
Abstract
Depression is one of the most frequent psychiatric symptoms observed in people during the development of Alzheimer’s disease (AD). We hypothesized that genetic factors conferring risk of depression might affect AD development. In this study, we screened 31 genes, which were located in 19 risk loci for major depressive disorder (MDD) identified by two recent large genome-wide association studies (GWAS), in AD patients at the genomic and transcriptomic levels. Association analysis of common variants was performed by using summary statistics of the International Genomics of Alzheimer’s Project (IGAP), and association analysis of rare variants was conducted by sequencing the entire coding region of the 31 MDD risk genes in 107 Han Chinese patients with early-onset and/or familial AD. We also quantified the mRNA expression alterations of these MDD risk genes in brain tissues of AD patients and AD mouse models, followed by protein-protein interaction network prediction to show their potential effects in AD pathways. We found that common and rare variants of L3MBTL2 were significantly associated with AD. mRNA expression levels of 18 MDD risk genes, in particular SORCS3 and OAT, were differentially expressed in AD brain tissues. 13 MDD risk genes were predicted to physically interact with core AD genes. The involvement of HACE1, NEGR1, and SLC6A15 in AD was supported by convergent lines of evidence. Taken together, our results showed that MDD risk genes might play an active role in AD pathology and supported the notion that depression might be the “common cold” of psychiatry.
INTRODUCTION
Alzheimer’s disease (AD), the most common neurodegenerative disease, is becoming a serious global health issue [1, 2]. It is generally considered as a cognitive disorder, resulting from accumulation of extracellular amyloid-β (Aβ) plaques and intracellular neurofibrillary tangles, neuron loss, cerebral atrophy, and neuroinflammation [1, 2]. Almost all AD patients have neuropsychiatric symptoms during the course of the disease [3]. Depression is one of the most frequent psychiatric symptoms observed in the development of mild cognitive impairment (MCI) and AD [3, 4]. The symptoms of depression, technically referred to as major depressive disorder (MDD), are clinically heterogeneous, with a lifetime prevalence of approximately 16% [5, 6]. Depression and AD are common conditions in older age, with impaired cognition [5]. However, the relationship between depression and AD is elusive. It is unclear whether the comorbidity of MDD in AD is the cause or just a byproduct. Besides the comorbidity, it has been reported that a history of depression might increase risk of developing AD, and people with both MCI and a history of depression would progress to AD at a much higher rate [7–10]. Other studies also suggested that depression may be a prodromal symptom of AD [11–13]. These reports indicated a potentially causal role of depression in the course of AD.
Genetic factors account for ∼37% of liability to MDD and depressive symptoms [14], while AD has a high heritability (∼79% ) [1, 15]. It is reasonable to hypothesize that genetic factors conferring risk of depression might also underlie the genetic basis of AD, with shared risk alleles for both MDD and AD. Indeed, our previous findings have shown that the MDD-risk allele CFH Y402H [16] also affects AD risk [17]. Moreover, the MEF2C gene, identified by the most recent and largest genome-wide association study (GWAS) for MDD [18], was a well-established AD risk gene in the largest GWAS for AD [19]. There might be more depression risk genes involved in AD, yet this kind of investigation was highly dependent on the reliability of recognized MDD risk genes. Numerous GWASs for MDD have been conducted in recent years, but no genome-wide significant risk locus has been identified due to clinical heterogeneity of the disorder [20–22]. Recently, two large scale GWASs for MDD had successfully identified several robust risk loci: the CONVERGE performed a GWAS for MDD in a phenotypically homogeneous sample of female Han Chinese and identified two loci exceeding the genome-wide significance [23]; the largest meta-analysis combining datasets of 23andMe and MDD GWAS from the Psychiatric Genomics Consortium (PGC) identified 17 single nucleotide polymorphisms (SNPs) showing the genome-wide significance [18]. The MDD risk gene list of these two studies [18, 23] offered a start point for us to test a hypothesis of common genetic basis of MDD and AD. In this study, we analyzed 31 genes that were located in the reported MDD risk loci [18, 23] in AD at the genomic and transcriptomic levels to test this hypothesis.
MATERIALS AND METHODS
Assignment of MDD risk genes
Though there were several GWASs for MDD [20–22], genome-wide hits were only observed in the most recent 23andMe-based GWAS in populations of European ancestry [18] and the CONVERGE study in Han Chinese populations [23]. The 23andMe-based GWAS meta-analysis combined three data sets: Discovery phase, 75,607 cases and 231,747 controls; PGC MDD, 9,240 cases and 9,519 controls; Replication stage, 45,773 cases and 106,354 controls, resulted in the largest sample size in MDD genetic research [18]. A total of 17 independent genome-wide significant SNPs from 15 loci, covering 21 surrounding genes (defined by a distance of <300 kb window within the GWAS loci), were identified and validated in the original study [18] (Supplementary Table 1). The CONVERGE Consortium performed whole genome sequencing of 5,303 MDD cases and 5,337 controls at a lower coverage and identified two loci reaching a genome-wide significance, which could be validated in 3,231 cases and 3,186 controls [23]. The two loci contained 10 surrounding genes in ∼500 kb of the signal as shown in the original study [23] (Supplementary Table 1). These 19 SNPs represented the most robust MDD risk loci ever identified, albeit it was yet unknown which gene is the causal gene targeted by the GWAS hits and more independent validations should be performed to confirm these risk loci. In this study, the 19 proxy MDD GWAS SNPs, as well as the 31 genes (including 21 for the 23andMe-based GWAS [18] and 10 for the CONVERGE [23]) within the 19 GWAS loci (as defined by the original studies [18, 23]) were subjected to comprehensive analyses in AD datasets (Fig. 1 and Supplementary Table 1).
Workflow and summary of the current study. We aimed to systematically investigate whether candidate genes located in the major depressive disorder (MDD)-associated loci were involved in Alzheimer’s disease (AD). Genome-wide significant hits from the largest meta-analysis combining 23andMe and MDD GWAS from the Psychiatric Genomics Consortium (PGC) in populations of European ancestry [18], as well as the CONVERGE study in Chinese population [23], were used to identify potential MDD candidate genes. We initially checked whether common variants within these MDD-associated GWAS loci were associated with AD by using summary data of the International Genomics of Alzheimer’s Project (IGAP) [19]. In addition to common variant association, we examined the entire coding regions of the 31 MDD candidate genes in 107 Han Chinese patients with early-onset and/or familial AD for rare variants. Transcriptomic profiling were performed using GEO data as processed in our recent report [33]. Protein-protein interaction network was constructed using online tool STRING (https://string-db.org/).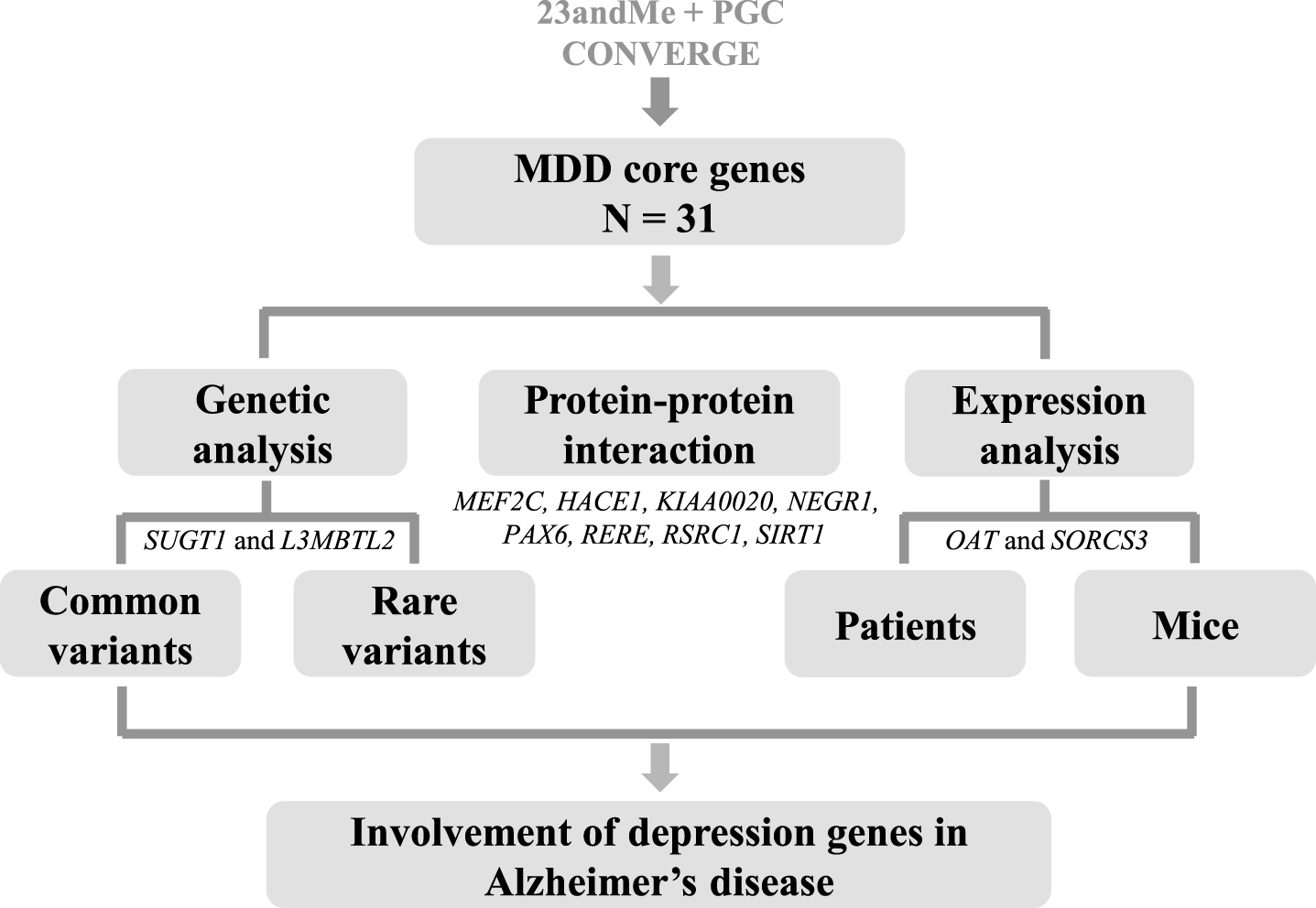
Association of common variants in MDD risk genes with AD using publicly available GWAS data
Involvement of MDD risk genes in AD was initially evaluated by using the publicly available International Genomics of Alzheimer’s Project (IGAP) GWAS data [19], which focused on the association of common variants with AD susceptibility. The IGAP data contained 7,055,881 SNPs in 17,008 AD cases and 37,154 controls [19]. Summary statistics were downloaded from http://web.pasteur-lille.fr/en/recherche/u744/igap/igap_download.php. Single-site association results of all variants within 10 kb of the MDD risk genes were retrieved. The single-site association results were subjected to the gene-based test using the online tool Versatile Gene-based Association Study (VEGAS2 v02, https://vegas2.qimrberghofer.edu.au/) [24, 25]. Detailed methodology of the gene-based test was described in the original study [24]. Briefly, this test considers a full set of markers within a gene and the linkage disequilibrium information between markers by using simulations [24].
Identification of coding variants in MDD risk genes in AD patients using next generation sequencing
The coding and flanking regions (UTRs and exons) of the 31 MDD risk genes were analyzed in 107 Han Chinese patients with AD by using next generation sequencing, to identify potentially causal variants. We focused on these AD patients that had an early onset age <55 years old and/or a positive family history (46.7% females, age 64.6±10.29 years, 37.4% APOE ɛ4 carriers), as these patients might be geneticically informative comparted with sporadic or late-onset AD. The targeted regions were captured by Nimblegene SeqCap Kit. Processed final libraries for each individual were pooled and sequenced on Illumina HiSeq2500 or 4000 (150-bp paired-end, Illumina, San Diego, CA, USA). Low quality raw reads were removed using Trimmomatic-0.32 [26]. Quality-filtered sequenced reads were aligned to the human genome reference assembly (build GRCh37/hg19) using Burrows-Wheeler Aligner (BWA) [27]. Picard Tools (http://broadinstitute.github.io/picard/) were used to flag duplicated reads. SNP call was performed through the canonical pipeline recommended by the Best Practice Variant Detection with the GATK (Genome Analysis Toolkit, https://www.broadinstitute.org/gatk/guide/best-practices) [28]. Variant Quality Score Recalibration (VQSR) from the GATK package was used to filter spurious variants due to sequencing errors and/or mapping artifacts. The ANNOVAR was used to annotate variants, which were assigned into different functional categories according to their locations and expected effects on the encoded gene products [29]. Missense, nonsense, frameshift, and splice site variants were defined as functional variants. We used the exome data of 160 Chinese individuals (40.6% females, age 52.6±16.5 years) showing no signs of memory loss and no familial history of neurodegenerative disorders [30] as a control sample. To increase the sample size of controls, the whole genome data of Han Chinese in Beijing (CHB, N = 103) and Southern Han Chinese (CHS, N = 105) from the 1000 Genome Project phase 3 [31] were pooled with the exome data of 160 Chinese individuals [30] as the population control (N = 368). Allele frequency data of 4327 East Asians from the ExAC (Exome Aggregation Consortium, http://exac.broadinstitute.org/) [32] was used as another control to validate the results in Chinese.
Expression changes of the MDD risk genes in AD brain samples and mouse models using our previously processed data
In addition to genomic analysis, we analyzed the mRNA expression profiles of the MDD risk genes in four AD-affected brain regions (frontal cortex tissues of 413 patient and 284 controls; temporal cortex tissues of 158 patients and 174 controls; hippocampus tissues of 74 patients and 65 controls; entorhinal cortex tissues of 39 patients and 39 controls), following the approach described in our recent study [33]. In brief, raw microarray data for each brain region was retrieved from the NCBI Gene Expression Omnibus database (GEO, http://www.ncbi.nlm.nih.gov/geo/) and renormalized as one combined dataset with an enlarged sample size. Details of data sources and processing were described in our previous study (Ref. [33] and references therein), and the expression data collection and renormalized expression profiles can be accessed through our database http://www.alzdata.org [33].
In addition to the differential expression analysis in brain tissues of AD patients, we investigated the expression alterations of these MDD risk genes in brain tissues of mouse AD models [34]. In brief, the transgenic mouse models with human mutant genes responsible for familial AD, which showed the presence of AD pathological features, were used for the genome-wide microarrays [34]. Hippocampus and cortex tissues were tested by using the MouseRef8 v2 (Illumina) microarray platform. Microarray data was processed and shared by the Mouse Dementia Network, available at Mouseac (http://www.mouseac.org). More details about this dataset were described in the original study [34]. Correlation between mRNA expression levels of genes of interest and the quantified level of pathology (Aβ plaques and tau burden) was measured based on the processed data, using the Pearson correlation test, as had been described in our recent study [33].
Protein-protein interaction network of the MDD risk genes with AD core genes
To evaluate the involvement of these MDD risk genes in the molecular network of AD, we performed the protein-protein interaction (PPI) analysis using proteins of the 31 MDD risk genes, together with 43 known AD genes (identified by GWAS and linkage studies [35]) using the online tool STRING (https://string-db.org/).
Convergent functional genomics (CFG) analysis
To cross-validate the involvement of target genes in AD at different levels, we used the convergent functional genomic (CFG) approach [36, 37] that integrates multiple lines of AD-related evidence, as we had done recently [33]. A gene was defined as AD-related if it: 1) showed a significant association with AD at the gene-based test using the IGAP data [19]; 2) had rare variants that were significantly associated with AD in 107 Han Chinese patients with AD; 3) was differentially expressed in any brain tissue of AD patients according to the AlzData.org [33]; 4) was correlated with AD pathology in AD mice based on the Mouseac (http://www.mouseac.org) [34]; or 5) was involved in the PPI network that was formed by the AD core genes. For each line of evidence, one point was assigned if the above fact was observed; otherwise zero point. This integration system led to CFG scores ranging from 0 to 5 points.
RESULTS
Associations of common and rare variants of the MDD risk genes with AD
Among the 19 genome-wide significant MDD-related SNPs (Supplementary Table 1), two SNPs (L3MBTL2 rs2179744 and intergenic rs4543289) showed positive associations with AD in IGAP dataset [19]. It is reasonable that other SNPs within the 31 MDD risk genes, rather than the 19 GWAS hits, might be associated with AD. We searched for other SNPs within the 31 MDD risk genes and found that 21 out of the 31 genes had SNPs showing nominally significant associations (p < 0.05) with AD (Table 1). Among them, three genes, SUGT1 (Gene-based p = 1.97×10−3), L3MBTL2 (Gene-based p = 9.60×10−3), and the known AD gene MEF2C (Gene-based p = 1.19×10 - 2), were significantly associated with AD at the gene level (Table 1).
List of MDD risk genes and association analysis of common variants in the MDD GWAS loci with AD
Note: No. of SNPs, number of SNPs included in the gene-based test. Start/Stop, range of target gene (position based on human genome reference assembly (build GRCh37/hg19)). The gene-based p-value was calculated by the online tool Versatile Gene-based Association Study (VEGAS2 v02, https://vegas2.qimrberghofer.edu.au/) [24, 25]. Top SNP and Top SNP p-value were retrieved from the summary statistics of the International Genomics of Alzheimer’s Project (IGAP) [19]. Significant p-values (<0.05) were marked in bold. One (MIR759) out of the 31 genes had no data available.
Besides associations of common variants with AD, we analyzed the entire coding region of the 31 genes in 107 Han Chinese patients with early-onset and/or familial AD using targeted sequencing, to investigate whether there was an enrichment of rare coding variants in these MDD risk genes in AD patients (Supplementary Table 2). Six genes, TMEM161B, L3MBTL2, SLC6A15, KIAA0020, LHPP, and MYPN, had a missense variant with nominally significance in AD (Table 2). Two of them (L3MBTL2 and SLC6A15) showed consistent associations when compared with both our in-house control [30] and the ExAC reference control [32]. Notably, L3MBTL2, which was also associated with AD in the common variant analysis, had a rare missense variant rs3804097 (p. I7V) that was enriched in AD (MAF = 4.7% ) compared with our in-house controls (MAF = 1.6% , p = 1.63×10 - 2, OR = 2.99) and the ExAC reference control (MAF = 2.7% , p = 8.60×10 - 2, OR = 1.78).
Significant coding variants in MDD risk genes in 107 AD patients
Note: Position was based on human genome reference assembly (build GRCh37/hg19); Allele, reference allele/alternative allele; AC, allele count; AN, allele number; OR, odds ratio; ExAC, Exome Aggregation Consortium (http://exac.broadinstitute.org/) [32]; AD, 107 subjects with Alzheimer’s disease; CN, 368 subjects combining 160 in-house non-dementia individuals [30] and 208 Chinese from the 1000 Genome project [31]. Significant p-values (<0.05) were marked in bold. The ANNOVAR was used to annotate variants, which were assigned into different functional categories according to their locations and expected effect on the encoded gene products [29]. Missense, nonsense, frameshift, and splice site variants were defined as functional variants. # Coding variants predicted to be damaging according to ANNOVAR annotation. All rare coding variants were listed in Supplementary Table 2.
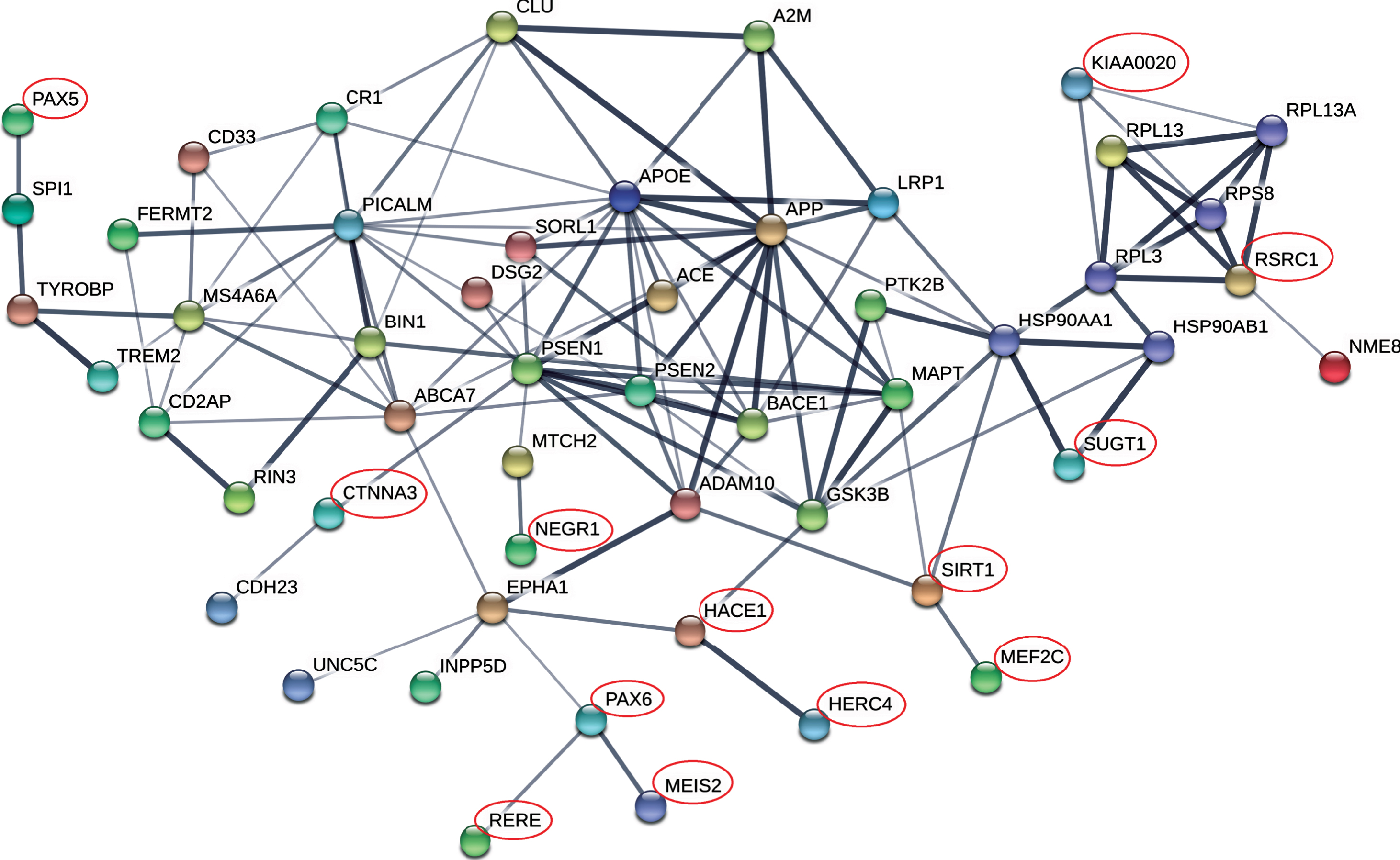
Protein-protein interaction network of MDD risk genes and AD core genes. Protein-protein interaction (PPI) network was constructed using online tool STRING (https://string-db.org/). AD GWAS genes from the IGAP list [19] and reported causal genes as reviewed by Guerreiro et al. [35], were defined as core AD genes. PPI interactions were defined using resources from text-mining, experiments, databases, co-expression, and co-occurrence, as the default setting by the software. Line thickness indicates the strength of confidence. MDD risk genes were marked by red circle.
Differential gene expression of MDD genes in brain tissues of AD patients and mice
To further investigate the involvement of MDD risk genes in AD development, we analyzed the mRNA expression profiles of the MDD genes in four brain tissues (hippocampus, entorhinal cortex, frontal cortex, and temporal cortex) of AD patients and controls ([33] and references therein). Eighteen genes showed a nominally significant (p < 0.05) differential expression in AD brain tissues compared with that of controls (Table 3). In particularly, five genes (MEF2C, SORCS3, OAT, DNAJC12, and SIRT1) were consistently altered in all four brain regions, with SIRT1 being upregulated and the other four genes being downregulated in AD brain tissues (Table 3). We then checked the differentially expressed pattern of these 18 genes in hippocampus tissues of AD mouse models [34]. Seven genes (FAM53B, HACE1, NEGR1, SORCS3, OAT, DNAJC12, and SLC6A15) were significantly correlated with AD pathology burden (number of amyloid plaques or tau tangles) (Table 3). Of note, mRNA expression levels of SUGT1 and L3MBTL2, which showed a genetic association with AD, had no expression alteration in AD brain tissues.
mRNA expression changes of MDD risk genes in brain tissues of AD patients and AD mouse models
Note: log2FC, log2 of fold change of gene expression level compared with that of control; r, Pearson correlation; Expression microarray data of the MDD genes in four brain tissues (hippocampus, entorhinal cortex, frontal cortex, and temporal cortex) in AD patients were retrieved from the NCBI Gene Expression Omnibus database (GEO, http://www.ncbi.nlm.nih.gov/geo/). Details were described in our previous paper [33] and the expression data collection and renormalized expression profiles can be accessed through our database http://www.alzdata.org. Expression data of AD mouse models were taken from the mouseac.org [34]. The number of amyloid-β (Aβ) plaques and the tau (Tau) burden were quantified and the correlation between mRNA expression levels of genes of interest, and the quantified level of pathology was measured based on the processed data, using the Pearson correlation test. Significant p-values (<0.05) were marked in bold. One (MIR759) out of the 31 genes was not available.
Protein-protein interaction of MDD genes with core AD genes
The AD GWAS genes from the IGAP study [19] and reported causal genes, as reviewed by Guerreiro et al. [35], were defined as core AD genes. The 31 MDD risk genes and the 43 core AD genes formed a network with significantly more interactions than expected (PPI enrichment p = 2.0×10 - 13) (Fig. 2). In total, 13 genes (PAX5, CTNNA3, KIAA0020, RSRC1, NEGR1, SIRT1, HACE1, HERC4, PAX6, RERE, MEIS2, SUGT1 and MEF2C), including the two genetically risk genes SUGT1 and MEF2C, were integrated into the AD network, suggesting an involvement of these genes in AD pathology (Fig. 2). In particular, the SIRT1 gene interacted with MAPT, ADAM10, and MEF2C, whereas HACE1 and PAX6 interacted with EPHA1 and GSK3B and were linked with RERE, MEIS2, and HERC4. The PAX5 interacted with the most recently recognized AD gene SPI1 [38] (Fig. 2). Notably, eight (MEF2C, HACE1, KIAA0020, NEGR1, PAX6, RERE, RSRC1, and SIRT1) of the 13 genes recognized from the PPI network were differentially expressed in AD brain (Table 3), suggesting functional roles of these genes in AD pathology.
Cross-validated targets using the CFG approach
Though we observed an apparent involvement of several MDD risk genes in AD at different levels, targets cross-validated by convergent lines of evidence might be more reliable. In our CFG analysis (Supplementary Table 3), we observed four genes (HACE1, MEF2C, NEGR1, and SLC6A15) being weighted with a score of 3, and 13 genes (CTNNA3, DNAJC12, FAM53B, HERC4, KIAA0020, L3MBTL2, LHPP, MEIS2, OAT, PAX6, SIRT1, SORCS3, and SUGT1) with a score of 2. These 17 genes were cross-validated by at least two independent lines of evidence, further supporting their active involvement in AD.
DISCUSSION
People with AD usually experience both cognitive and psychiatric symptoms [3, 4, 8]. Behavioral or psychiatric symptoms were thought to be more challenging effects of the disease [3, 11, 13, 39]. Understanding the relationship and cause of psychiatric symptoms in AD might be essential to improve the quality of life for people suffered. Depression, with high lifetime prevalence, is a common condition in older age. It has been recognized to be one of the most frequent psychiatric comorbidities in AD [3, 8, 40]. It is yet unclear whether depression in AD is causal or just a byproduct. Previous studies indicated that depression may be a prodromal feature and predict risk of AD [7, 11–13, 39, 40]. Therefore, genetic factors contributing to depression might affect risk to AD onset and development. We tested the hypothesis that MDD risk alleles might also contribute to AD risk in this study.
In fact, a previous study by Gibson et al. [41] assessed the genetic correlation of the MDD GWAS findings with neurodegenerative disease using linkage disequilibrium score regression. They observed no overlapping polygenic architecture between lifetime MDD and AD at the whole-genome level and suggested that the genetic overlap might be restricted to some specific genes [41]. Our previous findings showed that the MDD-risk allele CFH p.Y402H conferred AD risk [16, 17]. The MEF2C gene was identified as a risk gene of MDD by the most reliable MDD GWAS [18]; this gene was a previously established AD-risk gene [19, 35]. It is thus valuable to check systematically whether the top MDD risk loci were involved in AD. Taking advantage of the MDD risk gene list from two recently MDD GWAS [18, 23] with a large sample size, we analyzed these MDD GWAS hits in AD at the genomic variation, mRNA expression and PPI levels, to further explore the genetic correlation between depression and AD.
We initially checked whether the MDD GWAS SNPs were associated with AD using publicly available GWAS data [19]. Among the 19 MDD GWAS hits, only one SNP (L3MBTL2 rs2179744) within the genic region showed a positive association with AD; whereas 21 out of the 31 genes had nominally significant non-GWAS hit SNPs. Note that an intergenic MDD GWAS SNP rs4543289 also showed a positive association with AD, and further studies are needed to identify the underlying genes. Three genes (SUGT1, L3MBTL2, and MEF2C) were significantly associated with AD at the gene-based level. MEF2C was a recognized AD risk gene [19, 35] and was highly expressed in brain tissues according to the search at http://www.alzdata.org [33]. It is a transcription factor that functions in immune response and regulates synapse numbers and function [42]; both biological processes would contribute to the course of MDD and AD. The L3MBTL2 gene showed robust evidence for genetic association with AD in both common and rare variant analyses. Intriguingly, a recent large genome-wide analyses for personality traits also identified an association between L3MBTL2 SNPs and neuroticism [43]. The Schizophrenia Working Group of the PGC also identified association of L3MBTL2 SNPs with schizophrenia in the largest GWAS of schizophrenia [44]. These observations strongly suggested that L3MBTL2 might be a genetic risk factor common for neuropsychiatric disorders via its functions in neural development. However, data from the International Mouse Phenotyping Consortium (IMPC, http://www.mousephenotype.org/) showed that mice knocking out L3MBTL2 have decreased lumbar vertebrae number and increased sacral vertebrae number, but no abnormal behavior/neurological or nerve system phenotypes. Though it is highly expressed in neuron (cf. single cell expression data at our AlzData.org database [33]), its function in neural system remained unclear. Further functional assay is needed to characterize the underlying mechanism of L3MBTL2 in neuropsychiatric disorders.
We found that the SUGT1 gene was involved in the AD network and was genetically associated with AD. Previous report has shown that SUGT1 is highly expressed in temporal cortex and significantly decreased in the temporal, angular, and posterior cingulate cortex in AD brains as compared to aged controls [45]. Its decrease might be a result of neuron degeneration in AD, yet it is still unclear how it contributes to MDD and AD. Collectively, our results showed that SUGT1 and L3MBTL2, together with previously identified CFH [16, 17] and MEF2C [18, 19], might play a role in the neural system and serve as reliable common targets in MDD and AD.
Though only limited number of the MDD risk genes was found to be associated with AD genetic risk, 18 MDD risk genes were differentially expressed in AD brain tissues. In particularly, five genes (MEF2C, SORCS3, OAT, DNAJC12, and SIRT1) were consistently altered in all four brain regions of patients according to the AlzData [33]. What’s more, mRNA expression levels of SORCS3 and OAT were also significantly correlated with pathology burden in hippocampus of AD mice. These two genes might be highly involved in AD pathology. The OAT gene encodes the mitochondrial enzyme ornithine aminotransferase, which is a key enzyme in the synthesis of major excitatory and inhibitory neurotransmitters glutamate and GABA [46]. Its activities in brains of Huntington’s disease were found to be reduced compared to age-matched control brains [46]. However, no such changes were observed in AD or schizophrenia [46]. Though there was no doubt that OAT plays an essential role in neuron function, its involvement in MDD and AD needs further investigation. SORCS3 belongs to the sorting and signaling receptor family central in control of neuronal viability and function [47]. It is highly expressed in hippocampus and localizes to the postsynaptic density [47]. SORCS3-deficient mice (http://www.informatics.jax.org/marker/phenotypes/MGI:1913923) had deficits in behavioral activities and spatial learning [47]. Intriguingly, a previous study reported that SNPs of SORCS1, a family member of SORCS3, were associated with AD susceptibility [48]. Overexpression of SORCS1 reduced γ-secretase activity and Aβ level, while suppression of SORCS1 increased γ-secretase processing of AβPP and the level of Aβ [48]. Consistently, another important sorting related receptor SORL1 was an established AD top risk gene [19]. These observations suggested that SORCS3, together with other sorting related receptors, might be involved in both MDD and AD.
Besides the genomic and transcriptomic evidence, several MDD risk genes and AD core genes formed a network with high interactions than expected. Eight genes (MEF2C, HACE1, KIAA0020, NEGR1, PAX6, RERE, RSRC1, and SIRT1) in the PPI network were differentially expressed in AD brain (Fig. 2), suggesting potentially functional involvement of these genes in AD pathology. The genetically risk gene SUGT1 was also integrated into the AD network. Notably, the key MDD gene SIRT1 [23] interacted with AD core genes MAPT and ADAM10. In addition, though no single gene showed convergent lines of supporting evidence, our CFG analysis found that four genes (HACE1, MEF2C, NEGR1, and SLC6A15) (Supplementary Table 3) were cross-validated by data at different levels, including the well-known hit MEF2C. The other three genes (HACE1, NEGR1, and SLC6A15) were all highly expressed in neural system and might be core genes shared between MDD and AD.
By focusing on MDD risk genes locating in the susceptibility loci identified by GWAS, we found that: 1) two genes (SUGT1and L3MBTL2) showing nominally significant associations with AD; 2) SORCS3 and OAT were significantly correlated with AD pathology in both human AD brain tissues and AD mouse models; and 3) three new targets HACE1 , NEGR1, and SLC6A15 showing convergent lines of evidence to be involved in AD. However, there were several limitations in this study. First, the distilling of causal MDD risk genes is very challenging and the current list of candidate genes is incomplete. Due to the huge heterogeneity of depression, it is difficult to map the most reliable risk genes as shown by most of previous MDD GWASs [20–22]. Age at onset of depression was suggested to be bimodal at around 20 years old and 70– 80 years old [49]. It is reasonable to speculate that there might be different genetic bases for early-life and late-life depressions. Genetic correlation between AD and depression might be dependent on the onset age of depression, as AD mainly occurred in the elderly. However, there were some observations suggesting potentially similar biological processes underlying early-onset and late-onset depressions: 1) most of the MDD-risk genes shared by AD showed a stable expression level in prefrontal cortex from early adulthood (20 years old) to late-life (80 years old) (Supplementary Figure 1); 2) both early-life [10] and late-life depression [50] were indicated to increase risk of dementia. As the MDD-risk genes from the 23andMe-based GWAS meta-analysis were identified in subjects of all age groups [18], whereas the CONVERGE findings were based on subjects younger than 60 years old [23], the current MDD risk gene list might not be specific to early-onset and/or late-onset depressions, and refined MDD gene list are needed to understand the shared genetic basis between late-onset depression and AD. Second, the sample size to identify rare coding variants in these MDD risk genes was relatively small, and more samples will offer enhanced statistical power. Third, there was no well-designed functional assay to characterize these genes in the context of AD and depression in this study. In addition, considering the polygenic nature of depression [18, 23], it will be promising to test the correlation between polygenic risk score of MDD with depression and cognitive symptoms in AD patients in the future.
In short, our results were generally consistent with the report by Gibson et al. [41] that the comorbidity of MDD and AD might not be largely driven by genetic factors. However, our data showed that some MDD risk genes located in the GWAS regions might be involved in AD pathogenesis. It is currently unclear whether depression is a consequence of cognitive decline and neuronal loss affected by AD, or depression at early adulthood contributes to neuronal damage that leads to cognitive decline. Considering the report that APOE ɛ4, the major AD risk allele [19], has no contribution to depression [51], it is more possible that depression might be the upstream of AD or a byproduct event. Further research is needed to understand other non-genetic causes (e.g., common environmental factors) and underlying biological mechanisms that account for the complex relationship between neuropsychiatric disorders. This knowledge might help to reduce the psychiatric burden that AD patients and their caregivers suffered.
Footnotes
ACKNOWLEDGMENTS
This work was supported by the National Natural Science Foundation of China (Grant nos. 31730037 to YGY, 81560230 to HYJ, 81500940 to HZ, and 31601039 to DFZ), the Bureau of Frontier Sciences and Education, Chinese Academy of Sciences (CAS, Grant no. QYZDJ-SSW-SMC005 to YGY), and the West Light Foundation of the CAS (to DFZ). We thank the International Genomics of Alzheimer’s Project (IGAP) for providing summary results data for these analyses. The investigators within IGAP contributed to the design and implementation of IGAP and/or provided data but did not participate in analysis or writing of this report. IGAP was made possible by the generous participation of the control subjects, the patients, and their families. The i– Select chips were funded by the French National Foundation on Alzheimer’s disease and related disorders. EADI was supported by the LABEX (laboratory of excellence program investment for the future) DISTALZ grant, Inserm, Institut Pasteur de Lille, Université de Lille 2 and the Lille University Hospital. GERAD was supported by the Medical Research Council (Grant no. 503480), Alzheimer’s Research UK (Grant no. 503176), the Wellcome Trust (Grant no. 082604/2/07/Z) and German Federal Ministry of Education and Research (BMBF): Competence Network Dementia (CND) Grant no. 01GI0102, 01GI0711, 01GI0420. CHARGE was partly supported by the NIH/NIA grant R01 AG033193 and the NIA AG081220 and AGES contract N01– AG– 12100, the NHLBI grant R01 HL105756, the Icelandic Heart Association, and the Erasmus Medical Center and Erasmus University. ADGC was supported by the NIH/NIA grants: U01 AG032984, U24 AG021886, U01 AG016976, and the Alzheimer’s Association grant ADGC– 10– 196728.