
Abstract
Amyloid-β (Aβ) peptides, Aβ40, Aβ42, and recently Aβ25 - 35, have been directly implicated in the pathogenesis of Alzheimer’s disease (AD). We have previously shown that all three peptides decrease neuronal viability, but Aβ40 also promotes synaptic disassembling. In this work, we have studied the effects of these peptides on astrocytes in primary culture and found that the three Aβ peptides were internalized by astrocytes and significantly decreased astrocyte viability, while increasing ROS production. Aβ peptide internalization is temperature-dependent, a fact that supports the idea that Aβ peptides are actively endocytosed by astrocytes. However, inhibiting caveolae formation by methyl-beta-cyclodextrin or by silencing caveolin-1 with RNA interference did not prevent Aβ endocytosis, which suggests that Aβ peptides do not use caveolae to enter astrocytes. Conversely, inhibition of clathrin-coated vesicle formation by chlorpromazine or by silencing clathrin with RNA interference significantly decreased Aβ internalization and partially reverted the decrease of astrocyte viability caused by the presence of Aβ. These results suggest that Aβ is endocytosed by clathrin-coated vesicles in astrocytes. Aβ-loaded astrocytes, when co-incubated with non-treated astrocytes in separate wells but with the same incubation medium, promoted cell death in non-treated astrocytes; a fact that was associated with the presence of Aβ inside previously unloaded astrocytes. This phenomenon was inhibited by the presence of chlorpromazine in the co-incubation medium. These results suggest that astrocyte may perform Aβ transcytosis, a process that could play a role in the clearance of Aβ peptides from the brain to cerebrospinal fluid.
INTRODUCTION
Alzheimer’s disease (AD) is characterized by the formation of plaques composed of amyloid-β (Aβ) peptides [1]. The more frequent Aβ peptides found in these plaques are Aβ40 and Aβ42, which are formed from AβPP (amyloid-β protein precursor) by β- and γ-secretases [2]. Recently, it has been reported that these peptides can be detached from the plaques by a spontaneous mechanism that is not dependent on enzymatic catalysis [3]. In addition, Aβ25 - 35, the shorter Aβ peptide that has toxic effects, has also been found in the brain of AD patients, presumably coming from the cleavage of Aβ40 [4]. Moreover, normal aging also promotes the racemization of serine26 of Aβ40, an event that can result in the formation of the truncated forms of Aβ40 including Aβ25 - 35 [5]. This suggests that Aβ25 - 35, together with Aβ40 and Aβ42 can also play a role in the pathogenesis of AD. In this context, we have recently studied the effects of Aβ peptides on cell viability, reactive oxygen species (ROS) production and synaptic protein expression and localization in neurons in primary culture [6]. We found that Aβ25 - 35, Aβ40, and Aβ42 significantly decrease neuronal viability. In addition, Aβ40 elicits a clear delocalization of PSD-95 and synaptotagmin from prospective synapsis to the neuronal soma, suggesting Aβ40 has an immediate effect on synaptic disassembling.
Likewise, there is evidence that astrocyte function is also compromised in AD [7], a fact that may affect neuronal viability in vivo. In this context, Alois Alzheimer, in 1906, had already detected the presence of astrogliosis in the brain of the first patient diagnosed with AD [8], although this observation was considered to be a secondary response to the disease process. However, the presence of Aβ in human brain astrocytes is well-documented [9, 10] and Wyss-Coray and colleagues [11] provide clear evidence that astrocytes play an active role in the neurodegeneration observed in AD. Indeed, astrocytes may play a dual role in the pathogenesis of AD, where the response of astrocytes to Aβ could actively contribute to the disease process through the release of soluble inflammatory mediators, such as IL-1β, IL-6, and TNF-α [12], which promote astrocyte hypertrophy and subsequent loss of function [13, 14]. In addition, the death of Aβ-loaded astrocytes may give rise to some specific secondary plaques, the so-called glial fibrillary acidic protein (GFAP) positive plaques [15]. By contrast, astrocytes could also have a positive role in preventing disease progression, whereby the presence of astrocytes surrounding senile plaques could act as a barrier that restrains damage, at least in the initial stages of the disease [16]. Also, it is known that astrocytes mediate the clearance of Aβ by subjecting the peptide to degradation by a variety of degrading enzymes, such as insulin-degrading enzyme [17], neprilysin [18], and matrix metalloproteinase-9 [19]. Astrocytes may also contribute to the clearance of parenchymal Aβ by releasing degrading enzymes extracellularly [11, 20].
In this work, we investigated the effects of Aβ peptides on astrocyte viability and ROS production. We also studied the mechanism for Aβ internalization in astrocytes, and concluded that Aβ internalization occurred by clathrin-mediated endocytosis. In addition, our results suggest that Aβ may also undergo transcytosis by astrocytes, a finding that may contribute to the clearance of Aβ peptides from the brain to cerebrospinal fluid (CSF).
MATERIALS AND METHODS
Astrocyte cultures
Albino Wistar rats were obtained from the animal house of the University of Salamanca (Spain) and were used according to local and EU Ethics Committee guidelines. Cell cultures were prepared from the forebrains of 1-day-old Wistar rats as previously described [21]. Briefly, animals were decapitated and their brains immediately excised. After removing the meninges and blood vessels, the forebrains were placed in Earle’s balanced solution (EBS) containing 20μg/ml DNase and 0.3% (w/v) BSA. The tissue was minced, washed, centrifuged at 500×g for 4 min and incubated in 0.025% (w/v) trypsin (type III) and 60μg/ml DNase I for 15 min at 37°C. Trypsinization was terminated by the addition of DMEM containing 10% (v/v) FCS. The tissue was then dissociated by gently passing it eight times through a siliconized Pasteur pipette and the supernatant cell suspension was recovered. This procedure was repeated and the resulting cell suspension was centrifuged at 500×g for 5 min. The cells were then resuspended in DMEM containing 10% FCS and plated on Petri dishes coated with 10μg/ml of poly-L-lysine at a density of 1.0×105 cells/cm2. Astrocytes were maintained at 37°C and 5% CO2. Three days after plating, cytosine arabinoside (10 mM) was added to the culture medium for 2 days to prevent the growth of microglia and cells from the O-2 lineage. The culture medium was replaced with fresh medium twice a week. Experiments were carried out on confluent astrocytes after 18–21 days in culture.
Cellular treatments
Aβ peptides were purchased from Bachem (Bubendorf, Switzerland) and were prepared in sterile deionized water. The concentration used in the experiments was 30μM. Human Serum Albumin (HSA) was obtained from Grifols (Barcelona, Spain). In order to prepare the HSA-Aβ complexes, Aβ peptides were gently dissolved in an HSA solution.
For the endocytosis assays, astrocytes cultured for 18–21 days in vitro (DIV) were preincubated for 1 h at 37°C with chlorpromazine (10μg/ml) and methyl-beta-cyclodextrin (25 mM). These inhibitors were maintained at the same concentration in the medium throughout the experiments. Both products were purchased from Sigma Aldrich (Madrid, Spain).
Transcytosis assays
Proximal cultures were used in order to study transcytosis of Aβ in astrocytes. To prepare proximal cultures, primary astrocytes (18 DIV) were plated on transwell inserts (PIRP12R48, Merck Millipore, Darmstadt, Germany) at a density of 75,000 cells/200μl DMEM + 10% FCS, adding another 400μl to the well. After 72 h, the inserts were incubated for 5 min in Hanks medium in the absence or the presence of different Aβ peptides (30μM): Aβ25 - 35, Aβ40, or Aβ42. Then, cells were washed twice with fresh medium and inserts were co-incubated with non-treated astrocytes cultured on 24-well plates coming from the same culture, in such a way that there was no physical contact between them (see Supplementary Figure 1). The co-incubation was maintained in the absence or the presence of chlorpromazine, and then non-treated astrocytes (astrocytes located in the lower deck) were analyzed.
Cellular viability assay
Astrocytes were maintained in serum-free medium (Hanks medium, pH = 7.4) for 30 min or 1 h when carrying out the different treatments. Then, cellular viability was determined by the MTT reduction assay [22]. Briefly, MTT (Thermo Fisher, Waltham, USA) was diluted in Hanks medium (0.5 mg/ml) and added to the cells. After 75 min of incubation (37°C, 5% CO2, in darkness), the medium with MTT was replaced by dimethyl sulfoxide and the cells were gently shaken for 10 min in the dark. Finally, the absorbance was measured at 570 nm. Data are presented as percentages of cell viability as compared to non-treated cells.
Reactive oxygen species production
Production of ROS was measured using the fluorogenic 2’,7’-dichlorodihydrofluorescein-diacetate probe (H2DCFDA, Thermo Fisher) [23]. Astrocytes cultured for 18–21 DIV were incubated in Hanks medium containing 10μM H2DCFDA for 1 h treatments. Fluorescence at 535 nm was measured at the beginning and end of the experiment. The difference in fluorescence was normalized using cell viability data and was expressed as the percentage of ROS production as compared to non-treated cells.
Immunocytochemistry
Immunocytochemistry was essentially carried out as described by Domínguez-Prieto et al. [6]. After the treatments, cells were fixed in 4% paraformaldehyde for 20 min. Once fixed, cells were washed with PBS and permeabilized with 0.25% Triton X-100 for 1 h. Then, astrocytes were incubated overnight at 4°C with primary antibodies (1:200) against Aβ25 - 35 (LS-C51552, LSBio, Seattle, WA, USA), Aβ40 (NBP1-44047, Novus Biologicals, Littleton, CO, USA), Aβ42 (LS-C42699, LSBio) and GFAP (G3893, Sigma Aldrich), and then incubated for 2 h at room temperature with (1:1000) the secondary anti-rabbit or anti-mouse Alexa Fluor 488 or 647 (Thermo Fisher) antibodies. Images were taken using a Leica DM-IRE 2 TCS-SP2 confocal microscope with LCS Lite Software (Leica Microsystems, Wetzlar, Germany).
Astrocytes transfection
Astrocytes cultured for 18–21 DIV were transfected using Lipofectamine 2000 (Invitrogen, Thermo Fisher). A validated non-targeting siRNA (NT-siRNA) was used as a control of transfection, the Silencer® Negative Control No. 1 siRNA (AM4635, Invitrogen) for the caveolin-1 experiments and siGENOME Non-Targeting siRNA Pool #2 (D-001206-14-05, Dharmacon, GE Healthcare, Buckinghamshire, UK) for the clathrin studies. siRNA targeting caveolin-1 (Cav1-siRNA) was synthesized based on sequences from a previous report [24] (sense sequence: 5’ GGGACACACAGUUUCGACG, antisense sequence: 5’ CGUCGAAACUGUGUGUCCC). siRNAs targeting clathrin heavy chain (Clt-siRNAs) were purchased from Dharmacon (M-090659-01-0005). Cells were transfected with the double-strand siRNA complexed with 2.5μl/ml of Lipofectamine in culture medium without antibiotics. The cells were maintained in the presence of the oligonucleotides in culture medium without antibiotics for 24 h. Cav1-siRNA was used at 60 nM and the assays were performed 72 h post-transfection. A concentration of 100 nM was used for the Clt-siRNAs and astrocytes were treated 96 h after transfection. The extent of siRNA-mediated down-regulation of Cav1 or Clt expression was evaluated by Western blot analysis of parallel samples.
Western blot analysis
Cell proteins were extracted using a lysis buffer containing 5 mM Tris-HCl (pH 6.8), 2% SDS, 2 mM EDTA, 2 mM EGTA, 1 mM PMSF, and a cocktail of protease inhibitors (Calbiochem, Darmstadt, USA). Lysates were centrifuged at 14,000×g for 15 min at 4°C. Twenty μg of protein extract was analyzed in 10% precast commercial gels (NuPAGE Novex 10% Bis-Tris Midi Gel 1.0 mm). The buffer used for protein electrophoresis was NuPAGE MOPS SDS Running Buffer 20X. NuPAGE Sample Reducing Agent 10X and NuPAGE LDS Sample Buffer 4X were used to prepare the samples. Electrophoresis was run at room temperature using a constant voltage. After electrophoresis, the gels were washed in transfer buffer (10% methanol and 0.1% NuPAGE Antioxidant diluted in NuPAGE Transfer Buffer 2X) for 10 min. Then, the proteins were transferred to a nitrocellulose membrane (iBlot Gel Transfer Stacks Nitrocellulose) for 10 min and by applying a constant voltage. All products used for electrophoresis and subsequent electrotransfer were purchased from Invitrogen (Thermo Fisher). After blocking to prevent non-specific binding, the membranes were incubated overnight at 4°C with a rabbit polyclonal antibody against caveolin-1 1:1000 (Ab2910, Abcam, Cambridge, UK) or a mouse monoclonal antibody against clathrin heavy chain 1:1000 (61050, BD Biosciences, NJ, USA). A mouse monoclonal antibody against GAPDH (AM4300, Ambion, Thermo Fisher) was used to normalize and quantify protein expression. After several washes, the membranes were incubated with a secondary antibody against mouse immunoglobulin conjugated with peroxidase. Finally, membranes were incubated for 1 min with peroxidase substrates, which afforded a chemiluminescence reaction. The signal produced on the autoradiographic film was proportional to the amount of protein in the membrane. The bands were quantified using an image analysis program.
Statistical analysis
All results are presented as the mean±SEM of at least three independent experiments (n≥3). Data were analyzed for statistical significance using Student’s t-test when two groups were compared or one-way ANOVA, followed by an appropriate post-hoc test, for the comparison of three or more groups. Dunnett test was used to compare all the values with the control and Tukey test to compare all the values among themselves. Values were considered significant when p < 0.05.
RESULTS
Effects of Aβ peptides on cell viability and ROS production in astrocytes in primary culture
Recently, we studied the effects of Aβ peptides on neuronal viability, where it was found that Aβ25 - 35, Aβ40, and Aβ42 significantly decrease neuronal viability [6]. In this work, the effects of Aβ peptides on astrocytes in primary culture were assessed, and it was again observed that Aβ25 - 35, Aβ40, and Aβ42 strongly decrease astrocyte viability (Fig. 1A). In neurons, Aβ25 - 35 strongly decreased cell viability, while the effect of Aβ40 and Aβ42 was more moderate [6]. By contrast, in astrocytes, all three Aβ peptides caused a significant decrease in cell viability (Fig. 1). In addition, the presence of the three peptides significantly increased ROS production in astrocytes in primary culture (Fig. 1B).
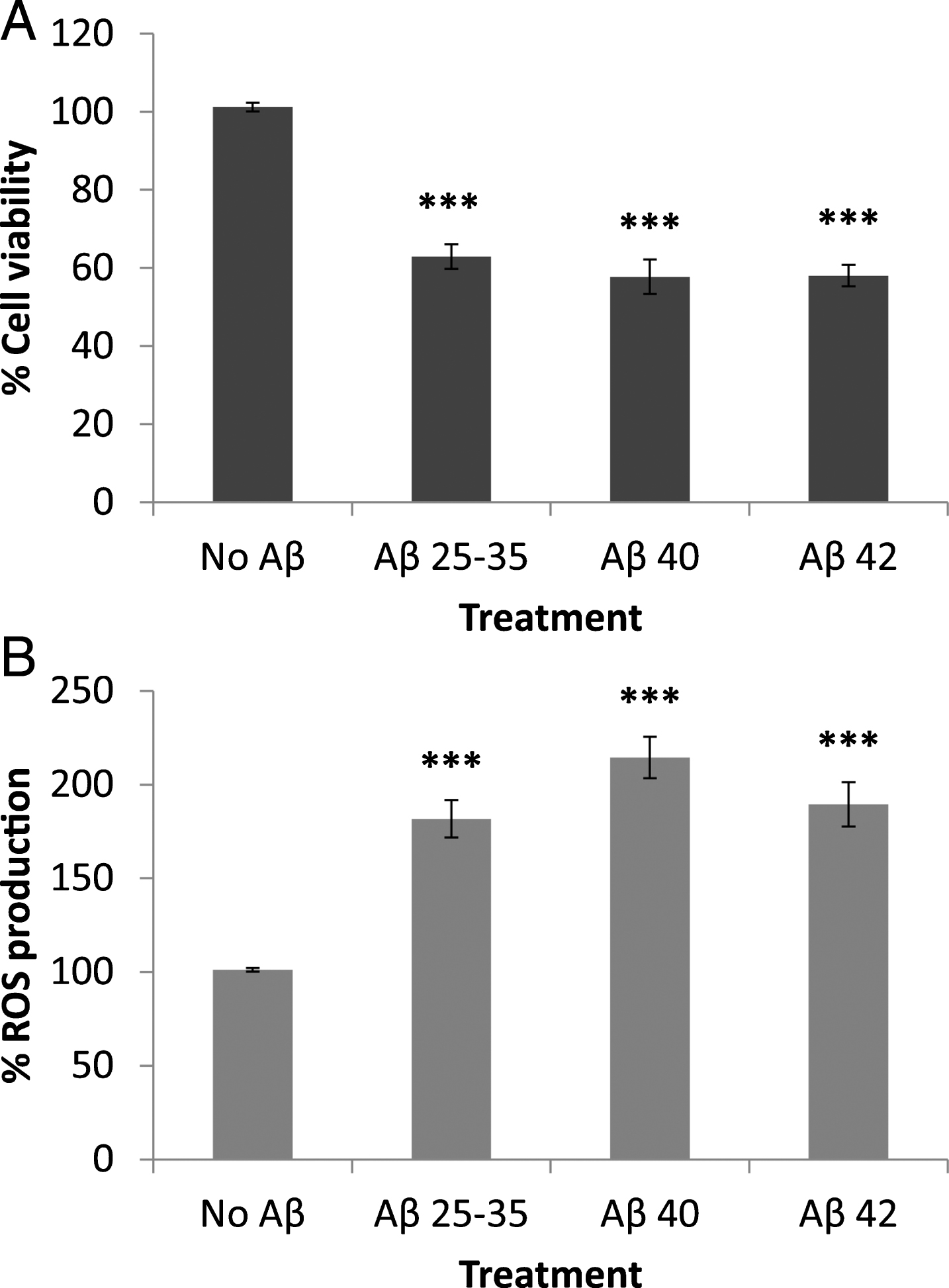
Effect of amyloid-beta peptides on astrocytes viability (A) and on ROS production (B). Astrocytes in primary culture (18–21 DIV) were incubated for 1 h in Hanks medium in the absence or the presence of three different Aβ peptides (30μM): Aβ25 - 35, Aβ40, or Aβ42. Results are expressed as percentages compared to non-treated cells and are means±SEM (n≥5). ROS production was normalized using the cell viability data. One-way ANOVA and Dunnet Test were applied in order to compare the different treatments to the controls. ***p < 0.001.
It should also be mentioned that unlike neurons [6], albumin-amyloid-β complexes still decreased astrocytes viability (see Supplementary Figure 2) suggesting that albumin is unable to prevent the deleterious effects of Aβ in astrocytes.
Cellular localization of Aβ peptides in astrocytes in primary culture
Aβ25 - 35 was internalized by astrocytes (Figs. 2 and 3) showing a diffuse intracellular distribution, while Aβ40 formed aggregates bound to the astrocytic membrane, which remained stained even after the immunochemistry preparations were washed (Figs. 2 and 3). However, discrete vesicular-like structures stained with Aβ40 were observed inside the cell (Fig. 3, arrows) and co-localized with GFAP (Fig. 3; yellow). In the case of Aβ42 no extracellular aggregates were observed, but the peptide was distributed in the cytoplasm and the nuclei of astrocytes (Figs. 2 and 3). It should be mentioned that Barucker et al. [25] claimed that Aβ42 specifically localizes in the nuclei, which suggests that Aβ42 could be a transcriptional regulator.
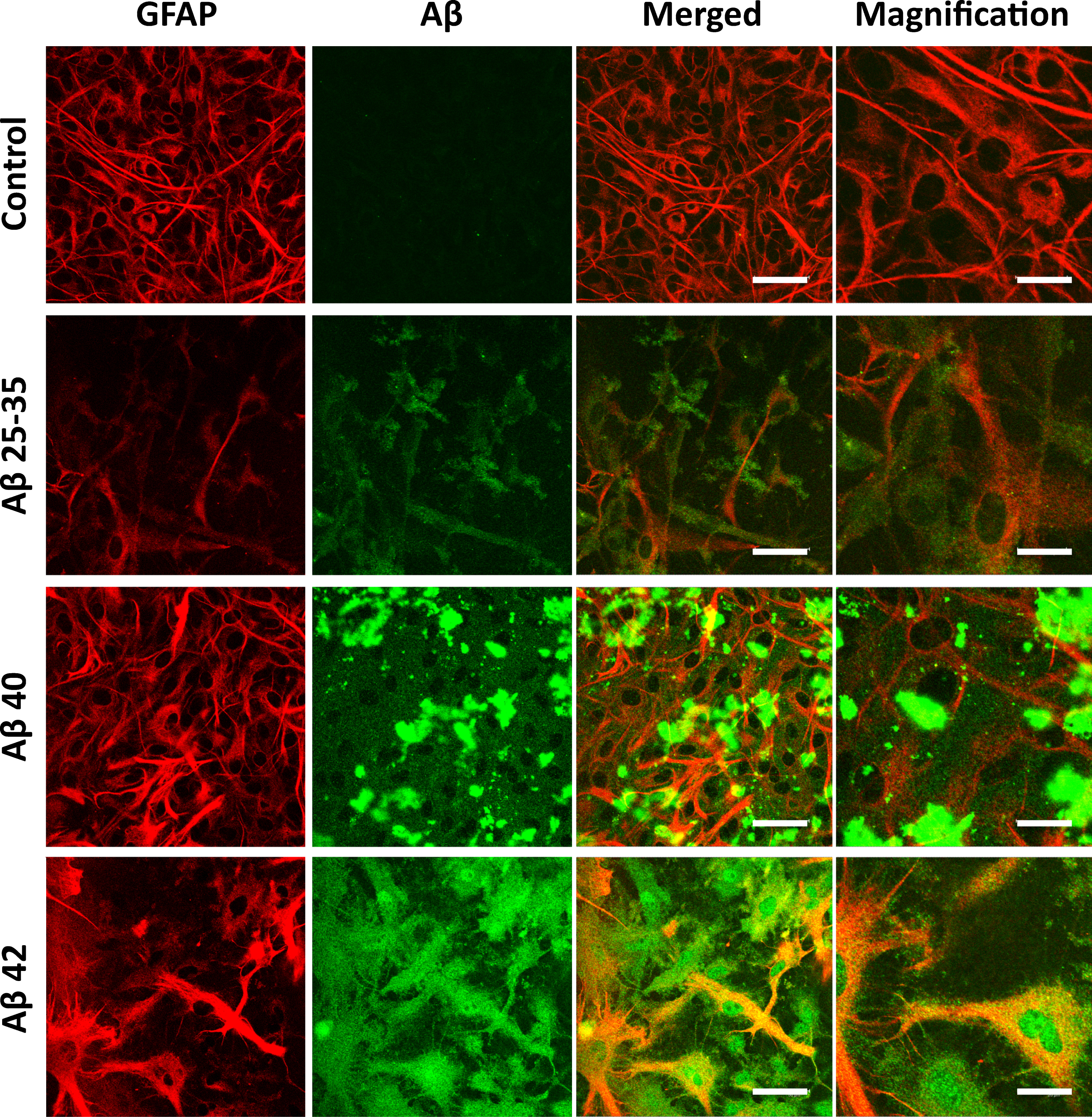
Cellular localization of amyloid-β peptides. Astrocytes in primary culture (21 DIV) were incubated for 1 h in Hanks medium in the absence or the presence of three different Aβ peptides (30μM): Aβ25 - 35, Aβ40, or Aβ42. After incubation, astrocytes were fixed and immunocytochemistry against GFAP (in red) and amyloid-beta (in green) were carried out. Images were taken using confocal microscopy. Scale bar: 50μm. Magnification scale bar: 20μm.

Internalization of amyloid-β peptides in astrocytes. Astrocytes in primary culture (21 DIV) were incubated for 1 h in Hanks medium in the presence of three different Aβ peptides (30μM): Aβ25 - 35, Aβ40, or Aβ42. After incubation, astrocytes were fixed and immunocytochemistry against GFAP (in red) and amyloid-beta (in green) were carried out. Images were taken using confocal microscopy. Orthogonal projections along the z-axis of the images are shown at the bottom and right.
Effect of temperature on the decreased cell viability caused by Aβ peptides and on Aβ peptide internalization in astrocytes in primary culture
Cellular viability was significantly enhanced in the presence of all three Aβ peptides by lowering the incubation temperature to 4°C (Fig. 4A). This result coincided with an observable decrease in peptide internalization (Fig. 4B), which suggested that a low temperature could inhibit the entry of Aβ peptides into astrocytes, and in turn decrease their deleterious effects.
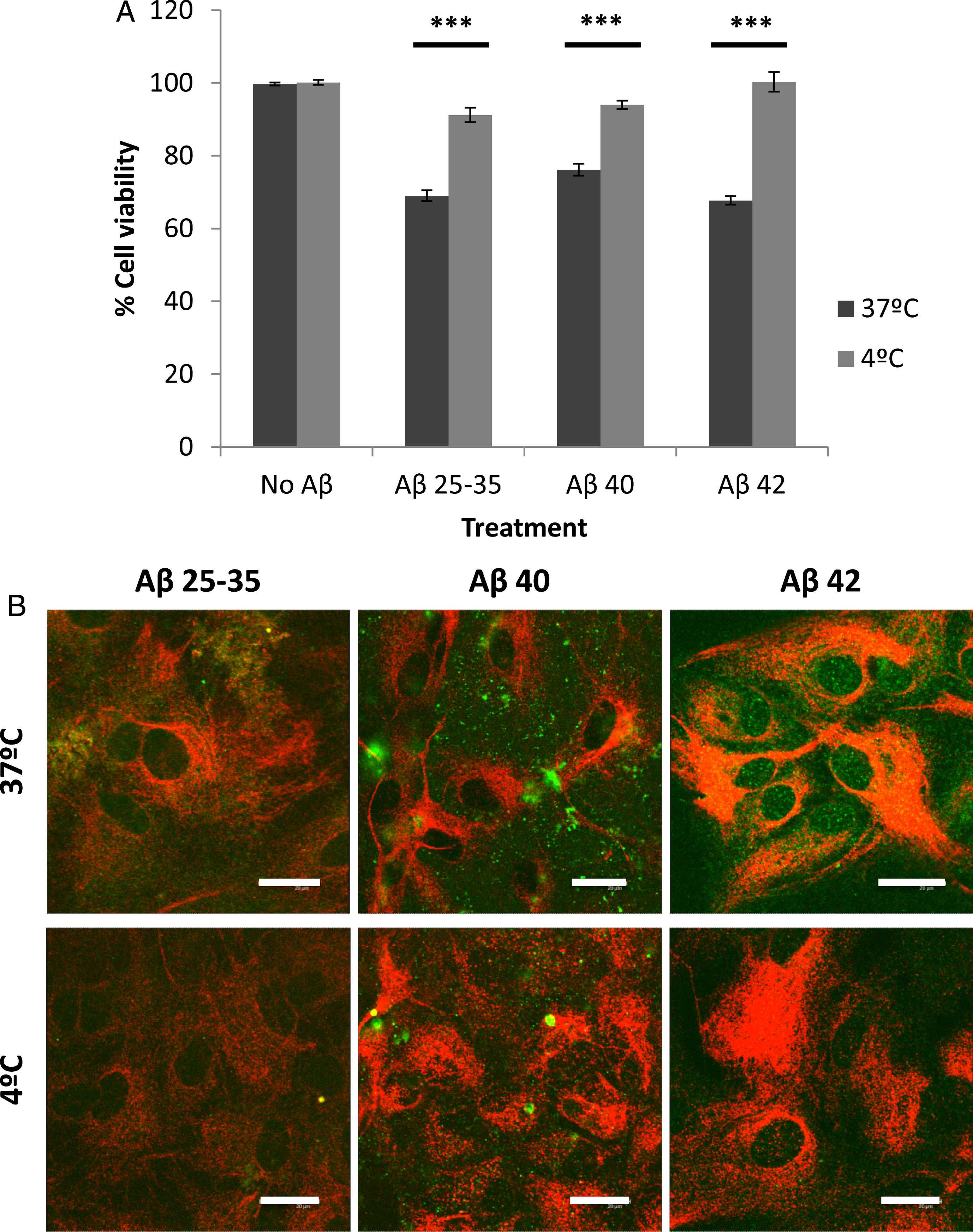
Effect of temperature on astrocyte viability (A) and amyloid-beta internalization (B). Astrocytes in primary culture (18–21 DIV) were incubated at 4°C or 37°C for 30 min in Hanks medium in the absence or the presence of three different Aβ peptides (30μM): Aβ25 - 35, Aβ40, or Aβ42. A) Results are expressed as percentages compared to non-treated cells and are means±SEM (n≥3). Student t-test was used to analyze differences between each treatment. ***p < 0.001. B) After incubation, astrocytes were fixed and immunocytochemistry against GFAP (in red) and amyloid-beta (in green) were carried out. Images were taken using confocal microscopy. Scale bar: 20μm.
Effect of methyl-beta-cyclodextrin (MBCD) or chlorpromazine (CPZ) on the decreased cell viability caused by Aβ peptides and on Aβ internalization in astrocytes in primary culture
The presence of methyl-beta-cyclodextrin, an inhibitor of caveolae-mediated endocytosis [26], did not significantly change the decrease in astrocyte viability caused by the Aβ peptides (Fig. 5A) or in their internalization (Fig. 5B). This suggested that Aβ endocytosis in astrocytes is not mediated by caveolae. Conversely, chlorpromazine (CPZ), a well-known inhibitor of clathrin-coated endocytosis [27], significantly prevented the decrease in cell viability caused by the peptides (Fig. 6A), and also decreased their internalization (Fig. 6B) in astrocytes. This suggested that chlorpromazine prevents Aβ peptide endocytosis and hence their deleterious effects in astrocytes.
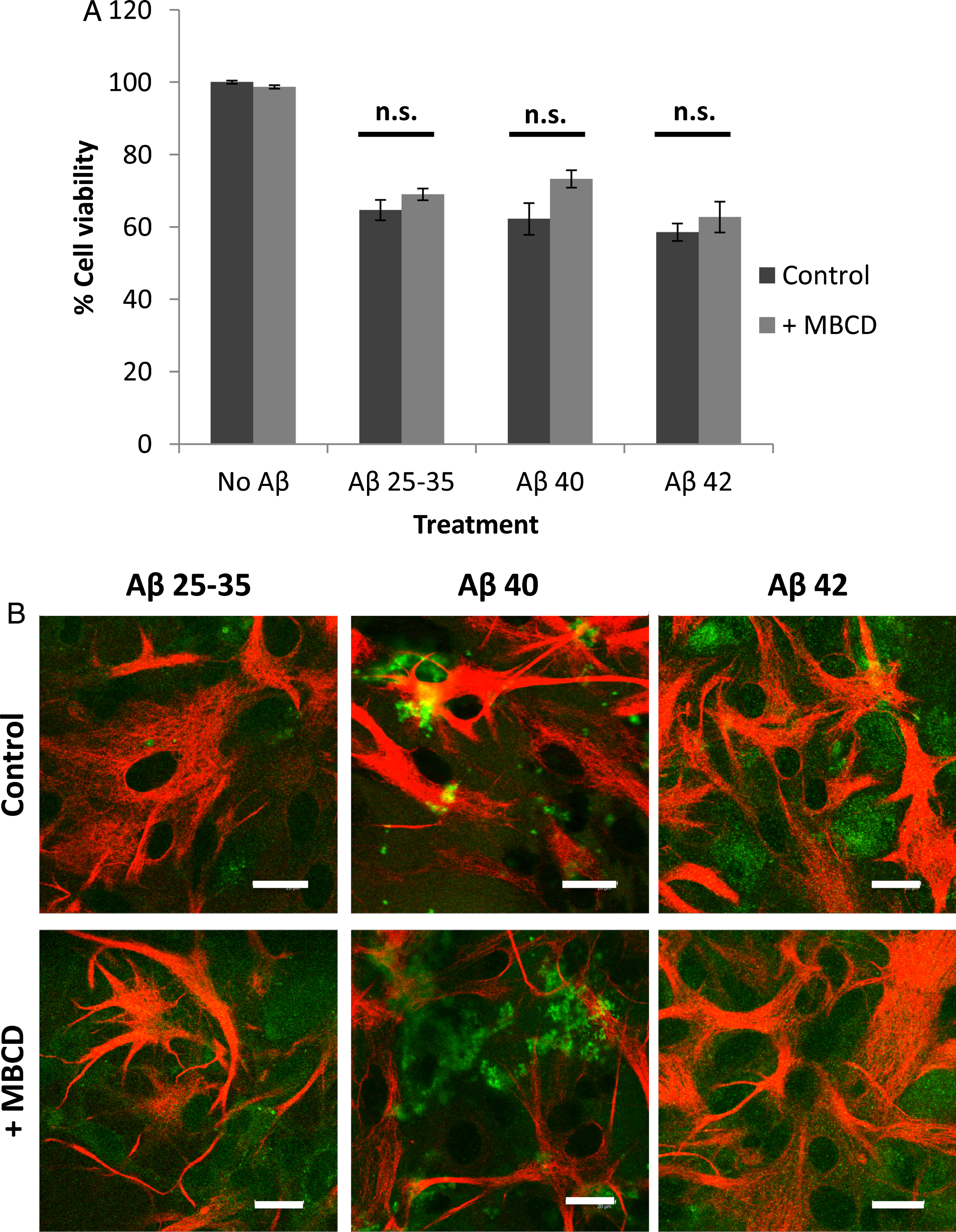
Effect of methyl-beta-cyclodextrin (MBCD) on astrocyte viability (A) and amyloid-beta internalization (B). Astrocytes in primary culture (18–21 DIV) were incubated for 1 h in Hanks medium in the absence or the presence of three different Aβ peptides (30μM): Aβ25 - 35, Aβ40, or Aβ42; and in the absence or the presence of MBCD (25 mM). A) Results are expressed as percentages compared to non-treated cells and are means±SEM (n≥4). Student t-test was used to analyze differences between each treatment. n.s.: no significant. B) After incubation, astrocytes were fixed and immunocytochemistry against GFAP (in red) and amyloid-beta (in green) were carried out. Images were taken using confocal microscopy. Scale bar: 20μm.
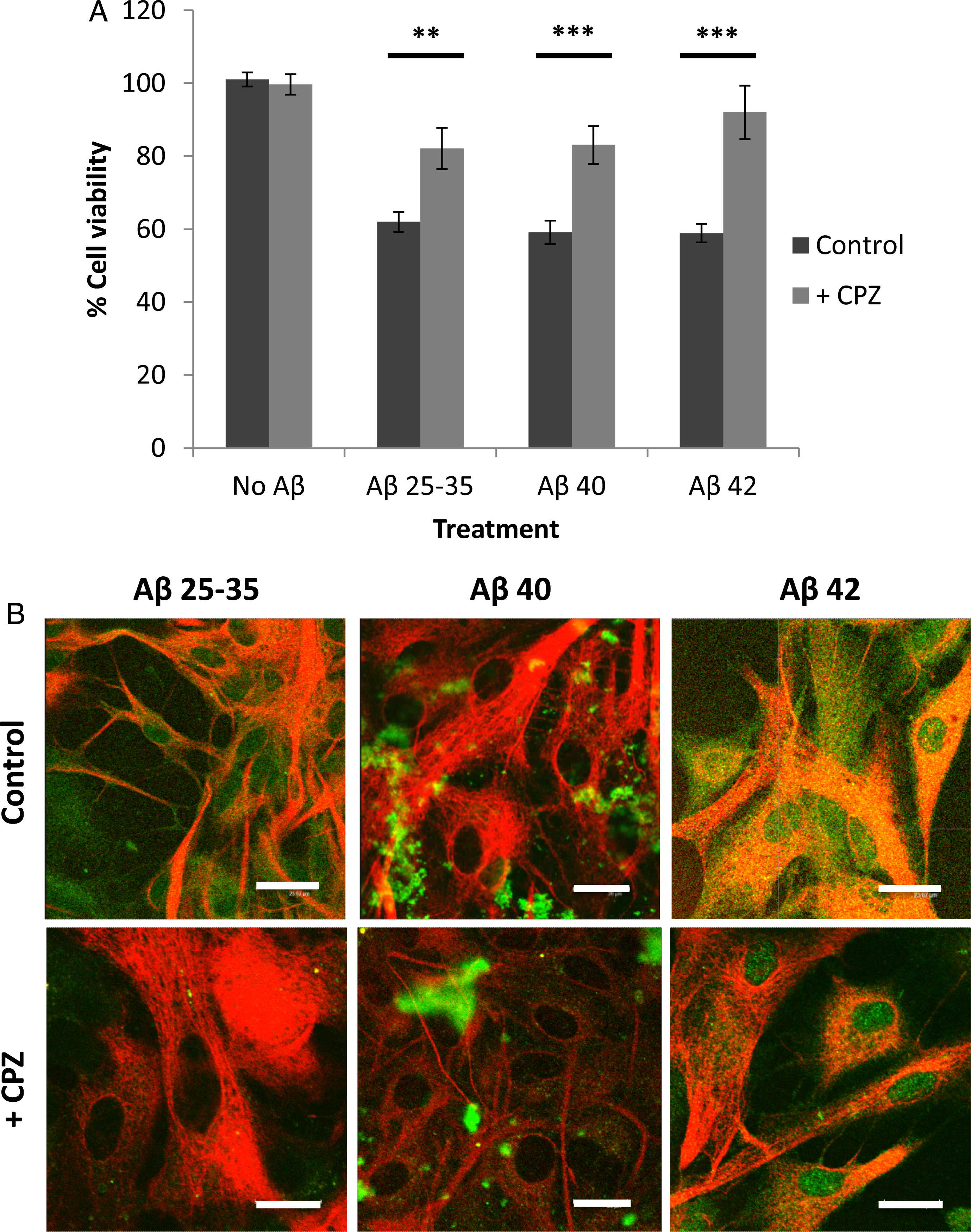
Effect of chlorpromazine (CPZ) on astrocyte viability (A) and amyloid-beta internalization (B). Astrocytes in primary culture (18–21 DIV) were incubated for 1 h in Hanks medium in the absence or the presence of three different Aβ peptides (30μM): Aβ25 - 35, Aβ40, or Aβ42; and in the absence or the presence of chlorpromazine (10μg/ml). A) Results are expressed as percentages compared to non-treated cells and are means±SEM (n≥4). Student t-test was used to analyze differences between each treatment (*). ***p < 0.001. B) After incubation, astrocytes were fixed and immunocytochemistry against GFAP (in red) and amyloid-beta (in green) were carried out. Images were taken using confocal microscopy. Scale bar: 20μm.
Effect of caveolin or clathrin silencing by RNA interference on the decreased cell viability caused by Aβ peptides in astrocytes in primary culture
Silencing caveolin-1 by Cav1-siRNA (Fig. 7B) did not change the effects of the Aβ peptides on cell viability (Fig. 7A), suggesting that the presence of caveolin-1 is not compulsory for Aβ internalization. Conversely, the silencing of clathrin by Clt-siRNA (Fig. 8B) significantly increased cell viability (Fig. 8A), suggesting that clathrin plays a key role in the internalization of Aβ peptides by astrocytes. It is worth noting that chlorpromazine exerted a higher inhibitory effect (Fig. 6A) than the silencing of clathrin by Clt-siRNA (Fig. 8A), a result that is consistent with the idea that other mechanisms may collaborate with the internalization of Aβ peptides by astrocytes. In this context, Vercauteren et al. [28] claim that chlorpromazine may also marginally inhibit clathrin-independent endocytosis.
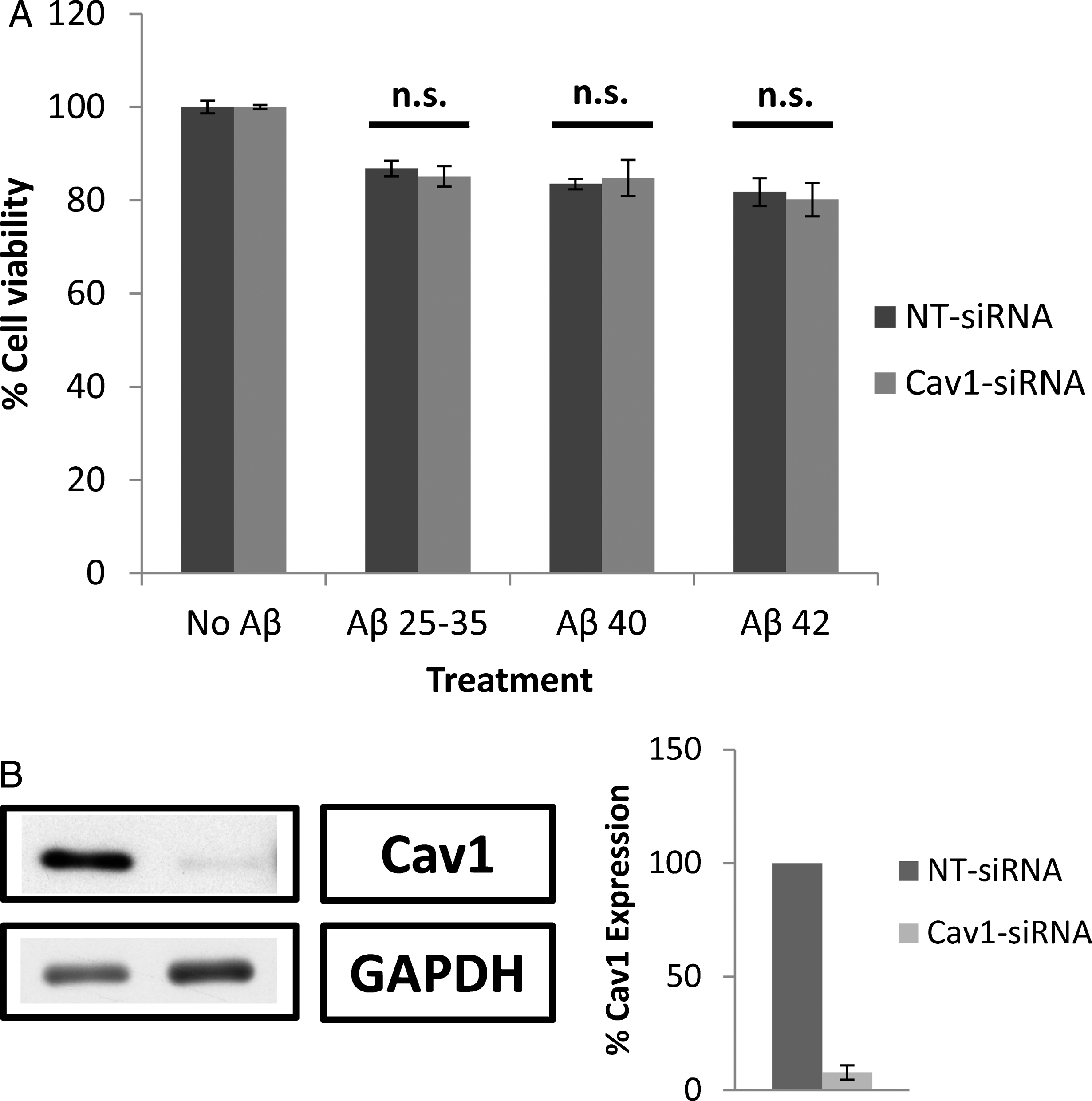
Effect of caveolin-1 (Cav1) silencing by Cav1-siRNA on astrocyte viability in the presence of amyloid-β. A) Astrocytes in primary culture (18–21 DIV) were transfected for 72 h with Cav1-siRNA and non-targeting siRNA (NT-siRNA). After that, cells were incubated for 1 h in Hanks medium in the absence or the presence of three different Aβ peptides (30μM): Aβ25 - 35, Aβ40, or Aβ42. Results are expressed as percentages compared to NT-siRNA transfected cells and are means±SEM (n≥5). Student t-test was used to analyze differences between each treatment. n.s.: no significant. B) Cav1 silencing was quantified by western blot. Results are expressed as percentages of NT-siRNA transfected cells.
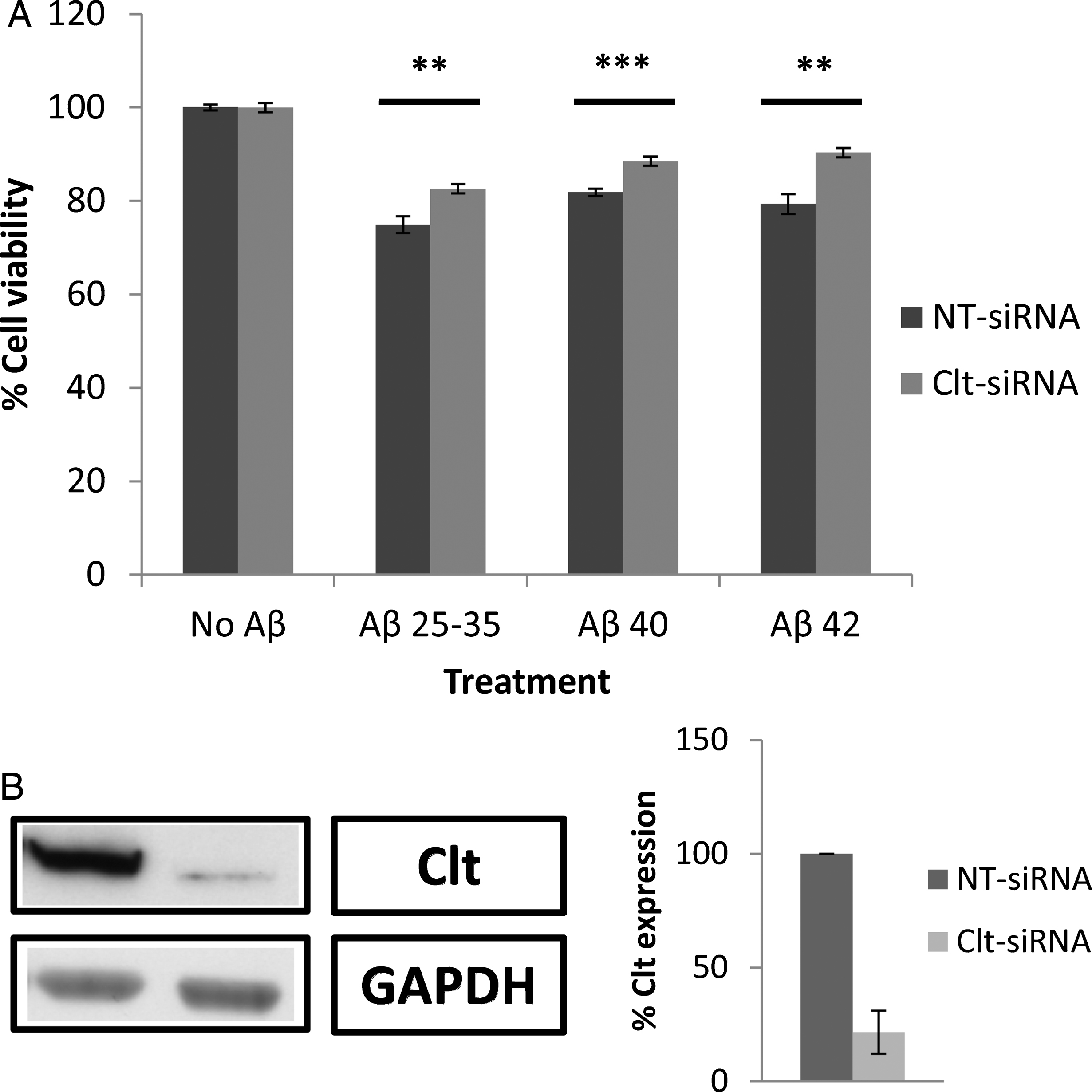
Effect of clathrin heavy chain (Clt) silencing by Clt-siRNAs on astrocyte viability in the presence of amyloid-β. A) Astrocytes in primary culture (18–21 DIV) were transfected for 96 h with Clt-siRNAs and non-targeting siRNA (NT-siRNA). After that, cells were incubated for 1 h in Hanks medium in the absence or the presence of three different Aβ peptides (30μM): Aβ25 - 35, Aβ40, or Aβ42. Results are expressed as percentages compared to NT-siRNA transfected cells and are means±SEM (n≥5). Student t-test was used to analyze differences between each treatment (*). ***p < 0.001, **p < 0.01. B) Clt silencing was quantified by western blot. Results are expressed as percentages of NT-siRNA transfected cells.
Effect of chlorpromazine on the transcytosis of Aβ peptides in astrocytes in proximal primary cultures
To ascertain whether Aβ peptides undergo transcytosis in astrocytes, (see Supplementary Figure 1) experiments were designed in which non-treated astrocytes were co-incubated with Aβ-loaded astrocytes without physical contact. Under these circumstances, cell viability and Aβ peptide internalization in previously non-treated astrocytes were assessed. After 4 h of co-incubation with Aβ-loaded astrocytes (upper deck), cell viability (Fig. 9A) decreased in the previously non-treated astrocytes (lower deck), an event that is associated with the internalization of Aβ peptides (Fig. 9B). However, the presence of chlorpromazine (CPZ) prevented the Aβ peptides from reaching the unloaded astrocytes, as shown by the lack of effect on cell viability observed in the astrocytes located in the lower deck under these experimental conditions (Fig. 10).
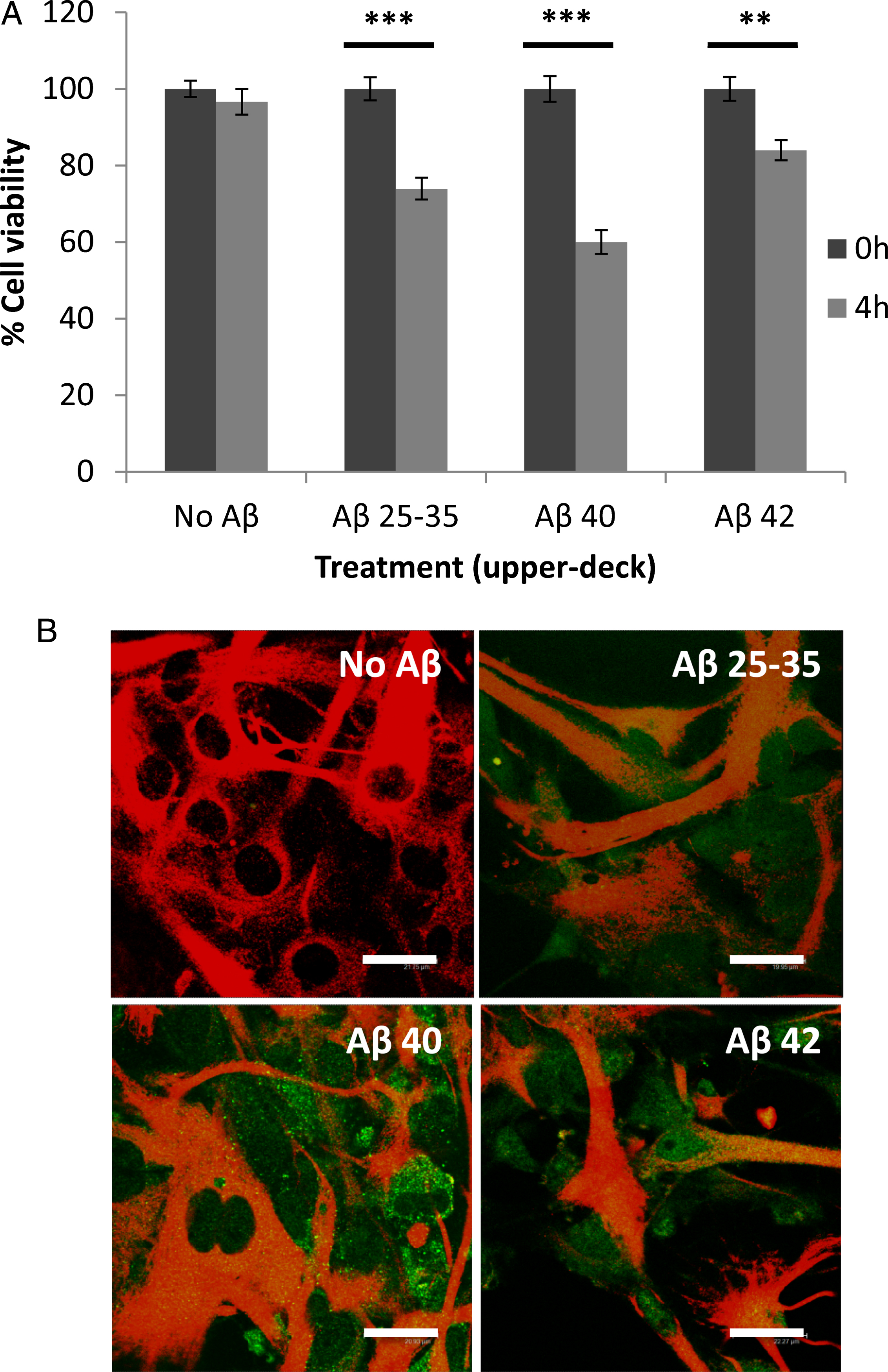
Transcytosis of amyloid-β peptides in co-cultured astrocytes. Primary astrocytes cultured on transwell inserts were incubated for 5 min in Hanks medium in the absence or the presence of three different Aβ peptides (30μM): Aβ25 - 35, Aβ40, or Aβ42. Then, the inserts were co-incubated with non-treated astrocytes cultured in 24-well plates coming from the same culture, with no physical contact among them. A) The viability of the astrocytes in the lower deck was analyzed after 4 h of co-incubation. Results are expressed as percentages compared to lower-deck astrocytes co-cultured with non-treated astrocytes and are means±SEM (n≥3). After 4 h of co-incubation, cell viability of non-treated astrocytes (lower deck) co-incubated with Aβ-treated astrocytes (upper deck) significantly decreased (p < 0.05). B) After 4 h of co-incubation, astrocytes in the lower deck were fixed and immunocytochemistry against GFAP (in red) and amyloid-beta (in green) were carried out. Images were taken using confocal microscopy. Scale bar: 20μm.

Effect of chlorpromazine (CPZ) on the transcytosis of amyloid-beta peptides in co-cultured astrocytes. Primary astrocytes cultured on transwell inserts were incubated for 5 min in Hanks medium in the absence or the presence of three different Aβ peptides (30μM): Aβ25 - 35, Aβ40, or Aβ42. Then, the inserts were co-incubated with non-treated astrocytes cultured in 24-well plates coming from the same culture, with no physical contact among them and in the absence or the presence of chlorpromazine (10μg/ml). The viability of the astrocytes in the lower deck was analyzed after 4 h of co-incubation. Empty transwell inserts were used as a control. Results are expressed as percentages compared to lower-deck astrocytes co-cultured with non-treated astrocytes and are means±SEM (n≥3). One-way ANOVA and Dunnet Test were applied in order to compare different conditions versus control. *p < 0.05; ***p < 0.001.
DISCUSSION
We previously reported that Aβ25 - 35, Aβ40, and Aβ42 significantly decrease cell viability in neurons in primary culture [6]. In the present study, the effect of Aβ peptides on astrocytes in primary culture has been assessed. The three Aβ peptides assayed significantly decreased cell viability and increased ROS production (Fig. 1), an event that is associated with the peptide internalization by the astrocytes. It should be mentioned that the enhancement of ROS production in astrocytes may be brought about by the activation of NADPH oxidase [29]. Moreover, Hettiarchchi et al. [30] recently reported that the toxic effects of Aβ peptides are due to the synthesis of peroxynitrite by combining nitric oxide with ROS.
To establish whether the entry of Aβ peptides into astrocytes is compulsory or not for the deleterious effects of Aβ peptides to occur, the mechanism for Aβ internalization was analyzed. The Aβ concentration used in our experiments was 30μM, which is significantly higher than concentrations previously reported in the literature. This concentration was chosen in order to ensure that endocytosis was completely saturated and therefore changes observed were associated to modifications in the endocytosis process and not to a lack of substrate. These Aβ concentrations were also used to load astrocytes with the peptides for transcytosis experiments. The results obtained show that by decreasing the incubation temperature to 4°C the decrease in astrocyte viability caused by the peptides is avoided, as well as most of the peptide internalization (Fig. 4). Since this low temperature was able to inhibit endocytosis [31, 32], this result is consistent with the idea that the three peptides are actively endocytosed by astrocytes and that internalization is necessary for their deleterious effects to occur. For this reason, the mechanism by which Aβ peptides are endocytosed by astrocytes was studied. The results showed that the presence of methyl-beta-cyclodextrin, an inhibitor of caveolin-mediated endocytosis (Fig. 5) or silencing caveolin-1 by caveolin1-siRNA (Fig. 7) did not prevent the cell death caused by the presence of the Aβ peptides, which suggests that these peptides do not use caveolae to enter astrocytes. Conversely, the presence of chlorpromazine, an inhibitor of clathrin-mediated endocytosis (Fig. 6) or silencing clathrin by clathrin-siRNA (Fig. 8) inhibited the decrease in astrocyte viability caused by the Aβ peptides, suggesting that Aβ peptides are endocytosed by clathrin-coated vesicles in astrocytes.
The presence of a specific mechanism for Aβ peptide internalization in astrocytes prompted us to investigate whether Aβ peptides underwent transcytosis in astrocytes. Hence, experiments were designed in which Aβ-loaded astrocytes were incubated with astrocytes not exposed to Aβ peptides, as they were kept in separate wells but with the same Aβ-free medium (for the experiment design see Supplementary Figure 1). Our results suggest that the Aβ-loaded astrocytes released the peptides into the medium, which reached the Aβ-free astrocytes and promoted cell death (Fig. 9). Additionally, chlorpromazine prevented cellular death in unloaded astrocytes (lower deck) when co-incubated with Aβ-loaded astrocytes, suggesting that the uptake of Aβ peptides by unloaded astrocytes (lower deck) accounted for the deleterious effects observed. This does not exclude the participation of inflammatory factors presumably released by Aβ-loaded astrocytes [13, 14], although it is unlikely that the effect of the inflammatory factors on the astrocytes in the lower deck depend on clathrin-coated vesicles and hence it might be sensitive to chlorpromazine.
These results indicate that astrocytes could perform an important role in the clearance of Aβ from the brain [20], where soluble forms of Aβ are released into CSF and transported to blood where they are disposed of [33–35]. Consequently, it is reasonable to suggest that astrocytes may play a role in the transport and release of Aβ in CSF. Under physiological circumstances, Aβ may be recruited by astrocytes and transported to CSF through the transcytosis of adjacent astrocytes. In such a way, low concentrations of Aβ are maintained inside the astrocytes which prevents cell death. However, accumulation of Aβ as observed in AD, may surpass Aβ clearance capacity by jamming astrocyte rows. This could lead to astrocyte death and eventually trigger astrocytosis [13, 14]. It is important to highlight that an alternative mechanism for Aβ transport using gap junctions (i.e., those fine regulated pores which connect adjacent astrocytes) should be discarded, because molecules can cross gap junctions [36] but must be smaller than 1.0–1.2 kDa [37].
Unlike neurons, complexing Aβ with human serum albumin did not escape the deleterious effects of Aβ in astrocytes (Supplementary Figure 2). However, we have reported that astrocytes use caveolae but not clathrin-coated vesicles to endocyte serum albumin [24]. Thus, the inability of albumin to prevent Aβ-deleterious effects in astrocytes (Supplementary Figure 2) might suggest that astrocytes use caveolae to uptake the albumin-Aβ complex. If so, Aβ per se, or as a Aβ-serum albumin complex, is able to reach the astrocytic cytoplasm using its own mechanism for active endocytosis. Nevertheless, Aβ uptake by astrocytes is enhanced, a finding that is consistent with the idea that astrocytes play a role in the disposal of Aβ peptides. Moreover, it has been proposed that Aβ peptides are released to CSF and finally transported to blood as Aβ-albumin complexes [38]. This fact explains why Aβ-albumin complexes are allowed to enter astrocytes where they participate in the transport and clearance of Aβ peptides.
Consequently, our results suggest that astrocytes play a key role in Aβ transport and disposal. Thus, astrocytes may actively internalize both free-Aβ or albumin-Aβ complexes, contributing to the clearance of Aβ from the intercellular space. Once inside the astrocytes, Aβ peptides can be degraded by specific enzymes [17–19] or transcytosed to neighboring astrocytes in order to be transported to CSF and eventually to plasma [39]. Altogether, our results support the idea that astrocytes play a key role in the disposal of Aβ in the CNS.
Footnotes
ACKNOWLEDGMENTS
This work was supported by a grant from Instituto Grifols S.A., Barcelona, Spain. M. Domínguez-Prieto was the recipient of a pre-doctoral grant (FPI) from the Spanish Ministry of Economy, Industry and Competitiveness (MINECO). We are grateful to T. del Rey for her technical assistance.