
Abstract
Extracellular accumulation of amyloid-β (Aβ) forming senile plaques is one of the hallmark pathologies in Alzheimer’s disease (AD), while the mechanisms underlying the neuronal toxic effect of Aβ are not fully understood. Here, we found that intracerebroventricular infusion of the aged Aβ42 in mice only induces memory deficit at 24 h but not at 7 days. Interestingly, a remarkably increased CREB (cAMP response element-binding protein) Ser133-phosphorylation (pS133-CREB) with microglial activation was detected at 24 h but not at 7 days after Aβ infusion. Aβ treatment for 24 h increased pS133-CREB level in microglia of the hippocampal non-granular cell layers with remarkably decreased pS133-CREB immunoreactivity in neurons of the hippocampal granular cell layers, including CA1, CA3, and DG subsets. Inhibition of microglia activation by minocycline or CREB phosphorylation by H89, an inhibitor of protein kinase A (PKA), abolished Aβ-induced microglia CREB hyperphosphorylation with restoration of neuronal function and attenuation of inflammatory response, i.e., reduced levels of interleukin-6 (IL6) and pCREB binding of matrix metalloproteinase-9 (MMP9) DNA. Finally, treatment of the primary hippocampal neurons with Aβ-potentiated microglia media decreased neuronal GluN1 and GluA2 levels, while simultaneous inhibition of PKA restored the levels. These novel findings reveal that intracerebroventricular infusion of Aβ only induces transient memory deficit in mice and the molecular mechanisms involve a stimulated microglial CREB phosphorylation.
INTRODUCTION
Alzheimer’s disease (AD) is the most common form of dementia in the elderly. Pathologically, AD is characterized by extracellular aggregates of the amyloid-β (Aβ) forming senile plaques and intraneuronal accumulation of the hyperphosphorylated microtubule-associated protein tau, forming neurofibrillary tangles [1–3]. Aβ is the main constituent of brain parenchymal and vascular amyloid, and it contributes to cerebrovascular lesions and is neurotoxic [4, 5]. In many different Aβ species, Aβ42 is much more toxic than Aβ40, possibly because Aβ42 has stronger tendency to aggregate [6]. Early studies suggest that Aβ can induce oxidative stress [7], mitochondrial dysfunction [8], glial responses [9], and lipid dysregulation [10] in AD.
As the resident macrophage cells, microglia act as the first and main form of active immune defense in the central nervous system. They are constantly scavenging plaques, damaged or unnecessary neurons and synapses, and infectious agents [11]. At the same time, microglia activation also damages the central nervous system in pathological condition. For instance, the pro-inflammatory microglia can produce and secret reactive oxygen species and cytokines in response to various environmental stimuli, and causes neuronal injury and death if this response is prolonged. In the early stages of neurodegeneration disease like AD, microglia are activated to the inflammatory responses [12].
As a ubiquitous and constitutively expressed transcription factor, cAMP response element-binding protein (CREB) has an important role in synaptic plasticity and long-term memory formation in the brain and has been shown to be integral in the formation of spatial memory [13], and as well as in neuronal survival [14]. CREB is downregulated in the patients of AD and increasing the expression of CREB is being considered as a possible therapeutic target for AD. The CREB protein has various functions in different brain areas [15], and its signaling pathway has been studied in both affective [16] and neurodegenerative [17, 18] disorders. To express a target gene, CREB requires phosphorylation at the critical serine-133, representing a point at which several major signal pathways converge [19]. Protein kinase A (PKA) is the upstream kinase of CREB and regulates the phosphorylation of CREB at Ser133. In neurons, the PKA/CREB signal pathway is a key regulator of synaptic plasticity and memory capacities [20–24].
To explore the mechanisms underlying the neuronal toxic effects of Aβ42 in the current study, we found unexpectedly that the brain ventricular infusion of aged Aβ42 only induced a transient learning and memory deficit. by increasing microglia CREB phosphorylation, which in term caused neuronal impairments.
MATERIALS AND METHODS
Animals and treatments
Wildtype C57BL mice (2 months old, male) were purchased from Beijing Huafukang, and the mice were housed under a 12-h light/dark cycle and kept with accessible food and water at 25°C. Aβ42 (Qiangyao Biotechnology) was aged according to the protocol described previously [25]. The Aβ42 (2μg/μl) was dissolved in dimethyl sulfoxide (DMSO), and then was incubated at 37°C for 7 days before use. The mice were anesthetized and injected through brain unilateral ventricle with the aged Aβ42 (5μl), and the control group were injected with same volume of sterile normal saline containing DMSO (1%). The coordinates for the injection were as follows: anterior-posterior: –0.3 mm; mediolateral: –1 mm; dorsoventral: –2.3 mm (from bregma and dura, flat skull). The behavior tests were carried out at 24 h or 7 days after the treatment, and then the mice were sacrificed for the following biochemical measurements. To inhibit microglia, the mice were received intraperitoneal injection of minocycline (50 mg/kg/day, Sigma) for 10 days before stereotactic brain infusion of Aβ42 and the other groups were received intraperitoneal injection with normal saline.
Novel object recognition
The test equipment is a white, square chamber with a bottom of 50×50 cm. Before each behavioral experiment was performed on each mouse, the test bench was sprayed with 75% alcohol and wiped clean. For 24 h groups, the mice were placed in the empty chamber to explore for 5 min before operation (habituation). At 6 h after the surgery, the mice were put back to the central area with two objects placed at the two corners of the chamber for 5 min (familiarization). After 24 h, one of the objects was replaced with the new one and the mice were put back in the central area of the chamber to explore for 5 min (test), and the times of the mice explored for the new object was recorded. For 7 days groups, the task started with a habituation trial on day 5 (5 days after the surgery) followed by a familiarization trial (day 6) in which two objects were presented to the animals and the test trial (day 7). To ensure the rigorous experiments, the order in which behavioral experiments were performed was kept as the same order as the familiarization on the first day and the test on the second day.
Western blotting
Western blotting was performed per the methods well established in our laboratory [26]. The hippocampi of the mice were dissected and homogenized with RIPA buffer (Beyotime) on ice. The proteins were separated by 10% SDS-PAGE, transferred onto nitrocellulose membranes and then blocked by 5% BSA for 1 h in room temperature. After washings by Tween-TBS, the membranes were incubated in primary antibodies overnight, and then incubated with secondary antibodies for 1 h. The blots were visualized by using Odyssey Infrared Imaging System (Licor Biosciences, Lincoln, NE, USA). The primary antibodies used in the present study include CREB (cell signaling, 1:1000), pS133-CREB (cell signaling, 1:1000), GluN2A (cell signaling, 1:1000), GluN2B (abcam, 1:1000), GluN1 (Millipore, 1:500), GluA1 (abcam, 1:1000), GluA2(Millipore, mab397), PSD93(abcam,ab2930), PSD95 (cell signaling, 1:1000), CaMKII (cell signaling,3362), synaptotagmin (abcam, ab13259), and β-actin (abcam, ab6276).
Golgi staining
After anesthetized, the mice were perfused with 0.9% NaCl and then the brains were immersed in the Golgi solution for 14 days. After dehydrated in 30% sucrose-PBS for 48 h, the brains were cut 100μm with a vibrating microtome (VT1000S, Leica, Germany). Then the slices were moved into CXA solution for staining.
Immunohistochemistry and immunofluorescence
After anesthetized, the mice were perfused by normal saline and 4% paraformaldehyde (PFA). The brains were removed and post-fixed in PFA for 24 h and then dehydrated in 30% sucrose-PBS for 48 h. After embedding, brains were cut into 30μm coronal sections. For immunohistochemistry (IHC) and immunofluorescence (IF), the experiments were carried out according to the established methods [27]. The brain slices were incubated with the same primary antibodies (Iba1, abcam, 1:200; pS133-CREB, cell signaling, 1:100; NeuN, Millipore, 1:50; GFAP, cell signaling, 1:200). For IHC, the images were observed using a microscope (Nikon, Tokyo, Japan). For IF, the images were captured using a Carl Zeiss LSM710 confocal microscope.
BV2 culture
Bv2 mouse microglia cells were grown in high glucose Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum and they were maintained in 95% humidified and 5 % CO2. After 6 h treated with the aged Aβ42 (2.5μM) or H89 2HCl (15μM), the cell extracts were used for western blotting and other tests. For studying the effects of microglia on the neurons, the bv2 cells were treated with the aged Aβ42 (2.5μM) or simultaneously with H89 (15μM) for 1 h, then the media were replaced with fresh DMEM without Aβ42 and H89 for 5 h. Then culture media were collected and added (1:9, v/v) into the primary hippocampal neurons cultured for 7 days in vitro (7 div). After 5 h, the neuronal extracts were prepared for the biochemical measurements.
Primary neuron culture
The hippocampal neurons were isolated from rats at embryonic day 15–18 (E15-18). The brain tissues were cut into small pieces and digested with 0.125% trypsin solution for 15 min. Neurons were cultured in dishes coated with 100μg/ml poly-D-lysine. The medium contains 97% neurobasal medium (Gibco), 2% B-27 (Gibco) and 1× GlutaMAX (Gibco). After cultured for 7 div, the bv2 media were added (1:9, v/v) at day 8 and the neurons were cultured for another 5 h.
Chromatin immunoprecipitation assay
Chromatin immunoprecipitation (ChIP) was analyzed using a kit (#17–371, Merk Millipore) by following the manufacturer’s instructions. To quantify CREB-bound level of MMP-9 DNA, qPCR was performed with following primers: MMP-9- ChIP-F: GGATTCAGGAGTCAGAGCAC, and MMP-9-ChIP-R: CATTGCTTCAAAGCACAGAC.
Statistical analyses
Data were analyzed by Student’s t–test or one-way ANOVA (GraphPad Software) and presented as mean±SEM. The statistically significance was set at p < 0.05.
RESULTS
Intracerebroventricular infusion of Aβ42 only induces transient memory deficit with a remarkably increased CREB phosphorylation in hippocampus
To study the effect of Aβ on learning and memory in mice, we injected stereotaxically the aged Aβ42 into the lateral ventricle of 2-month-old wildtype mice. The control group was injected with the same volume of normal saline (containing 1% DMSO, v/v). The novel object recognition test was performed at 24 h and 7 days after the surgery. We observed that the Aβ42 injected mice showed significantly decreased exploring times to the novel object compared with the control group at 24 h after infusion (Fig. 1A). However, no significant difference was detected between Aβ42 and the control groups at 7 days after the infusion (Fig. 1B). To explore the molecular bases underlying the observed phenomenon, we examined the activity or expression level of CREB and synapse-associated proteins at 24 h and 7 days after the infusion. Compared with the control group, PSD93 was significantly increased and GluA2 was decreased in Aβ42 mice at 24 h after the infusion, while no difference of GluN2A, GluN2B, GluN1, GluA1, and PSD95 was detected between Aβ42 and the control group (Fig. 1C, E). Interestingly, we observed that the phosphorylation level of CREB at Ser133 (pS133-CREB) was significantly increased in Aβ42 mice at 24 h after the infusion (Fig. 1C, E). No significant difference of pS133-CREB and the synapse-associated proteins was found between the two groups at 7 days after the infusion (Fig. 1D, F). We also observed that number of dendritic spines in mouse brain slices treated with Aβ42 for 24 h was decreased compared with the control group (Fig. 1G, H), but this difference was not shown in 7 days group (Fig. 1I, J). These data suggest that Aβ42 can cause acute damage to the brain leading to learning and memory impairments in mice, while this damage may be recovered naturally after 7 days.
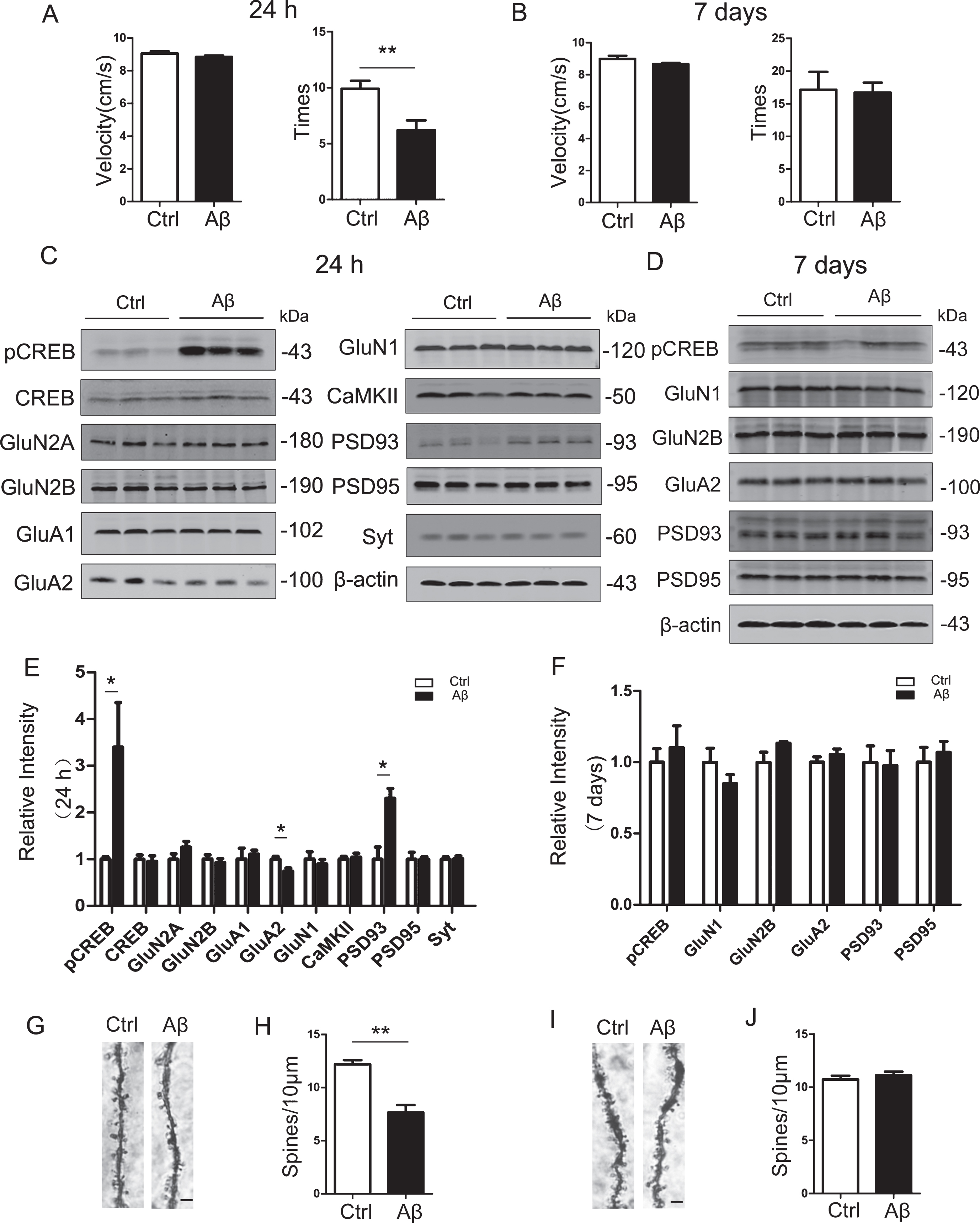
Intracerebroventricular infusion of aged Aβ only induces transient memory deficit with remarkable elevation of pCREB. The mice (2 months old) received unilateral ventricular infusion of the aged Aβ. After 24 h or 7 days, the memory ability was tested by novel object recognition (NOR). Then, the mice were sacrificed and the hippocampi were collected for measurement of the memory-associated proteins and spine densities. A) The exploring times for novel object was decreased in mice exposed to Aβ for 24 h with no difference in movement speed (n = 10 per group). B) No difference of exploring times between Aβ and control mice at 7 days (n = 7 per group). C, E) Levels of Ser133-phosphorylated CREB (pCREB) and PSD93 were significantly increased and GluA2 decreased, while no changes in total CREB and other synapse-associated proteins were detected in 24 h Aβ group measured by western blotting (n = 6 for each group). D, F) No difference of the synapse-associated proteins and CREB were detected between Aβ and Ctrl groups at 7 days (n = 5). G, H) The spine density was decreased in 24 h Aβ group (Scale bar = 2μm). I, J) No difference of spine densities was detected between two groups at 7 days. Data were presented as mean±SEM (unpaired t test, *p < 0.05, **p < 0.01).
Intracerebroventricular infusion of Aβ only transiently activates microglia
Using microglia marker Iba-1 for immunohistochemical staining, we observed that microglia were extensively activated shown by the increased number and enlarged diameter of microglia cells in the hippocampus of 24 h Aβ42 treated mice (Fig. 2A). Quantitative analyses showed that the average diameter of microglia was increased to 1.5-fold of the control group (Fig. 2C) with a remarkably increased number of microglia at 24 h after Aβ42 treatment (Fig. 2D), while these abnormalities were not detected at 7 days (Fig. 2B, E, F). These data suggest that Aβ42 may damage neurons through activation of microglia in the acute phase (24 h) after Aβ42 treatment. Immunohistochemical staining with the astrocyte marker GFAP showed that the number and morphology of astrocytes in the Aβ42 group was not changed compared with the control group at 24 h (Fig. 2G, H), indicating that Aβ does not significantly affect astrocytes at the acute phase. Furthermore, the number of neurons in the hippocampus of the Aβ42 group was also not significantly changed compared with the control group (Fig. 2I, J), suggesting that Aβ42 treatment does not cause neuron death in acute phase.
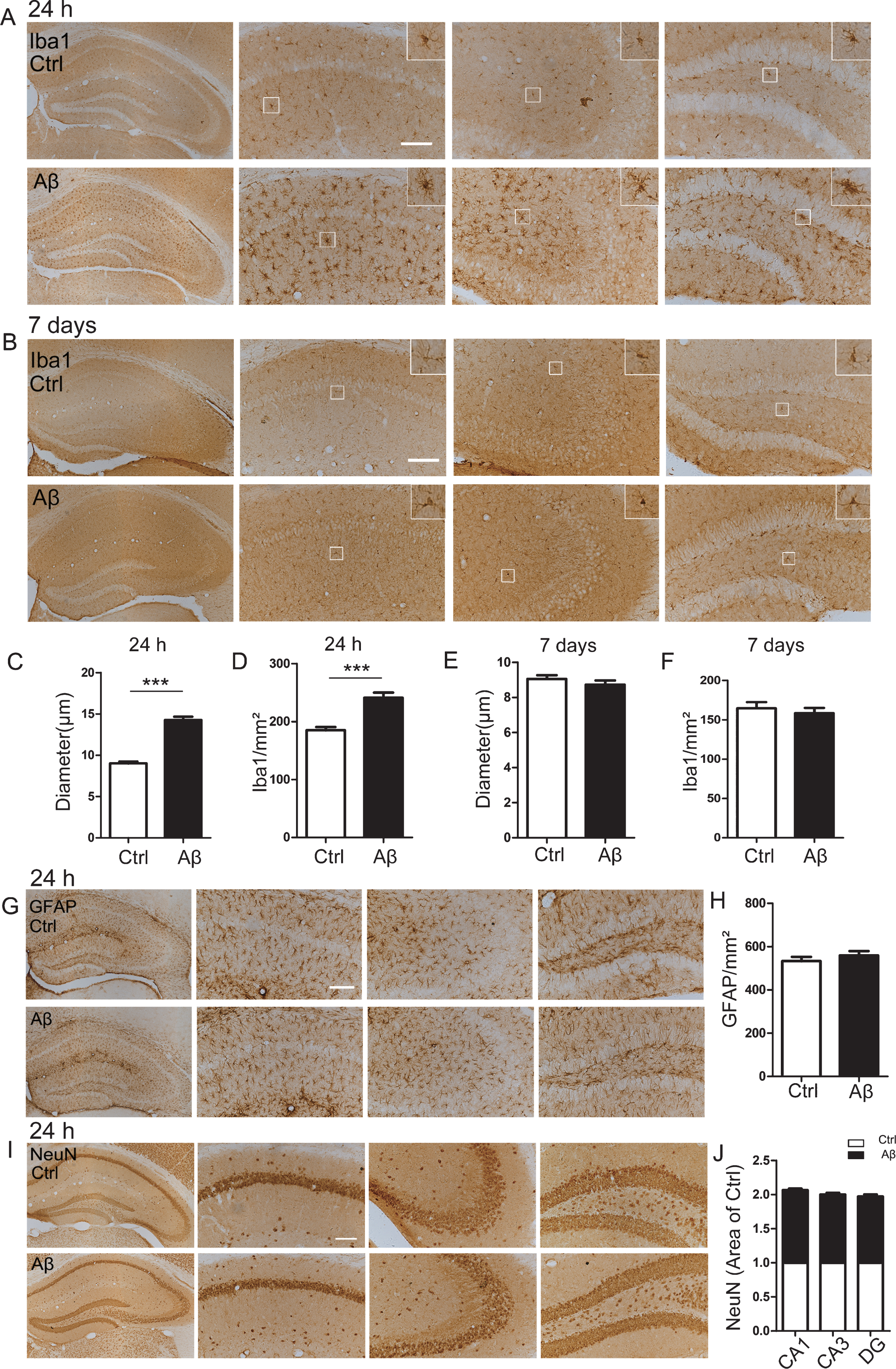
Intracerebroventricular infusion of aged Aβ only transiently activates microglia. A, C, D) Aβ exposure activates microglia demonstrated by the increased Iba1 immunoreactivity and the enlarged diameter of the microglia in hippocampus of the 24 h group. B, E, F) No activation of microglia cells was detected at 7 days after Aβ administration. G, H) No difference of GFAP staining (astrocytes marker) in hippocampus of 24 h group. I, J) No difference of NeuN staining (neuron marker) in hippocampus of 24 h group. Scale bar = 100μm, n = 4 for each group. Data were presented as mean±SEM (unpaired t test).
Intracerebroventricular infusion of Aβ increases microglial pS133-CREB level
The aforementioned results showed that level of pS133-CREB in hippocampus of the 24 h Aβ42 group increased significantly (Fig. 1C, E). However, the results of immunohistochemical staining showed that the expression level of pS133-CREB in the hippocampal granule cell layer and pyramidal layer (including DG, CA3, CA1) was significantly lower than that of the control group at 24 h after Aβ42 infusion; however, in other non-granular cell layer areas of the hippocampus, the expression of pS133-CREB was significantly increased compared with the control group (Fig. 3A), suggesting that Aβ42 has different activation states of CREB in different cell types. To further explore the enhanced expression of pS133-CREB cell types, we performed immunofluorescence co-localization staining. The immunoreactivity of pS133-CREB was significantly reduced in the hippocampal granule cell layer, while extensive co-localization of pS133-CREB with Iba-1, a marker of microglia, was detected in the non-granular cell layer at 24 h after Aβ42 treatment (Fig. 3B). By co-immunofluorescence staining of NeuN and pS133-CREB, we found that the expression of pS133-CREB in the NeuN-positive cells of the Aβ42 group was significantly lower than that of the control group in the hippocampus (Fig. 3C). At day 7, no significant change of pS133-CREB in the granule cell layer was detected in Aβ42 group (Fig. 3D). These results reveal that Aβ42 may activate microglia by increasing the phosphorylation of CREB, by which it causes neuronal damages in acute phase.
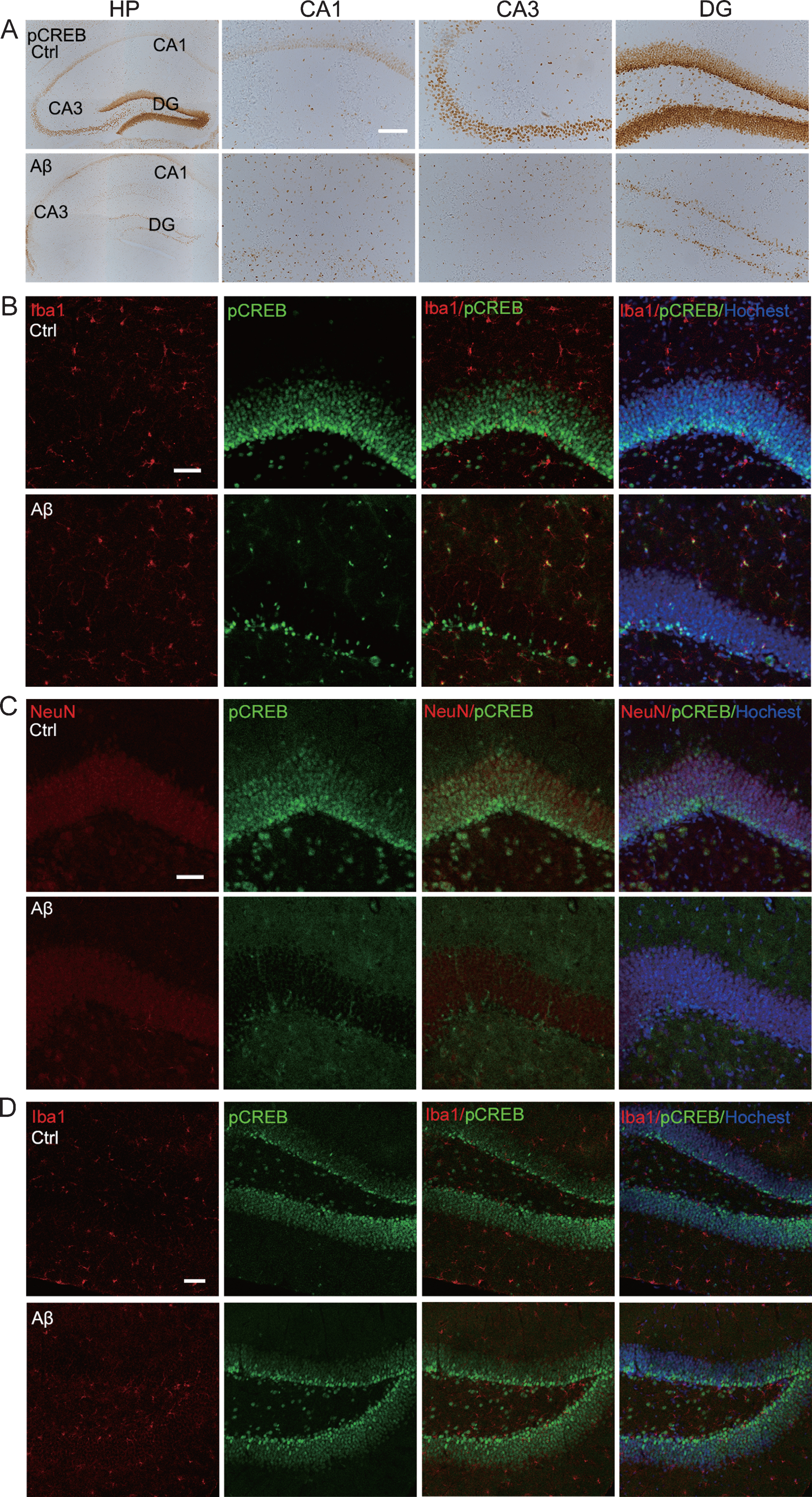
Intracerebroventricular infusion of aged Aβ remarkably increases microglial pS133-CREB with reduced neuronal immunoreactivity at 24 h. A) Aβ remarkably increased pS133-CREB immunoreactivity in hippocampal non-granular cell layers and non-pyramidal layer with significantly decreased staining in granule cell layers (DG) and pyramidal layer (including CA1, CA3), measured at 24 h by immunohistochemical staining (Scale bar = 100μm), or (B) by co-immunofluorescent staining of Iba1 and pS133-CREB (Scale bar = 50μm). C) Aβ significantly decreased neuronal pS133-CREB immunoreactivity measured by co-immunofluorescent staining of NeuN and pS133-CREB in hippocampal of 24 h group (Scale bar = 50μm). D) Aβ does not significantly change glial pS133-CREB immunoreactivity by co-immunofluorescent staining of Iba1 and pS133-CREB in hippocampal CA1, CA3, DG of the 7 days group (Scale bar = 50μm).
Inhibition of microglial activity attenuates Aβ-induced microglia elevation of pCREB with improved memory
Minocycline is a potent microglial inhibitor. After intraperitoneal infusion of minocycline for 10 days, the aged Aβ42 was infused into the lateral ventricle (Fig. 4A). The novel object recognition test showed that minocycline restored the exploring times to the novel object (Fig. 4C) without changing the movement speed of the mice (Fig. 4B), suggesting improvement of cognitive function by minocycline. Immunohistochemistry data showed that minocycline inhibited Aβ42-induced microglia activation (Fig. 4D), shown by decreasing number and diameter of the microglia (Fig. 4E, F). By co-immunofluorescence staining of pS133-CREB and Iba-1, we observed that inhibition of microglia by minocycline attenuated Aβ42-induced elevation of pS133-CREB co-stained with Iba-1 (Fig. 4G) with reduction of IL-6 (Fig. 4H). Among several synapse-associated proteins, we observed that GluA2 was remarkably decreased in Aβ42 treated mice at 24 h after the infusion, while minocycline restored the level of GluA2 (Fig. 4I, J). These data suggest that inhibiting microglial activation attenuates its CREB phosphorylation with attenuation inflammation and synapse impairment and restoration of learning and memory.

Inhibition of microglia prevents Aβ-induced microglial pS133-CREB expression and impairments of memory. A) The mice were received intraperitoneal injection of minocycline (Mino) for 10 days before Aβ infusion. B) There was no difference in movement speed between the four groups of mice (n = 10 per group). C) The exploring time for novel object was decreased in mice exposed to Aβ for 24 h while Mino reversed the reduction. D-F) Mino inhibits microglia activation demonstrated by reduced Iba1 immunoreactivity and the diameter of the microglial cells in hippocampus (Scale bar = 25μm, n = 4). G) Mino attenuates Aβ-induced pCREB expression in microglia measured by co-immunofluorescent staining of Iba1 and pS133-CREB in the hippocampus at 24 h after Aβ infusion (Scale bar = 10μm). H) Mino attenuates Aβ-induced elevation of IL-6 in hippocampus measured by ELISA (n = 5). I, J) Mino restored Aβ-induced reduction of GluA2 in hippocampal extracts measured by western blotting (n = 6). Data were presented as mean±SEM (one-way ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001).
Aβ activates microglia through CREB phosphorylation
To further verify the effect of Aβ on microglia, we treated the mouse microglial cell line bv2 with aged Aβ42 for 6 h. The results showed that the level of pS133-CREB in Aβ42-treated bv2 cells was significantly increased compared with the control group with a simultaneous upregulation of PKA, an important upstream kinase for CREB (Fig. 5A, B). The level of IL-6 in the bv2 culture medium was also significantly increased after Aβ treatment (Fig. 5C). Matrix metalloproteinase-9 (MMP-9) is an important protease secreted by microglia that participates in inflammatory reactions. Chromatin immunoprecipitation (ChIP) results showed that the binding of MMP-9 DNA to pCREB was significantly increased in Aβ42 treated bv2 cells (Fig. 5D). After treatment of bv2 with PKA inhibitor (H89 2HCl) and inhibition of CREB phosphorylation (Fig. 5A, B), the level of IL-6 in bv2 culture medium was decreased (Fig. 5C), and the binding of MMP-9 DNA and pCREB was also decreased (Fig. 5D). These results suggest that Aβ42 activates microglia PKA-CREB signaling pathway, and thus increases the expression of MMP-9 and promotes IL-6 secretion by increasing CREB phosphorylation.
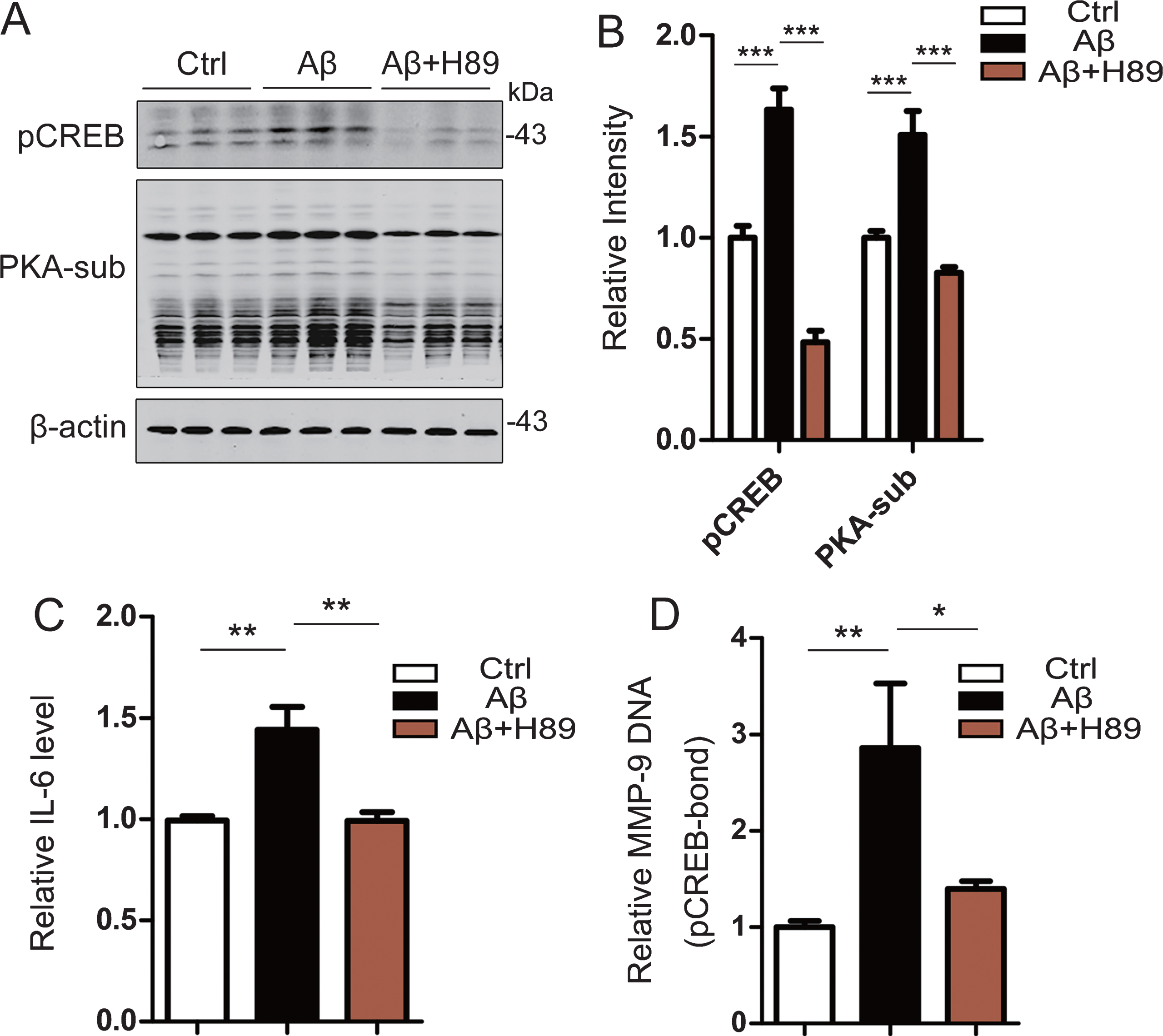
Aged Aβ activates microglia and induces inflammation through CREB phosphorylation in Bv2 cells. Bv2 cells were treated with Aβ or simultaneously Aβ and H89 for 6 h. A, B) Aβ treatment increased CREB phosphorylation at Ser133 (pCREB) with activation of PKA measured by using PKA-substrate assay and western blotting, while inhibition of PKA by H89 attenuated Aβ-induced CREB phosphorylation (n = 6). C) Aβ increased IL-6, while inhibition of PKA-associated CREB phosphorylation by H89 restored IL-6 level in the culture medium measured by using an ELISA kit (n = 6 per group). D) Aβ increased binding level of MMP-9 DNA and pCREB detected by using a ChIP kit (n = 6 per group). Data were presented as mean±SEM (one-way ANOVA, *p < 0.05, **p < 0.01, ***p < 0.001).
Aβ affects neuronal synaptic protein expression and development through microglial PKA-dependent manner
To further verify whether Aβ42 causes neuron damages through microglia, we treated bv2 cells with Aβ42 for 1 h, and then replaced the old medium with fresh DMEM and cultured for 5 h. The media were collected and added to the primary cultured hippocampal neurons at a ratio of 1:9 (v/v, 10% bv2 medium) for 5 h. Culture of the neurons with Aβ-potentiated media decreased expression level of synaptic proteins GluN1, GluA2, and synaptotagmin (Syt) without affecting GluN2A and GluN2B, while simultaneous inhibition of PKA in microglia by H89 attenuated Aβ-induced reduction of GluN1 and GluA2 (Fig. 6A, B). Immunofluorescent staining data also showed Aβ-potentiated media remarkably inhibited development of neuronal processes, while simultaneous inhibition of PKA in microglia attenuated neuronal impairments induced by Aβ-potentiated media (Fig. 6C). These data demonstrate that Aβ induces neuronal damage through microglia PKA-mediated CREB phosphorylation.

Aβ-potentiated microglial media affect neuronal development and synaptic protein expression via PKA-dependent manner. The bv2 cells were treated with aged Aβ (2.5μM), or Aβ plus H89 (15μM) or vehicle (DMSO, 1:1000, v/v) for 1 h, then the culture media were removed. After thorough washing, the bv2 cells were cultured with new medium without Aβ and H89 for 5 h. Then the culture media were collected and added (1:9, v/v) into the primary hippocampal neurons. After 5 h, the neuronal extracts were used for measurement of synapse-associated proteins by western blotting (n = 6). C) The neuronal morphology changes measured by immunofluorescent staining of anti-MAP2 antibody (scale bar = 25μm). Data were presented as mean±SEM (one-way ANOVA, *p < 0.05, **p < 0).
DISCUSSION
Alzheimer’s disease (AD) is the most common neurodegenerative disorder. As one of the two major pathological hallmarks of AD, Aβ is toxic to the neurons. However, due to the morphological diversity and the diversity of targets, the mechanisms underlying the toxic effects of Aβ on neurons become very complicated. Tremendous studies show that Aβ can induce neuroinflammation, oxidative stress, and mitochondrial dysregulation [28]. There are also increasing reports demonstrated that Aβ can enhance neuronal activity and synaptic plasticity [29], improve hippocampal LTP with positive regulation of neurotransmission and memory [30], and maintain the balance of lipid metabolism by binding and transporting cholesterol [31]. Aβ is generated by β-secretase [32–34], γ-secretase [35] and δ-secretase [36] cleavage of amyloid-β protein precursor (AβPP), while the mice with AβPP of β-site APP-cleaving enzyme 1 (BACE1) knockout exhibit memory impairment [37, 38]. We speculate that the time window (i.e., acute or chronic) we choose to do the measurement may at least contribute the observed protective or detrimental role of Aβ in the nervous system. For instance, we found in the present study that intracerebroventricular infusion of Aβ only caused neuronal damages and memory deficit at 24 h. By 7 days after the treatment, the Aβ-induced damages were no longer detected. These data suggest that the short-term exposure to Aβ is transient and reversible, and neuronal function may be recovered by breaking Aβ overproduction in early stages of the AD progression. Although we observed elevation of PSD93 protein level at 24 h after Aβ infusion, we currently do not understand why only PSD93, but not others, was selectively increased. We speculate that expression of PSD93 may be more actively regulated by pCREB, or PSD93 expression may be more predominant in microglia than the neurons, or it may be just a non-specific stress response because the elevation of PSD93 was not detected in 7 days group or in the brains.
Regarding the effects of the exogenously infused Aβ42 on learning and memory, diverging observations have been reported. By using novel object recognition test as used in the current study, some reported that oligomer Aβ42 could cause learning and memory impairments in mice after being injected for 24 h but the memory deficit was no longer detectable at 10 days if Aβ42 was not injected again [39]; rats had no social memory impairment at 7 days after being injected with aged Aβ42 [40], which were in agreement with the observations in the current paper. Other studies using Morris water maze and Y-maze tests showed that the mice injected with aged Aβ42 had learning and memory damages and loss of neurons at 14 days after the injection [41, 42]; the cognitive impairments of the mice were even detected at 21 days after single injection of the aged Aβ42 [43]. In our current study, we could also see reduction of some synapse-associated proteins at 14 days after Aβ42 exposure, but we did not detect memory deficits at 7 days and 14 days. The reason causing these discrepancies is currently not fully understood. We speculate that the paradigms applied for behavioral tests may be one of them.
Microglial cell is the first line of defense of the nervous system by secreting many inflammatory mediators, including complement proteins, cytokines, and chemokines [44–46]. In general, microglia can sense and monitor the alterations in surrounding environment, and then they change the morphology into amoebic-like and migrate to the damaged sites bring protective or meanwhile toxic effects [47]. Aβ can activate the microglia, making microglia to phagocytose and clear Aβ, and by which it brings protective effects. When microglia attempt to remove toxic substances from nervous systems, it also secretes a large number of inflammatory factors that can damage the neurons. For example, incubating mouse microglia with Aβ promotes mitogenesis and secretion IL-1 and TNF-α [48]. We also observed in the current study that Aβ treatment for 6 h significantly increased levels of IL-6 and MPP-9 in bv2 cells. In the brain of AD patients, microglia cells have been detected to be largely accumulated in areas where amyloid plaques are deposited. Aβ can bind to the receptor complex containing the pattern recognition receptor CD36 and a heterodimer composed of the Toll-like receptors (TLR) to promote nuclear transport of nuclear factor κB (NFκB) [49], and thus activate the cAMP/PKA/CREB signaling pathway [50], resulting in expression of inflammatory factors [51].
As a widely expressed transcription factor in neurons, CREB is at the core of various signaling pathways for synaptic plasticity and memory formation. CREB is mainly regulated by phosphorylation, and the Ser133 site is the most critical site of the transcriptional activity regulation. The Ser133 of CREB can be phosphorylated by many kinases, including PKA, protein kinase C (PKC), Ca2 +/calmodulin-dependent kinase II and IV (CaMKII and CaMKIV), mitogen/stress-activated kinase (MSK), ribosomal S6 kinase (RSK), AKT, and MAPKAP kinase-2 (MK2) [52–58]. We found with surprising here that Aβ treatment for 24 h induced Ser133-CREB hyperphosphorylation in total hippocampal extracts of mice but with an impaired memory. Therefore, we further investigated the pS133-CREB level in neurons and microglia, respectively. We found that Aβ treatment for 24 h significantly decreased the CREB phosphorylation in the neurons with a remarkably increased pS133-CREB in microglia. These results can explain the Aβ-induced neuronal impairments and memory deficits. By co-culturing the primary neurons with Aβ-potentiated bv2 media, we further confirmed that Aβ treatment in the acute phase could induce neuronal impairments by pS133-CREB-dependent microglial secretion of pro-inflammatory cytokines.
In summary, we found in the present study that intracerebroventricular infusion of aged Aβ induces transient memory deficit with neuronal impairments, and the molecular mechanisms involve pS133-CREB-dependent microglial activation.