) for kindly providing the progenitors of the Spanish colonies of 3xTg-AD and non-transgenic mice. A. LL. is the recipient of a fellowship from the Basque Government (BFI 2012-119). All authors have seen and approved the manuscript being submitted.