
Abstract
Angiotensin converting enzyme (ACE) is involved in proteolytic processing of the amyloid-β(Aβ) peptide implicated in the development of Alzheimer’s disease (AD) and known products of ACE-based processing of Aβ42 are characterized by reduced aggregability and cytotoxicity. Recently it has been demonstrated that ACE can act as an arginine specific endopeptidase cleaving the N-terminal pentapeptide (Aβ1-5) from synthetic Aβ peptide analogues. In the context of proteolytic processing of full length Aβ42, this suggests possible formation of Aβ6-42 species. The aim of this study was to test a hypothesis that some N-terminally truncated Aβ peptide(s) could retain aggregability and neurotoxic properties typical for Aβ42. We have investigated aggregability of two amyloid-β peptides, Aβ6-42 and isoD7-Aβ6-42, mimicking potential proteolytic products of Aβ42 and isoD7-Aβ42, and evaluated their effects on the repertoire of brain Aβ binding proteins, and cytotoxicity towards neuroblastoma SH-SY5Y cells. Aggregability of isoD7-Aβ6-42 and Aβ6-42 was higher than that of full-length peptides Aβ42 and isoD7-Aβ42, while the repertoire of mouse brain Aβ binding proteins dramatically decreased. Aβ6-42 and isoD7-Aβ6-42 exhibited higher neurotoxicity towards SH-SY5Y cells than Aβ42 and isoD7-Aβ42, respectively. They effectively stimulated production of ROS and NO, and also TNFα secretion by cells. Thus, our results suggest that ACE-dependent processing of full-length Aβs could result in formation of more pathogenic peptides.
Keywords
INTRODUCTION
It becomes increasingly clear that angiotensin converting enzyme (ACE; EC 3.4.15.1) is involved in proteolytic cleavage of amyloid-β peptide (1–42) (Aβ42), a key player in the development and progression of Alzheimer’s disease (AD) [1–5]. Although good evidence exists that ACE prevents two major events, Aβ aggregation and Aβ-induced cytotoxicity [1–5], it is known that in some groups of patients (e.g., in the absence of apolipoprotein E4 allele) therapy with ACE inhibitors reduces risk of AD [6]. However, results obtained in different laboratories are often contradictory [7]. Evident discrepancy also considered in a recent review [7] may be attributed not only to existing data on the primary role of ACE inhibitors on in vivo levels of important ACE peptide substrates other than Aβ; it is also possible that we do not know much about effects of appropriate Aβ42-derived peptides that can be formed during Aβ42 processing catalyzed by ACE.
Somatic ACE consists of a single polypeptide chain (1277 residues), in which N- and C-domains have been recognized [8, 9]. In the context of Aβ42 proteolytic processing, the N-domain is especially interesting as it can cleave N-terminal penta- or heptapeptides from Aβ [2, 10–12]. Cleavage of the N-terminal heptapeptide by ACE significantly weakened aggregability and cytotoxicity of the resultant Aβ, while the heptapeptide itself, Aβ1-7, did not demonstrate aggregability and cytotoxicity at all [2]. Recently it has been demonstrated that the N-terminal domain acts as an arginine specific endopeptidase cleaving the N-terminal pentapeptide (Aβ1 - 5) from synthetic Aβ peptide analogues [11, 12]. It cleaved more efficiently the analogue containing isoD7 than the analogue containing D7 [11]. It was hypothesized that the N-terminally truncated Aβ peptide(s) in vivo could retain aggregability and neurotoxic properties typical for Aβ42 [12]. In this context it is important to elucidate, whether removal of the N-terminal pentapeptide has any impact on aggregability and cytotoxic characteristics of resultant Aβ6-42? The definite answer to this question would provide additional evidence for possible involvement of ACE in production of toxic Aβ species.
In this study, we have investigated two truncated amyloid-beta peptides, Aβ6-42 and isoD7-Aβ6-42, mimicking putative proteolytic products of Aβ42. The isoD7-Aβ6-42 peptide was chosen due to frequent isomerization of the D7 residue in Aβ species found in the brain of AD patients [13, 14]. Besides aggregability we have also investigated the effect of this proteolytic modification on the repertoire of mouse brain Aβ binding proteins and cytotoxicity towards human neuroblastoma SH-SY5Y cells.
MATERIALS AND METHODS
Materials
Synthetic peptides Aβ42: [H2N]DAEFRHDSGYEVHHQKLVFFAEDVGSNKGAIIGLMVGGV VIA-[COOH], isoD7-Aβ42: [H2N]-DAEFRH [isoD] SGYEVHHQKLVFFAEDVGSNKGAIIGL MVGGVVIA-[COOH] - and their truncated forms (Aβ6-42 and isoD7-Aβ6-42) were synthesized by Biopeptide Co., Inc (San Diego, CA, USA, custom made order). Their purity exceeded >98% as checked by HPLC and amino acid sequences were confirmed by mass spectrometry. Acetic acid (sodium salt), boric acid, formic acid, sodium tetraborate, and sodium hydroxide were from Acros Organics (Geel, Belgium). Affi-Gel 10 was purchased from BioRad (Hercules, CA, USA). All other materials and chemicals were from Sigma-Aldrich (St. Louis, MO, USA).
Aggregation studies
For aggregation studies the peptides pretreated as described in [15] were dissolved in 10 mM NaOH. Aβ solutions (0.5 mM) in 10 mM NaOH were adjusted with 100 mM HEPES-buffer (pH 5.0) to pH 7.4 and centrifuged (15 min, 16,000 g, 4°C) to remove insoluble peptide aggregates. Peptide concentration in the supernatant was evaluated spectrophotometrically at 280 nm using the molar extinction coefficient ∈280= 1490 M–1cm–1 [16]. This stock solution was used to prepare 30 μM Aβ solutions in buffer H containing 150 mM NaCl, 10 mM HEPES, pH 7.4. Aβ solutions were kept on ice before use. Zinc-induced Aβ aggregates were prepared by mixing 30 μM Aβ solution with buffer H containing 300 μM ZnCl2 (final Aβ concentration of 25 μM and a zinc/Aβ molar ratio of 2). The mixture was immediately used for turbidity measurements performed at 405 nm (A405), using an Agilent 8453E spectrophotometer (Agilent Technologies, USA). Dynamic light scattering (DLS) measurements were performed using a Zetasizer Nano ZS apparatus (Malvern Instruments Ltd., Malvern, UK) at 25°C after incubation for 20 min at room temperature as described in [17].
Fluorescence measurements of self-aggregation of Aβ isoforms were carried out on the Infinite M200 PRO microplate reader (TECAN, Switzerland) using excitation and emission wavelengths of 450 nm and 482, respectively [17]. Aliquots of Aβ isoform solutions (100 μL, peptide concentration of 30 μM) were mixed in Corning 96-well plates with 20 μL of the thioflavin T (ThT) solution in buffer H (ThT concentration of 150 μM) to provide the final Aβ and ThT concentrations of 25 μM. The fluorescence measurements were started immediately after preparation of Aβ/ThT mixtures and carried out in triplicates. Aβ/ThT mixtures were incubated at 37°C under constant stirring. The relative ThT fluorescence has been calculated as (F - F0)/F0, where F and F0 are the ThT fluorescence in the presence and the absence of Aβ peptides at a given time, respectively.
Proteomic profiling of mouse brain aβ-binding proteins
Male 3-4-month-old C57BL/6 mice (20–25 g) obtained from the Stolbovaya nursery (Moscow region) were used in the experiments performed at least one week after their arrival from the nursery. Animals received a standard laboratory chow and water ad libitum and their decapitation was performed between 11.00 h and 13.00 h under light ether anesthesia. All procedures conformed to the Russian version of the Guide for the Care and Use of Laboratory Animals (Washington, 1996) were approved by the Animal Care and Use Committee of the Institute of Biomedical Chemistry. The brains were immediately dissected and homogenized at a low speed in 0.05 M potassium-phosphate buffer, pH 7.4 (1:3, w/v), using an Ultra-Turrax T 10 homogenizer. In each experiment, pooled brain homogenates from three mice were used.
Homogenates were lysed with 3% Triton X-100 (final concentration) at 4°C for 1 h, diluted three-fold with 50 mM potassium phosphate buffer, pH 7.4, and centrifuged at 16000 rpm (Eppendorf 5415 R centrifuge) for 20 min at 4°C.
Aβ42, Aβ6-42, and isoD7-Aβ6-42 were immobilized on the Affi-Gel 10 resin in accordance with a recently developed protocol [18]. Nonspecific binding was evaluated using the control Affi-Gel resin lacking Aβ peptides.
The cleared supernatant of brain homogenate (5 mg of protein/mL) was added to the suspension of each affinity sorbent and control resin (1:1 v/v). Bacitracin, aprotinin, and PMSF were added up to the final concentrations of 0.005%, 0.01%, and 1 mM, respectively, and the suspension was incubated overnight at 4°C at a gentle stirring. The same incubations were also performed with the control Affi-Gel 10 without the immobilized peptide ligands.
After the incubation the sorbent was washed with the same buffer up to the absence of the protein in the washing (evaluated by baseline at A280) and then packed into the columns (1 × 0.75 mL). Proteins were eluted from the column by 1 M glycine buffer, pH 2.8, containing 150 mM NaCl at a flow rate of 0.5 mL/min. The eluate was concentrated up to 0.200 mL using Amicon Ultra centrifugal filter devices (Millipore, Hessen, Germany). Proteins were extracted, modified, and digested on filters using the FASP protocol (filter-aided sample preparation) [18].
The peptide samples were analyzed using high resolution Orbitrap Fusion mass spectrometer (Thermo Scientific, Waltham, MA USA) exactly as described in [19]. Peak lists obtained from MS/MS spectra were identified using OMSSA version 2.1.9 and Novor version 1.05.0573. The search was conducted using SearchGUI version 3.2.20. Protein identification was conducted against a custom version of the Uniprot-based complement database for the Mus musculus (Mouse) taxonomy group (release November 2017). The decoy sequences were created by reversing the target sequences in SearchGUI. The identification settings were as follows: trypsin as a specific protease, with a maximum of 1 missed cleavages, ±5ppm as MS1 level and ±0.05 Da as MS2 level tolerances; variable modifications: oxidation of M (+15.994915 u), deamination of N (+0.984016 u), deamination of Q (+0.984016 u), carbamidomethylation of C (+57.021464 u). Peptides and proteins were inferred from the spectrum identification results using Peptide Shaker version 1.16.15. Peptide Spectrum Matches (PSMs), peptides and proteins were validated at a 1.0% false discovery rate (FDR) estimated using the decoy hit distribution (examples of representative mass-spectra of some identified proteins are given in Supplementary Material, see Supplementary Figure S). Raw data files of mass spectrometry determination of proteins have been deposited via the PRIDE repository (Submission Reference: 1-20180328-104264)
Cell culture studies
The SH-SY5Y neuroblastoma cell line was obtained from the European Collection of Authenticated Cell Culture (ECACC, Public Health England, UK). Cells were cultivated in Dulbecco’s modified Eagle’s medium (DMEM) (Sigma Aldrich) supplemented with 10% heat-inactivated fetal calf serum (FCS, HyClone), 2 mM L-glutamine, 100 units/mL penicillin and 100 μg/mL streptomycin (complete DMEM). The cultivation was performed at 37°C in a humid atmosphere with 5% CO2. Cells were then collected using trypsin-versene, washed with complete DMEM, counted, and seeded into four-well plates (Nunc, Thermo Fisher Scientific, USA), coated with 0.01% Poly-L-lysine, at 400,000 cells per well in 1 mL of DMEM supplemented with 5% FCS. After cultivation at 37°C in a humid atmosphere with 5% CO2 for 24 h the medium containing 5% FCS was replaced for FCS-free DMEM. The Aβ peptides (10 μM) dissolved as described in [20] or equivalent amounts of DMSO were added to the wells, and the cells were cultivated at 37°C and 5% CO2 for 24 h (for evaluation of ROS and TNFα production) or 48 h (for evaluation of apoptosis and necrosis).
Apoptotic cells were detected using fluorescent microscopy and Hoechst 33342 dye; necrotic cells were detected using propidium iodide (PI). Briefly, after removal of FCS-free DMEM, the cell layer was stained with the Hoechst 33342 dye (10 μg/mL) in phosphate buffer for 30 min at 37°C in the darkness. After addition of PI (the final concentration of 30 μM) cells were visualized using an inverted fluorescent microscope (Keyence BZ8100, Japan). The number of apoptotic cells was calculated as a proportion of cells with fragmented DNA (and unstained with PI) to the total number of cells (defined as 100%). Cells stained with PI were considered as necrotic ones. For each variant, at least 20 fields of view (each of which containing 150–250 cells) were analyzed. TNFα production by SH-SY5Y was evaluated by the cytotoxic effect on target L-929 cells as described in [21]. Formation of reactive oxygen species (ROS) in SH-SY5Y cell was evaluated using the nitroblue tetrazolium test as described in [21].
Data of cells culture studies are expressed as the (mean±SEM) of at least four independent experiments performed in quadruplicates. The differences among the groups analyzed using the Shapiro-Wilk’s test of One Way ANOVA with Tukey’s pairwise comparisons were considered as statistically significant at p < 0.05.
RESULTS
Spontaneous aggregability, evaluated by the thioflavin T fluorescence assay, was higher in Aβ6-42 and isoD7-Aβ6-42 as compared with parent full-length peptides (Fig. 1A). Aβ6-42 demonstrated markedly higher zinc-dependent aggregability evaluated by turbidimetry (Fig. 1B). According to our data, these differences reached the level of statistical significance (p < 0.02) after incubation with zinc ions for 20 min (Supplementary Figure 1). Zinc-dependent aggregability of isoD7-Aβ6-42 was somewhat higher than in the case of isoD7-Aβ42 but this difference did not reach the level of statistical significance. The characteristic sizes of zinc-induced aggregates were significantly higher (p < 0.05) in Aβ6-42 and isoD7-Aβ6-42 than in the case of the full length peptides (Fig. 1C).
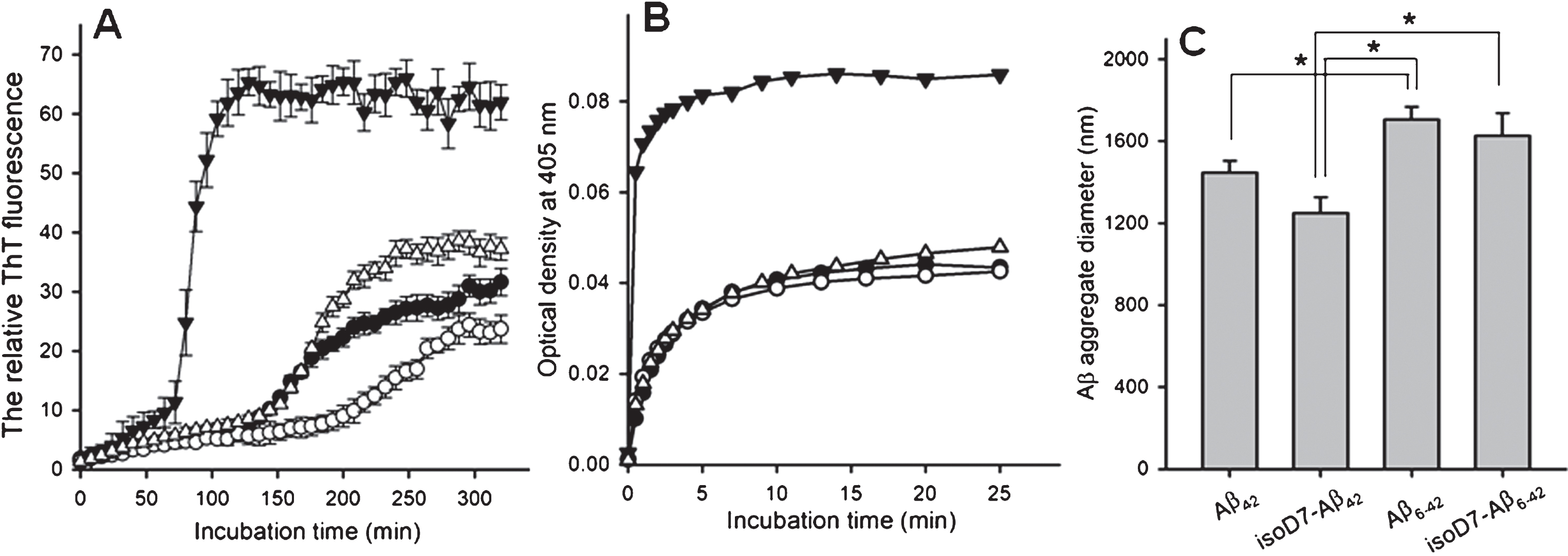
A) The time-dependence of relative ThT fluorescence in the presence of various Aβ isoforms. Closed circles – Aβ42; open circles – isoD7-Aβ42; closed triangles – Aβ6-42; open triangles – isoD7-Aβ6-42. Peptides (25 μM) were incubated in 10 mM HEPES, pH 7.4, containing 150 mM NaCl at 37°C under constant agitation. The peptide/ThT molar ratio was 1:1. Data represent the mean values±SEM of three measurements. B) The representative time-dependence of turbidity of Aβ solutions as a function of incubation time after addition of zinc ions. Symbols are the same as in (A). The zinc/peptide molar ratio was 2. Peptides (25 μM) were incubated in 10 mM HEPES, pH 7.4, containing 150 mM NaCl at room temperature under quiescent conditions. C) The characteristic diameter of zinc-induced Aβ aggregates formed by the Aβ isoforms studied. DLS measurements were carried out in 20 min after the addition of zinc ions to the peptide solution. Peptide concentrations, zinc/peptide molar ratio and incubation conditions are the same as in (B). Data represent the mean values±SEM of three measurements. Parenthesis and asterisk indicate statistically significant differences (p < 0.05; two-tailed t-Student test).
Removal of the N-terminal pentapeptide dramatically influenced the proteomic profile of mouse brain Aβ-binding proteins. Our previous studies revealed more than 80 individual proteins in the rat [22] or mouse brain [19] that specifically bound to an affinity sorbent with Aβ42 as the affinity ligand. In this study, the number of mouse brain proteins bound to Aβ42 (74; Supplementary Table 1) was consistent with the previously reported results. In contrast to the full length peptide we found only 12 individual proteins specifically bound to the affinity sorbent containing Aβ6-42 as the affinity ligand (Fig. 2). The same situation was also observed in the case of the affinity sorbent with isoD7-Aβ6-42 as a ligand: we found only 16 individual proteins specifically bound to this affinity sorbent (Fig. 2). In terms of GO the identified proteins, specifically bound to full-length Aβ, are involved in various cell processes, including regulation and metabolism (Table 1). Proteins bound to Aβ6-42 and isoD7-Aβ6-42 do not participate in metabolic processes at all and play some role in regulations. Since removal of the N-terminal pentapeptide dramatically decreases the number of Aβ-binding proteins it appears that possible interaction with intracellular targets does not play a significant role in the effect of the truncated peptides on the cells.
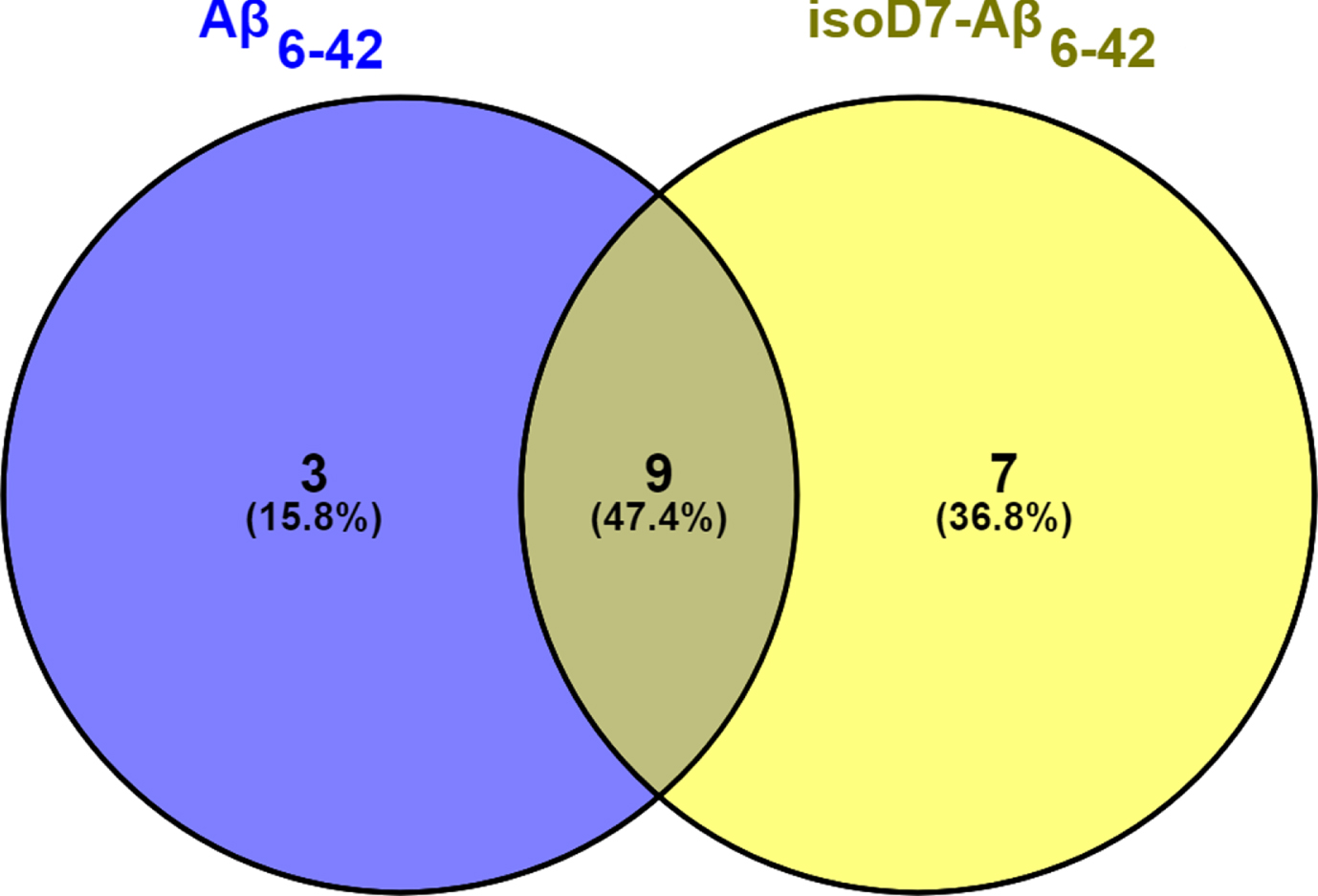
The Venne diagram illustrating total number of Aβ-binding proteins identified in the mouse brain using Aβ6-42 and isoD7-Aβ6-42 as the affinity ligands. The total number of all identified proteins (28) was defined as 100%. The proportion of identified proteins common for both sorbents was 47.4%. The proportion of proteins specific for Aβ6-42 and isoD7-Aβ6-42 was 15.8% and 36.8%, respectively.
Functional characteristics of brain proteins bound Aβ42, Aβ6-42, isoD7-Aβ6-42
*Since each identified protein may be involved in more than one biological process the number of GO annotations exceeds the total number of identified proteins.
Removal of the N-terminal pentapeptide slightly enhanced apoptogenic properties of resultant Aβ6-42 and isoD7-Aβ6-42 (Fig. 3A) and increased necrosis of SH-SY5Y cells as compared with the effects of the parent peptides (Fig. 3B). The truncated peptides demonstrated somewhat higher efficacy in inducing generation of ROS (Fig. 3C) and also in releasing the proinflammatory cytokine, TNFα, in comparison with the full-length peptides (Fig. 3D).

The effect of Aβ peptides (10 μM) on apoptosis (A), necrosis (B) of SH-SY5Y cells and their production of ROS (C) and TNFα (D). OD is optical density. Cell were cultured with Aβ peptides during 48 h (A, B) or 24 h (C, D). Data represent the mean values±SEM of at least four independent experiments performed in quadruplicates; *p < 0.05.
DISCUSSION
ACE is involved in proteolytic processing of peptide substrates related to AD [5, 7]. In the context of proteolytic degradation of Aβ, the key player in the development and progression of AD [1–5], ACE and its domains, particularly, its N-domain, can cleave various synthetic Aβ analogues [1–5, 23]. The use of shorter Aβ analogues as potential ACE substrates is often explained by the fact that “full-length Aβ (especially its human form) is difficult to handle” [23]. Since the ACE N-domain acted as an arginine specific endopeptidase cleaving the N-terminal pentapeptide from the Aβ analogue (corresponding to human Aβ1-16) it was assumed that in vivo the full length Aβ could be hydrolyzed by ACE in the same endopeptidase mode [12]. The latter suggests possible formation of Aβ6-42 species. Taking into consideration higher efficiency of ACE-based cleavage of the isoD7-Aβ analogue [11] and higher toxicity of isoD7-Aβ toward human neuronal cells [20] it was thus important to investigate the neurotoxic effects of Aβ6-42 and isoD7-Aβ6-42.
In this study, we have used three approaches for evaluation of metabolic consequences of N-terminal pentapeptide cleavage from Aβ: 1) oligomer formation; 2) interaction with intracellular targets; and 3) viability of cells treated with the truncated Aβ peptides and production of ROS and release of the proinflammatory cytokine, TNFα.
In the context of oligomer formation, the truncated Aβ peptides demonstrated either the same or even higher aggregability (Fig. 1) than their parent peptides, Aβ42 and isoD7-Aβ42. The latter suggests their potential involvement in formation of extracellular oligomers.
Results of mouse brain proteomic profiling performed in this study indicate that the repertoire of proteins bound to Aβ6-42 and isoD7-Aβ6-42 as affinity ligands is much poorer than in the case of full length peptides, Aβ42 (Supplementary Table 1) and isoD7-Aβ42 investigated earlier [19]. In this study we were able to identify only 12 individual proteins bound to Aβ6-42 and 16 proteins bound to isoD7-Aβ1-42 (Fig. 2). Since interaction with intracellular targets may also represent the primary pathogenic effect that precedes formation of extracellular pathogenic oligomers/aggregates of the Aβ peptide [19], proteolytic modification of this peptide decreases possibility of interaction of truncated Aβ with potential intracellular targets. The latter is also supported by the GO annotation: none of the truncated Aβ peptides binds proteins involved in metabolic processes (See Table 1 and Supplementary Table 1). Taking into consideration the fact that ACE active sites are exposed to the extracellular space [24], proteolytic products are obviously released outside the cell; this means that their interaction with putative intracellular targets requires additional steps, including translocation of truncated peptides inside the cell. In our viewpoint this decreases probability of realization of the pathogenic scenario, associated with interaction of Aβ peptides with intracellular targets because other competitive processes (e.g., oligomer formation) occur outside the cells.
Cytotoxicity studies revealed that removal of the N-terminal pentapeptide from Aβ42 and isoD7-Aβ42 did not abolish their effects on SH-SY5Y cells. Moreover, apoptosis and especially necrosis were even higher in the presence of isoD7-Aβ6-42 as compared with both Aβ42 and isoD7-Aβ42 (Fig. 3A, B). The increase of cell death via necrosis suggests a nonspecific mode of the Aβ peptide action, which can be attributed to formation of pores and channels in the plasma membrane induced by Aβ oligomers [25, 26]. It is known that ROS and TNFα play an important role in the molecular mechanisms underlying neuronal death in neurodegeneration [27]. In this context it is important to emphasize that their production by SH-SY5Y cells was even higher in the presence of truncated peptides in comparison with their full length analogs (Fig. 3C, D). The fact that truncation of peptides does not reduce their toxic effects (induction of apoptosis, generation of ROS, and release of TNFα) suggests that these effects can be attributed to formation of Aβ oligomers rather than to Aβ interaction with particular target proteins.
Thus, results of our study demonstrate that some of the potential products of proteolytic cleavage of Aβ catalyzed by ACE exhibit increased cytotoxic activity typical for the full length Aβ42 and isoD7-Aβ42 peptides. Earlier it was demonstrated that ACE-based removal of the N-terminal heptapeptide significantly attenuated aggregability and cytotoxicity of the resultant Aβ8-40 [2]. This obviously illustrates importance of the charged N-terminal H6D7 residues for both Zn2 +-induced aggregability and cytotoxicity. Although the repertoire of Aβ-binding proteins has not been investigated in this context (comparison of Aβ species with removal of N-terminal pentapeptide versus heptapeptide), significantly increased number of proteins bound to Aβ42 containing phosphorylated S8 seems to support this viewpoint as well [19].
The presence of the N-terminally truncated Aβ peptide(s) in vivo has been neither demonstrated nor investigated yet; molecular dynamics simulation together with analysis of catalytic conversion of synthetic Aβ peptide analogues suggest possibility of the removal of the N-terminal pentapeptide from Aβ42 by ACE [11]. In our viewpoint, this situation resembles the history of D-serine discovery in the brain: initially a synthetic compound, N-methyl-D-aspartate, was found to be a glutamate receptor agonist [28] and only later, D-serine was found in the brain [29], and identified as an endogenous ligand for the glycine site of the N-methyl-D-aspartate receptor [30]. Thus, there is a clear need of targeted search of Aβ6-42 peptides in vivo.
In conclusion, the results of our study support the viewpoint that some truncated Aβ peptides formed during ACE-based proteolytic processing of Aβ1-42 could be even more (neuro)toxic than the parent peptide [12]. This suggests that the use of ACE inhibitors would be beneficial for therapy of AD patients at least in the context of Aβ1 - 42 metabolism. However, it should be noted ACE plays a key role in the formation of regulatory peptides (first of all angiotensin II) of the renin-angiotensin system (RAS) and there is a growing interest in altered RAS functioning in AD [7]. Certain evidence exists that the overactive RAS recognized in AD could also induce negative (angiotensin II mediated) effects on the cholinergic system and “RAS-acting drugs may thus potentially supplement existing anti-cholinesterase treatment strategies in AD” [7]. This suggests a multifunctional role of ACE inhibitors in the therapy of AD and one of them consists in inhibition of Aβ metabolism resulting in the formation of more toxic truncated Aβ peptides.