
Abstract
HSPA6 (Hsp70B’) is an inducible member of the Hsp70 (HSPA) family of heat shock proteins that is present in the human genome and not found in mouse and rat. Hence it is lacking in current animal models of neurodegenerative diseases. To advance knowledge of the little studied HSPA6, differentiated human neuronal SH-SY5Y cells were treated with the proteotoxic stress-inducing agent MG132. A robust induction of HSPA6 was apparent which localized to the periphery of MG132-induced protein aggregates in the neuronal cytoplasm. Components of the protein disaggregation/refolding machine that co-operate with Hsp70 also targeted the periphery of cytoplasmic protein aggregates, including DNAJB1 (Hsp40–1), HSPH1 (Hsp105α), and HSPB1 (Hsp27). These data suggest that HSPA6 is involved in the response of human neuronal cells to proteotoxic stress that is a feature of neurodegenerative diseases which have been characterized as protein misfolding disorders. Constitutively expressed HSPA8 (Hsc70) also localized tothe periphery of cytoplasmic protein aggregates following the treatment of differentiated human neuronal cells with MG132. HSPA8 could provide a rapid response to proteotoxic stress in neuronal cells, circumventing the time required to upregulate inducible Hsps.
Keywords
INTRODUCTION
Despite numerous clinical trials of potential therapeutics for Alzheimer’s disease (AD) that appeared promising in animal models, there is currently a paucity of effective therapies [1–4]. This suggests that current animal models may not encompass all aspects of the complex human disease process. Neurodegenerative diseases, such as AD have been characterized as ‘protein misfolding disorders’ in which misfolded, aggregation-prone proteins accumulate, triggering neuronal dysfunction leading to premature cell death [5–9]. There is a critical need to improve our understanding of components of the protein quality control machinery and how they co-operate to maintain proteostasis in human neuronal cells in order to better design therapeutic compounds to treat neurodegenerative diseases. A key feature of these diseases is proteotoxic stress caused by the inability of the protein quality control machinery to manage misfolded proteins and the formation of toxic oligomers [7–12]. A strategy that has been proposed is to upregulate heat shock proteins (Hsps) that are protein ‘repair agents’ involved in maintaining protein homeostasis by refolding misfolded proteins [7, 12–14].
Hsp70 (HSPA) is a multigene family that is a key player in the machinery involved in managing misfolded cellular proteins. HSPA1A (Hsp70–1) has been widely studied; however, HSPA6 (Hsp70B’) has received comparatively little attention [15, 16]. Interestingly, HSPA6 is present in the human genome but not the genomes of mouse and rat [15, 17–19]. Hence current animal models of neurodegenerative diseases are missing a component of the protein quality control machinery. Upregulation of HSPA6 has recently been reported in the brains of patients with AD, Parkinson’s disease, and dementia with Lewy bodies [20–22]. HSPA6 expression in AD patients was increased 6.3-fold and 30.4-fold in patients with Parkinson’s disease [22]. A 9.4-fold increase in HSPA6 expression was observed in the brains of patients with Parkinson’s disease with dementia, compared with a 3.4-fold increase of HSPA1A [21]. To advance knowledge of HSPA6, differentiated human neuronal SH-SY5Y cells were treated with the proteotoxic stress-inducing agent MG132. A robust induction of HSPA6 was apparent which localized to the periphery of MG132-induced protein aggregates in the neuronal cytoplasm. In addition, we report that constitutively expressed HSPA8 may provide a rapid response to proteotoxic stress, circumventing the time required to upregulate inducible Hsps.
MATERIALS AND METHODS
Cell culture and differentiation
Human neuronal SH-SY5Y cells (American Type Culture Collection, Manassas, VA, USA) were cultured in Dulbecco’s modified Eagle’s medium (DMEM; Wisent, QC, Canada) supplemented with 10% fetal bovine serum (FBS; Wisent) at 37°C in a humidified 5% CO2 atmosphere. Cells were plated onto polystyrene tissue culture plates (for Western blotting and viability assays) or glass-bottom plates (for immunofluorescence) at 4.5×104 cells per cm2. Cells were left to adhere to the growth surface for 24 h before replacing medium with fresh serum free DMEM containing 10 μM all-trans-retinoic acid (R2625; Sigma Aldrich, St. Louis, MO, USA) to induce neuronal differentiation for 72 h [23].
Treatment with MG132, celastrol, and arimoclomol
Following differentiation, media was replaced with fresh serum free DMEM containing MG132 (at the indicated concentrations), celastrol alone (0.3 μM), or celastrol (0.3 μM) plus arimoclomol (50 μM) for 12 h. MG132 (BML-PI102; Enzo Life Sciences, Farmingdale, NY, USA) or celastrol (70950; Cayman Chemical, Ann Arbor, MI, USA) dissolved in DMSO was added directly to the media. Arimoclomol (gift from Professor Michael Cheetham, Institute of Ophthalmology, University College London, UK) was prepared fresh for each experiment by dissolving in serum free DMEM and filtering. DMSO was used as a vehicle control for MG132 and celastrol. We have previously reported that celastrol and arimoclomol are neuronal inducers of Hsps whereas classical heat shock is not [16, 24].
Western blotting
Cells were harvested, dissolved in Laemmli buffer and boiled for 10 min. Protein quantification was carried out using the RC DC Protein Assay Kit from Bio-Rad Laboratories (Hercules, CA, USA) according the manufacturer’s protocol. Thirty μg of protein was separated on a 12% SDS-PAGE gel using a Mini-PROTEAN 3 electrophoresis apparatus (Bio-Rad Laboratories, Hercules, CA, USA) with a 4% stacking gel. A Mini Trans-Blot® Module (Bio-Rad Laboratories) was employed to transfer proteins to nitrocellulose membranes. HSPA1A (SPA-810), HSPA6 (SPA-754), HSPA8 (SPA-815), DNAJB1 (SPA-400), and HSPB1 (SPA-803) primary antibodies were purchased from Enzo Life Sciences (Farmingdale, NY, USA). We have previously reported the specificity of the Hsp70 antibodies to HSPA6, HSPA1A, and HSPA8 [24]. HSPH1 (ab109624) primary antibody was obtained from Abcam (Cambridge, MA, USA) and β-tubulin antibody (MAB3408) from EMD Millipore (Billerica, MA, USA). Horseradish peroxidase-conjugated secondary antibodies (Sigma-Aldrich) were detected using enhanced chemiluminescence (Amersham, Piscataway, NJ, USA). Densitometry was performed using Quantity One® 1-D Analysis software (Bio-Rad Laboratories). Data was normalized to a β-tubulin control and represented the mean density relative to the DMSO control ± standard error of the mean (SEM) for three independent replicates.
Viability assay
An equal volume of cell suspension was mixed with 0.4% trypan blue (T10282; Thermofisher Scientific). After 2 min, the cell suspension was loaded into a disposable Countess® cell counting chamber slide and the percent of viable cells quantified using a Countess® automated cell counter (C10281; Invitrogen, Thermofisher Scientific). Percent viability after 12 h incubation with the indicated concentration of MG132 was normalized to 100% for the DMSO control and presented as the mean ± SEM for three independent replicates.
Immunofluorescence
For the microscopic investigation of the subcellular localization of Hsps, cells were fixed in 4% paraformaldehyde for 30 min subsequent to a 12 h treatment with 2.5 μM MG132. Cells were permeabilized in 0.1% triton X-100 in PBS with 100 mM glycine for 30 min and blocked in 5% FBS for 1 h. The same Hsp primary antibodies used for Western blotting were also employed for immunofluorescence. The α-tubulin primary antibody (ab18251) was purchased from Abcam (Cambridge, MA, USA). Cells were incubated with primary antibodies overnight at 4°C in 1% FBS followed by incubation with Alexafluor-conjugated secondary antibodies (Molecular Probes, Thermofisher Scientific, Burlington, ON, Canada) in 1% FBS for 2 h. Cells were then counterstained with 300 nM DAPI (Invitrogen, Thermofisher Scientific). For staining aggregated proteins, the Proteostat® Aggresome Detection Kit (Enzo Life Sciences) was used according to the manufacturer’s protocol.
Fluorescence images were captured using a Quorum Wave FX-X1 spinning disk confocal microscope (Quorum Technologies, Guelph, ON, Canada) equipped with a high resolution Humamatsu Orca R2 camera (Humamatsu Photonics, Japan). Excitation lasers: 405, 491, 561, 644 nm. Emission filters (nm/bandpass): 460/50, 525/50, and 593/40.
Volocity 3D image analysis software (Perkin Elmer, Waltham, MA, USA) was used for image analysis, processing, generation of 3D opacity renderings from z-stack images and 3D movies. Expression of specific Hsps was determined for 100 individual cells. ImageJ software (http://imagej.nih.gov/ij/) was employed to generate fluorescence intensity line scans using TIFF images exported from Volocity. Background subtracted images were used to generate intensity profile plots representing the fluorescence signal intensities for the indicated channels in a defined linear region using the RGB (red-green-blue) Profiler plugin. Fluorescence intensity line scans are representative of at least 3 cells that were scanned in 3–4 linear regions across structures of interest.
Heat shock
Following 12 h treatment with 2.5 μM MG132, cells were immersed in a circulating water bath calibrated at 43°C (±0.2°C) for 20 min and then either fixed for immunofluorescence (20 min) or returned to 37°C until fixation at a later time point (1 or 3 h). The 0 time-point equaled the start of heat shock.
Statistical analysis
Two-way ANOVA with Bonferroni’s post hoc analysis was used to test for statistical significance of Western blot densitometry and viability data using Graphpad Prism 5 software (La Jolla, CA, USA).
RESULTS
Proteotoxic effects of MG132 on differentiated human neuronal cells
Ubiquitin-tagged neuronal proteins accumulated in differentiated human SH-SY5Y neuronal cells as the MG132 concentration was elevated (Fig. 1A) and localized to proteostat-positive neuronal protein aggregates (Fig. 1B). Misfolded, aggregation-prone proteins are targeted for proteasomal degradation by ubiquitin, and these proteins have been reported to increase after treatment with MG132 that inhibits the proteasome [25, 26]. Ubiquitinated proteins were detected in neuronal cytoplasmic protein aggregates identified by the proteostat marker (encircled in the magnified images on the right in Fig. 1B) and confirmed by signal co-localization in the fluorescence intensity line scan of the encircled proteostat-positive protein aggregates (ubiquitin- green line, proteostat- red line). Proteostat has been employed as a marker to identify protein aggregates that form following MG132 in differentiated SH-SY5Y cells [25–27]. Proteostat is a molecular rotor dye that is non-fluorescent in solution and becomes highly fluorescent on binding to aggregated proteins where it intercalates into the strands that hold beta sheets together [27].
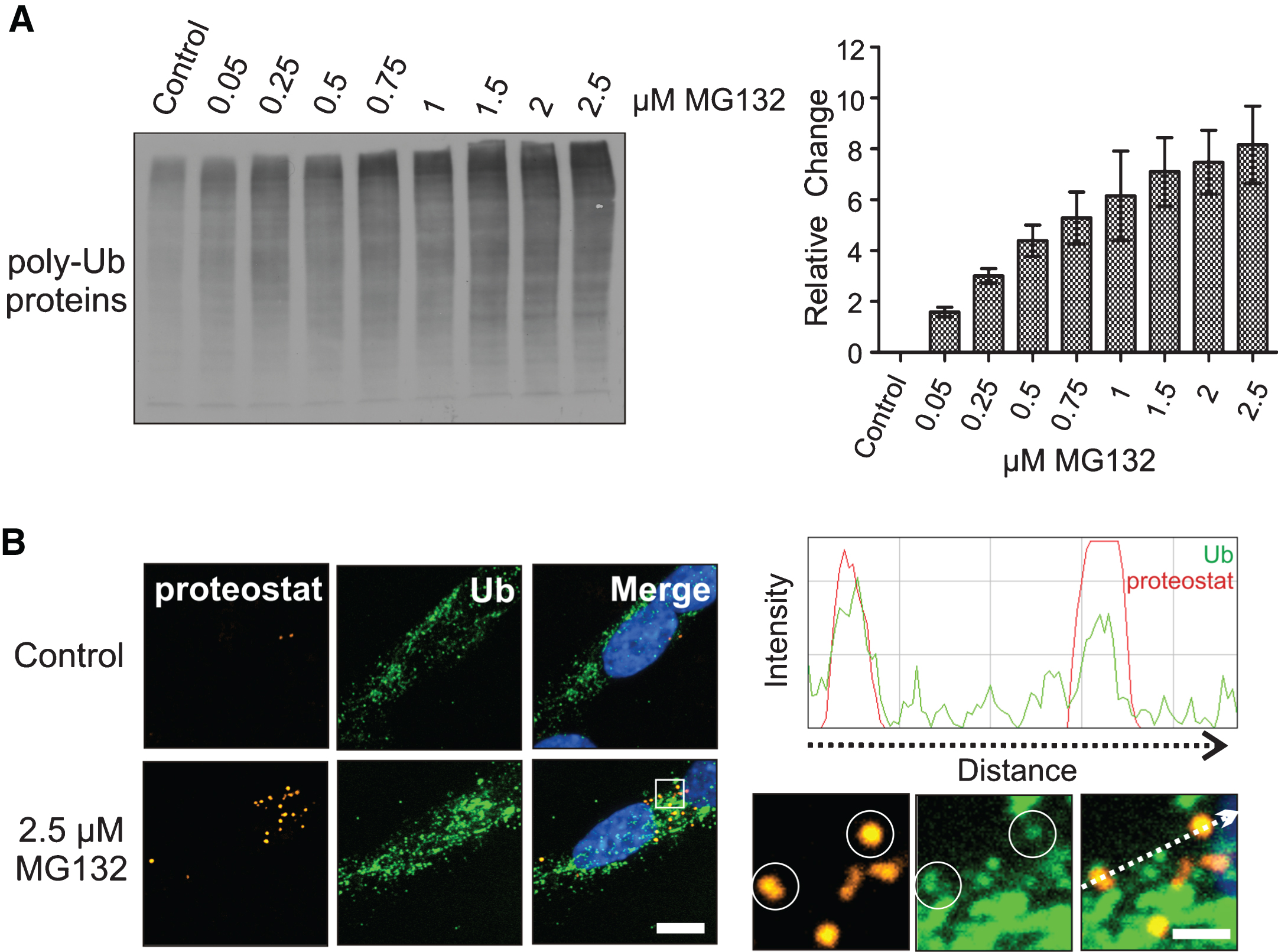
Elevation of neuronal poly-ubiquitinated proteins and protein aggregation following proteotoxic stress. A) Effect of MG132 dosages on poly-ubiquitinated (poly-Ub) neuronal proteins. B) Co-localization of ubiquitin-tagged neuronal proteins (green) with proteostat-positive neuronal proteins (orange, encircled), confirmed by a fluorescence intensity line scan demonstrating ubiquitinated protein peaks (green) overlap with proteostat peaks (red). DAPI (blue) was used to visualize nuclear DNA. The line scan was generated using the ImageJ RGB (red-green-blue) Profiler plugin from a TIFF image displaying the proteostat signal in red and the ubiquitin signal in green. Scale bar represents 5 μm. Scale bar for magnified images is 1 μm.
Induction of HSPA6 in differentiated human neuronal cells by MG132
As shown in Fig. 2, HSPA6 (Hsp70B’) was induced in differentiated human SH-SY5Y neuronal cells following treatment with the proteotoxic agent MG132. Robust induction of HSPA6 was apparent at 2.5 μM MG132 compared to HSPA6 levels that were induced by treatment with celastrol, or celastrol plus arimoclomol, which we have previously reported as neuronal inducers of HSPA6, whereas classic heat shock does not induce HSPA6 in differentiated human SH-SY5Y neuronal cells [16, 24]. Interestingly, MG132 also triggered the induction of other key components of the protein disaggregation/refolding machinery, namely: DNAJB1 (Hsp40–1), HSPH1 (Hsp105α). and HSPB1 (Hsp27). As shown in Fig. 2, MG132 induced the widely studied HSPA1A (Hsp70–1). Levels of constitutively expressed HSPA8 (Hsc70) were not significantly affected. Induction of HSPA6 in differentiated human neuronal cells required higher concentrations of MG132 compared to HSPA1A (Fig. 3A), suggesting that HSPA6 is induced by severe proteotoxic stress. Induction of HSPA6 was observed with the low dose of 0.5 μM MG132 at the longer incubation time of 24 h compared to 12 h of incubation. Viability was reduced after 24 h incubation with MG132, but was not significantly affected after 12 h over the range of 0.05 to 2.5 μM MG132 (Fig. 3B).
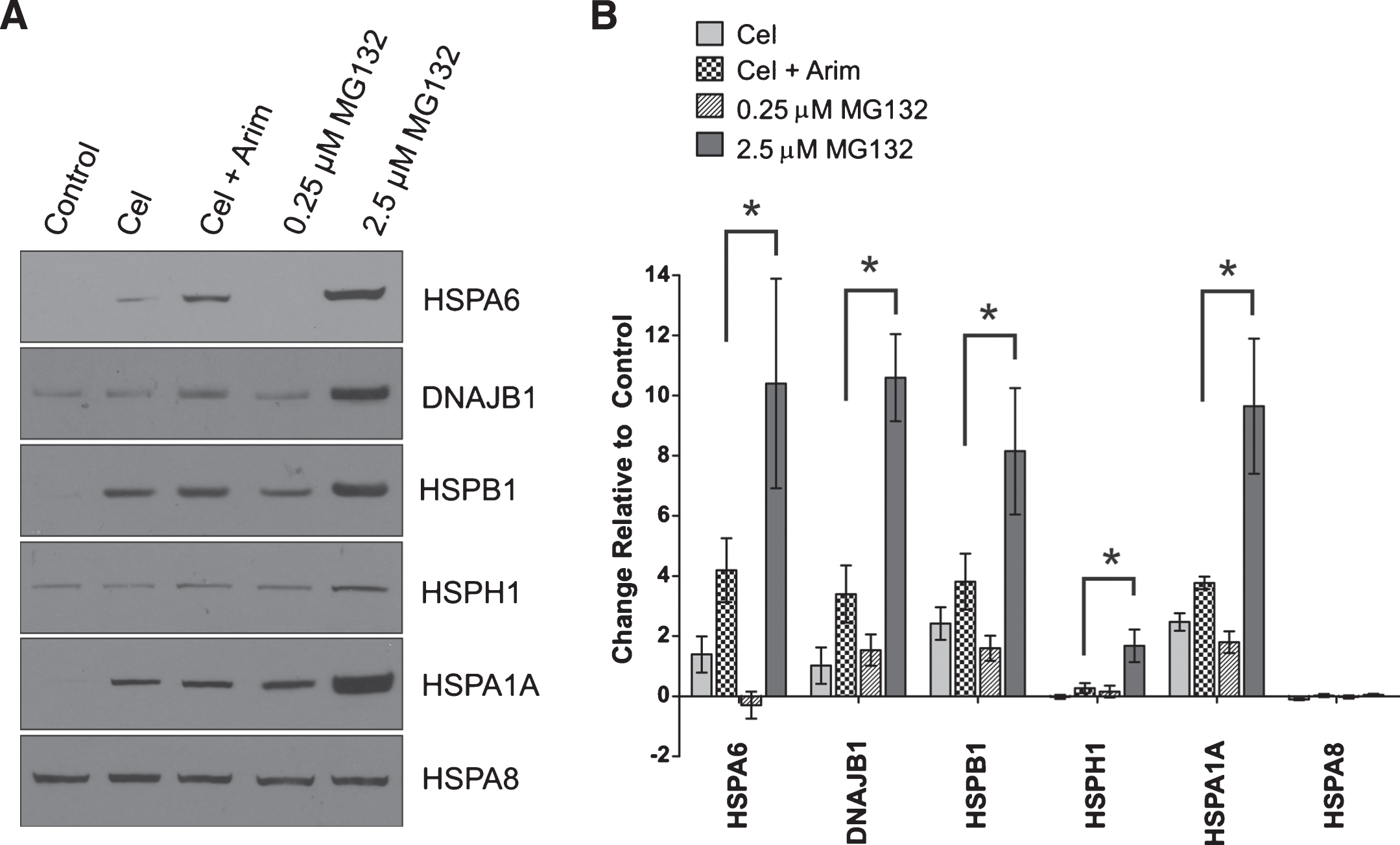
Induction of HSPA6 (Hsp70B’) and components of the protein disaggregation/refolding machine by proteotoxic stress. A) Differentiated human SH-SY5Y neuronal cells were treated with the proteotoxic stressor MG132, or celastrol, or celastrol plus arimoclomol, followed by Western blot analysis. β-tubulin was used as a loading control. B) Change in Hsp band intensity relative to the control lane (*p < 0.01; n = 3). Band densities were first normalized to the β-tubulin loading control and then expressed relative to the control lane. Induction of HSPA6, DNAJB1, HSPB1, HSPH1 and HSPA1A was apparent after 2.5 μM MG132. The level of constitutively expressed HSPA8 did not change significantly.
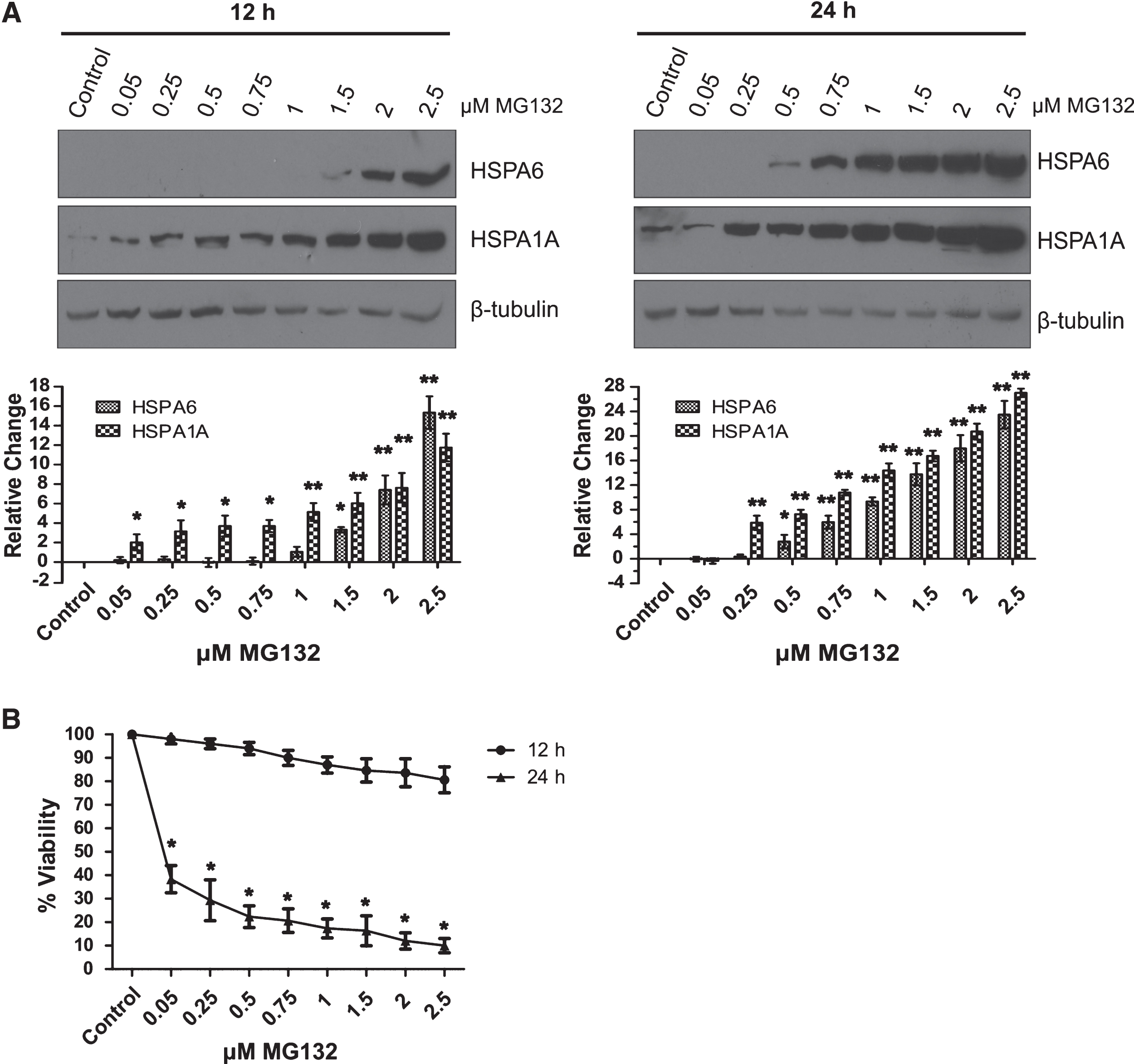
Effect of MG132 concentration on neuronal induction of HSPA6. A) HSPA6 and HSPA1A levels were examined by Western blotting following 12 h (left panel) and 24 h (right panel) incubation with MG132. Induction of HSPA6 required higher doses of MG132 compared to HSPA1A (*p < 0.01; **p < 0.05; n = 3). Band densities were normalized to the β-tubulin loading control and then expressed relative to the control lane. B) Effect of MG132 dosage on the viability of differentiated human neuronal cells after 12 h and 24 h (*p < 0.01).
Intracellular localization of MG132-induced HSPA6 in human neuronal cells
Immunocytochemistry revealed a cytoplasmic localization of MG132-induced HSPA6 at the cytoplasmic poles of differentiated human neuronal cells (indicated in Fig. 4A top panels by arrows pointing to green HSPA6 signal). Neuronal cellular processes were detected by α-tubulin (orange signal) and neuronal nuclear DNA by DAPI (blue signal). Recently we have reported that thermal stress targets HSPA6 to the neuronal nucleus following induction by celastrol plus arimoclomol [28]. We have also observed that YFP-tagged HSPA6 translocates to nuclear sites following heat shock [29–31]. However, MG132-induced HSPA6 did not translocate to the nucleus following heat shock (Fig. 4B, green signal in left panels), suggesting that it is committed to proteotoxic response mechanisms in the neuronal cytoplasm. HSPA1A exhibited similar results (Fig. 4A and B, bottom panels and right panels).
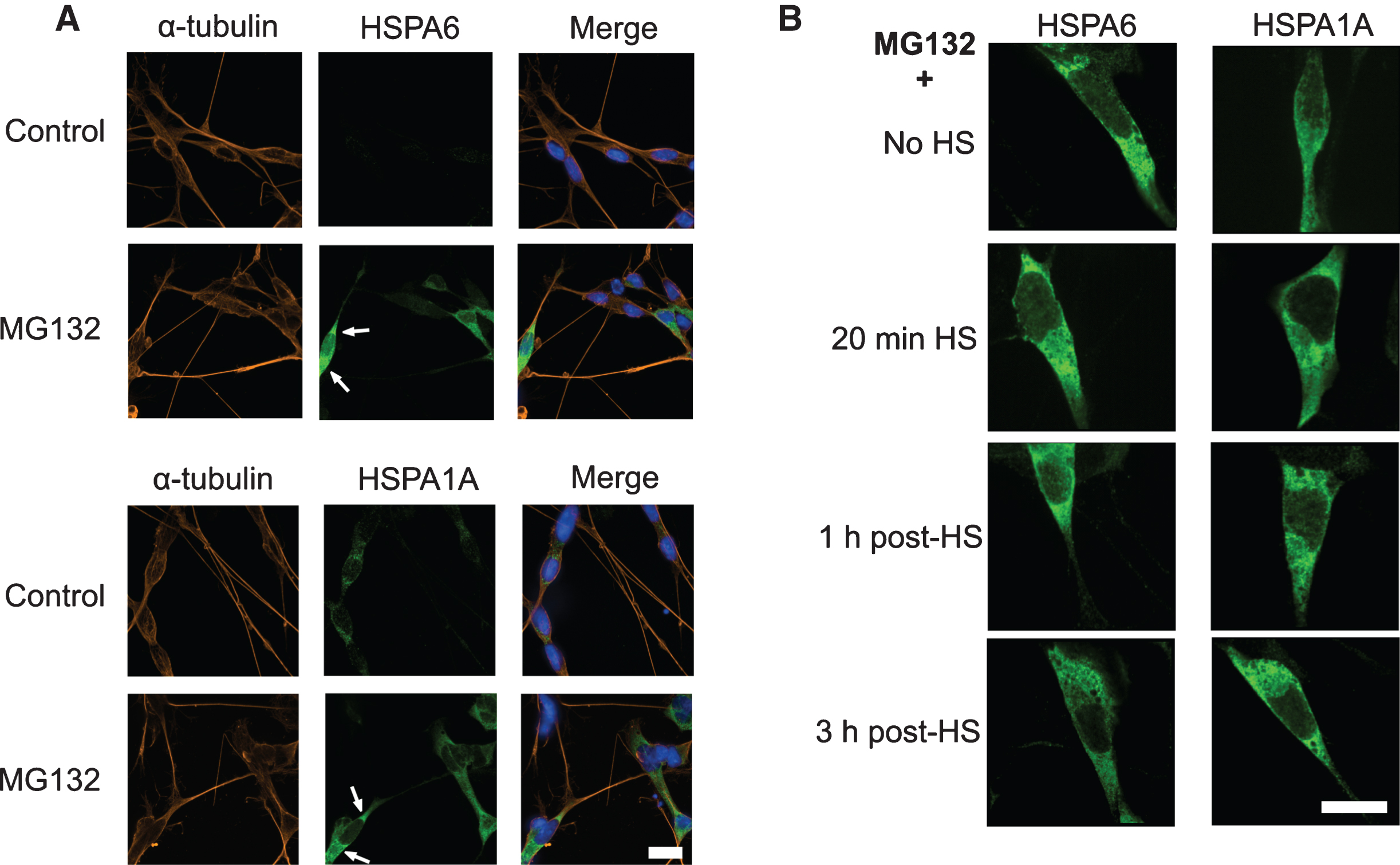
MG132-induced HSPA6 localized to the neuronal cytoplasm. A) Immunofluorescence localization of HSPA6 (green signal indicated by arrows in upper panel) and HSPA1A (green signal, lower panels) to the cytoplasm of differentiated human neuronal SH-SY5Y cells after treatment with 2.5 μM MG132. DAPI (blue) was used to visualize neuronal nuclear DNA; α-tubulin (orange) identifies neuronal processes. After MG132 treatment 66% of the cells were HSPA6 positive and 95% were HSPA1A positive. Scale bar represents 15 μm. B) Effects of heat shock on the localization of MG132-induced HSPA6 and HSPA1A. HS, heat shock. Scale bar represents 15 μm.
Targeting of HSPA6 to the periphery of protein aggregates in the neuronal cytoplasm
Higher resolution analysis (Fig. 5A, shown in boxed magnified insert) revealed that the cytoplasmic signal for MG132-induced HSPA6 (red) was localized around the periphery of proteostat-positive protein aggregates (orange). Fluorescence intensity line scans (indicated by dashed arrow in the magnified insert shown in Fig. 5A and Supplementary Figure 8A) confirmed that HSPA6 fluorescence intensity (red line) was concentrated at the periphery of proteostat-positive cytoplasmic aggregates (green line). As shown in the lower panels of Fig. 5A, the HSPA6 signal (red) surrounded proteostat-positive cytoplasmic protein aggregates (orange signal; indicated by top and bottom views of the cytoplasmic aggregate labelled with the white star). MG132-induced HSPA1A (red) surrounded proteostat-positive protein aggregates (orange signal at white star), shown in Fig. 5B and Supplementary Figure 8B.

Targeting of MG132-induced HSPA6 to the periphery of cytoplasmic protein aggregates in differentiated human neuronal cells. Cytoplasmic protein aggregates (orange signal) were detected with the proteostat marker. Fluorescence intensity line scans shown on the right demonstrated (A) HSPA6 and (B) HSPA1A fluorescence intensity peaks (red) at the periphery of proteostat peaks (green). Lower panels in (A) and (B): The HSPA6 and HSPA1A signals (red) surrounded cytoplasmic protein aggregates (orange; indicated by white star) presented as high magnification top and bottom views. Scale bar represents 5 μm. In high magnification images the scale bar represents 1 μm.
Components of the mammalian disaggregation/refolding machine also localize to the periphery of cytoplasmic protein aggregates after MG132 treatment
As shown in Fig. 6, the localization of other key components of the disaggregation/refolding machine in relation to proteostat-positive protein aggregates was investigated following treatment of the differentiated human neuronal cells with MG132. DNAJB1 (Hsp40–1; red signal in Fig. 6A and Supplementary Figure 8C) and the small Hsp, HSPB1 (Hsp27; red signal in Fig. 6B and Supplementary Figure 8D), localized at the periphery of the proteostat-positive cytoplasmic aggregates (orange signal in Fig. 6A and B and Supplementary Figure 8C and D), as was observed for HSPA6 (Fig. 5A). This was confirmed by fluorescence intensity line scans (proteostat- green line; DNAJB1 and HSPB1- red lines). Signal for HSPH1 (Hsp105α; red signal in Fig. 6C and Supplementary Figure 8E) was also observed at the periphery of the proteostat-positive cytoplasmic aggregates (orange), indicated by the arrows in the higher magnification panels, and in the fluorescence intensity line scan shown in the right panels in Fig. 6C (HSPH1- red line, proteostat- green line). In addition, the HSPH1 signal overlapped with the proteostat-positive protein aggregates (arrowheads), confirmed by fluorescence intensity line scan (right panels in Fig. 6C).

Localization of components of the protein disaggregation/refolding machine to the periphery of neuronal cytoplasmic protein aggregates (A) DNAJB1 (red) and (B) HSPB1 (red) were observed at the periphery of proteostat-positive cytoplasmic aggregates (orange), confirmed by fluorescence intensity line scans on the right. C) The disaggregase HSPH1 localized to both the periphery (arrows) and core (arrowheads) of proteostat-positive aggregates in the cytoplasm, confirmed by the fluorescence intensity line scan on the right. After MG132 treatment 91% of the cells were DNAJB1 positive, 98% were HSPB1 positive and 83% were HSPH1 positive. Scale bar represents 5 μm. For high magnification images the scale bar represents 1 μm.
Targeting of constitutively expressed HSPA8 (Hsc70) to the periphery of protein aggregates after MG132
HSPA8 (Hsc70) that is constitutively expressed in differentiated SH-SY5Y cells localized to the periphery of proteostat-positive cytoplasmic protein aggregates after MG132 treatment, confirmed by fluorescence intensity line scans (Fig. 7 and Supplementary Figure 9).
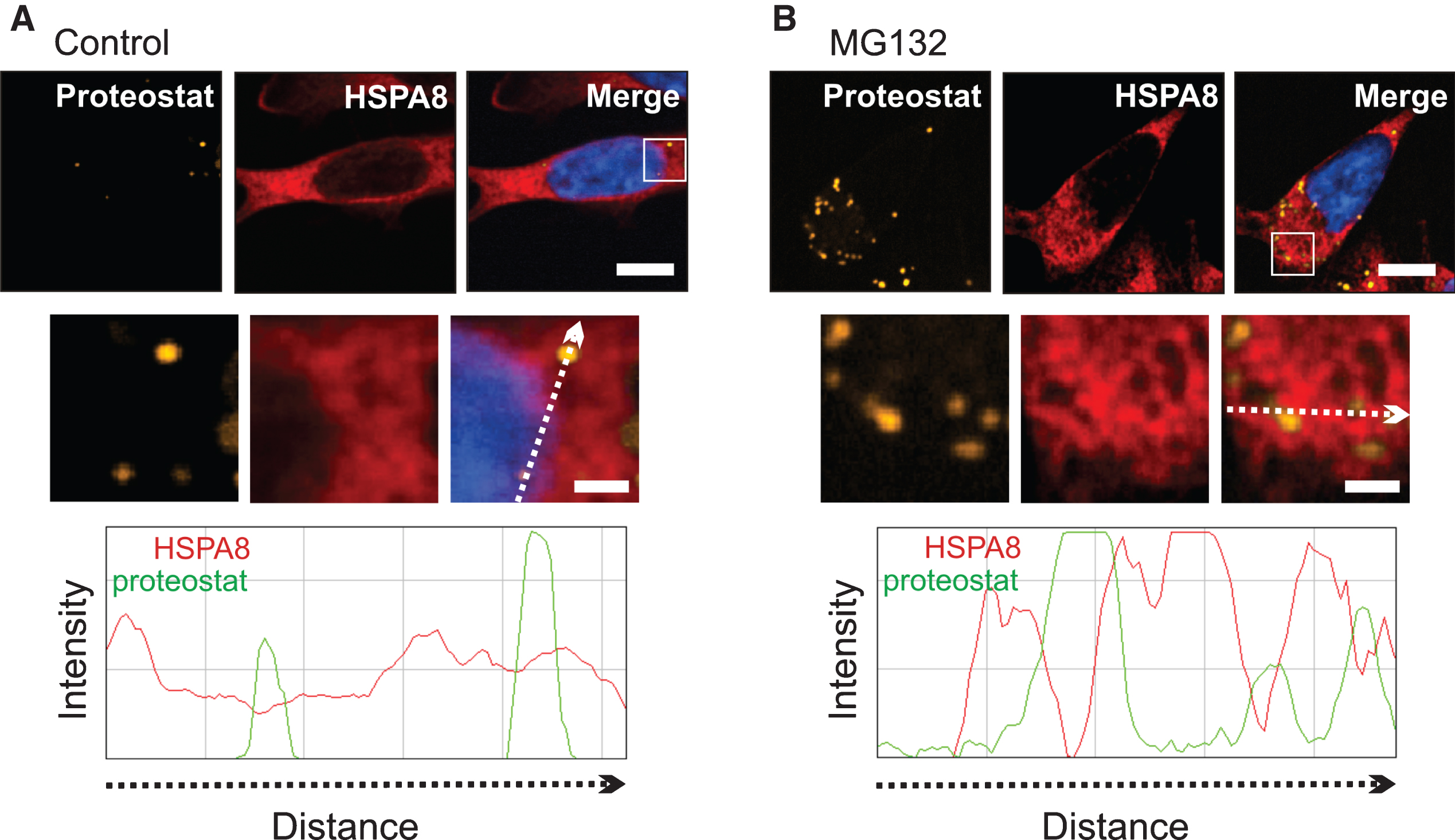
Constitutively expressed HSPA8 (Hsc70) targeted the periphery of cytoplasmic protein aggregates after MG132. A) Control. B) MG132 treatment. HSPA8 surrounded proteostat-positive cytoplasmic protein aggregates after MG132, confirmed by a fluorescence intensity line scan showing HSPA8 fluorescence peaks (red line) at the periphery of proteostat-positive protein aggregates peaks (green line). 100% of the cells were positive for constitutively expressed HSPA8 in both control and MG132-treated cells. Scale bar represents 5 μm. In high magnification images of the boxed area, the scale bar represents 1 μm.
HSPA8 translocates to nuclei after heat shock in differentiated human neuronal cells following proteotoxic stress by MG132
Unlike MG132-induced HSPA6 and HSPA1A (Fig. 4), constitutively expressed HSPA8 translocated after heat shock to nuclei in differentiated human neuronal cells (Fig. 8), in addition to localizing to the periphery of cytoplasmic protein aggregates (Fig. 7). Immediately after heat shock, HSPA8 (red), targeted nuclear speckles (arrowheads in magnified inserts of Fig. 8A), that were identified by co-localization with the nuclear speckle marker protein SON (orange) [32, 33]. The fluorescence intensity line scan on the right (scan line indicated by the dashed arrow in the magnified insert) confirmed that HSPA8 fluorescence peaks (red line) co-localize with SON fluorescence peaks (green line). Nuclear speckles are rich in RNA splicing factors [34–36]. As shown in Fig. 8B, later at 1 h post-heat shock, HSPA8 co-localized with nucleophosmin (NPM, orange) a marker of the granular component layer of the nucleolus (arrows) that is involved in processing of ribosomal RNA and ribosomal subunit assembly [37–39]. Overlap of HSPA8 fluorescence (red line) and NPM fluorescence was confirmed by the fluorescence intensity line scan on the right. Later during recovery at the 3 h time-point, HSPA8 no longer targeted nuclear structures (Fig. 8C), confirmed by the line scan on the right where the HSPA8 fluorescence (red line) no longer coincided with SON fluorescence peaks (green line) at nuclear speckles (arrowheads). SON and NPM were employed as nuclear markers as we have previously reported that heat shock targets HSPA8 first to nuclear speckles and subsequently to the granular layer of the nucleolus [28]. At 1 h post-heat shock, when HSPA8 targeted the nucleolus (Fig. 8D, arrow), a strong HSPA8 signal was also apparent in the cytoplasm that surrounded proteostat-positive aggregates (orange, confirmed by the line scan panel on the right).

HSPA8 translocated to nuclear structures after thermal stress in MG132-treated human neuronal cells. Following MG132 for 12 h, cells were subjected to heat shock for 20 min followed by recovery for 1 or 3 h. A) At 20 min, HSPA8 (red) co-localized with SON (orange) at nuclear speckles (arrowheads), confirmed by the fluorescence intensity line scan on the right. B) At 1 h HSPA8 (red) co-localized with the nucleolar marker nucleophosmin (orange; arrow). C) Three hours post-HS HSPA8 (red) was no longer apparent at nuclear structures. The line scan on the right confirmed that HSPA8 (red line) fluorescence did not overlap with SON fluorescence (green line). D) During recovery, when HSPA8 targeted the nucleolus (arrow), HSPA8 signal was also apparent at the periphery of protein aggregates (orange), confirmed by the line scan on the right. SON, nuclear speckle marker; NPM, nucleophosmin, nucleolar marker; HS, heat shock. Scale bars represent 5 μm.
Video representation of the localization of heat shock proteins at the periphery of MG132-induced neuronal cytoplasmic protein aggregates
The localization of stress-induced HSPA6 and HSPA1A, and also constitutively expressed HSPA8, at the periphery of MG132-induced cytoplasmic protein aggregates was visualized in 3D videos presented in Supplementary Figures 1–4. The video in Supplementary Figure 1 tracks through an image stack to show HSPA6 (red signal) surrounding cytoplasmic protein aggregates (orange signal, white star). Supplementary Figure 2 is a rotational view of HSPA6 around the same protein aggregate. Rotational views of HSPA1A and constitutively expressed HSPA8 surrounding MG132-induced cytoplasmic protein aggregates are presented in Supplementary Figures 3 and 4.
DNAJB1 (red) and HSPB1 (red) that act co-operatively with HSPA (Hsp70) proteins in the protein disaggregation/refolding machine can be seen surrounding neuronal cytoplasmic protein aggregates (orange, white star) in Supplementary Figures 5 and 6, respectively. Supplementary Figure 7 demonstrates that the disaggregase HSPH1 (red) is localized at both the periphery and the core of neuronal cytoplasmic protein aggregates after MG132 treatment. The yellow signal observed in protein aggregates represents the overlap of the HSPH1 (red) and proteostat (green) signals.
DISCUSSION
Potential therapies to counter AD that appeared promising in animal models have repeatedly been found to be ineffective in human clinical trials [1–4]. This has led to the suggestion that current mouse models of the disease may not reflect the full complexity of the human neurological disease [40, 41]. A key feature of neurodegenerative diseases, including AD, is proteotoxic stress caused by the inability of the cellular protein quality control machinery to manage misfolded proteins and the formation of toxic oligomers [7–12]. There is a critical need to improve our understanding of protein quality control in human neurons to better design effective therapeutic compounds to treat neurodegenerative diseases which have been characterized as protein misfolding disorders.
HSPA6 is an inducible member of the Hsp70 (HSPA) family that is present in the human genome and not found in mouse and rat [15, 17–19]. The present study seeks to advance knowledge of HSPA6 by investigating its expression and localization in differentiated human neuronal cells following exposure to MG132, a proteotoxic stress-inducing agent [25, 27]. The results suggest that severe proteotoxic stress triggers HSPA6 induction in differentiated human neuronal cells, whereas the widely studied HSPA1A is induced by lower dosages of MG132. Proteotoxicity from persistent protein misfolding and aggregation is relevant to the chronic nature of AD. HSPA6 induction was observed following incubation with lower dosages of MG132 over a longer time frame, suggesting that chronic proteotoxicity may be a trigger for HSPA6 expression. Current animal models of neurodegenerative diseases lack the HSPA6 gene which is present in the human genome, and hence may be hampered in their response to severe proteotoxic stress [15, 17–19]. It has been reported that severe stress induces HSPA6 protein expression in human colon cancer cells and that HSPA6 induction is transient [15, 18]. Knockdown of HSPA6 reduced the viability of colon cancer cells in response to MG132 [15]. Recently it has been reported that HSPA6 siRNA knockdown decreased the viability of differentiated human SH-SY5Y neuronal cells following thermal stress and that other Hsps were not upregulated in compensation for the loss of HSPA6 [16]. Interestingly, some human colon cancer cell lines induce HSPA6 in response to MG132 while others do not [15]. HSPA6 expression was observed in 66% of differentiated human neuronal cells after MG132. This could reflect heterogeneity in the SH-SY5Y cell line that has been reported [42, 43].
HSPA6 and key components of the mammalian protein disaggregation/refolding machinery, namely DNAJB1 (Hsp40–1), HSPB1 (Hsp27), and HSPH1 (Hsp105α), were observed to surround protein aggregates that form in the neuronal cytoplasm after severe proteotoxic stress induced by MG132. DNAJ (Hsp40) proteins act as ‘holdases’ that detect and bind to misfolded proteins and present them to members of the Hsp70 family that serve as ‘foldases’ to refold proteins to a biologically active state [44–46]. HSPH1 co-operates with the Hsp70/40 machine and acts as a ‘disaggregase’ to dissociate protein aggregates [46, 47]. HSPB1 is a member of the small heat shock protein family that enhances the activity of the protein disaggregation/refolding machinery [48–50]. The machine has been shown to be capable of dissociating fibrils of the neurodegenerative disease-associated protein α-synuclein under in vitro conditions [47]. Our study demonstrates the localization of HSPA6 and components of the protein disaggregation/refolding machine to the periphery of neuronal cytoplasmic protein aggregates following proteotoxic stress. Interestingly, the disaggregase HSPH1, but not other Hsps, localized not only to the periphery but also the core of protein aggregates where it may facilitate the disentanglement of aggregated proteins prior to the action of the disaggregation/refolding machine at the periphery of the protein aggregate. Line scan and 3D analyses support a peripheral localization for the disaggregation/refolding machine components. However, it cannot be excluded that the HSPA6, DNAJB1, and HSPB1 antibodies may not reach epitopes buried within the aggregate.
We have previously demonstrated that heat shock induces nuclear translocation of HSPA6 in differentiated human SH-SY5Y neuronal cells that are treated with celastrol plus arimoclomol to induce HSPA6 [28]. The present study indicates that MG132-induced HSPA6 is targeted to the periphery of proteostat-positive cytoplasmic protein aggregates. Hence different stressors target HSPA6 to different intracellular sites as different stressors likely affect different cellular mechanisms. The lack of redistribution of HSPA6 to the nucleus following thermal stress suggests that MG132-induced HSPA6 is committed to proteotoxic response mechanisms in the neuronal cytoplasm associated with cellular reactions to protein aggregation. This could hamper the ability of neuronal cells to respond effectively to additional stressors.
The current results also increase knowledge of constitutively expressed HSPA8 (Hsc70) which has received comparatively little attention compared to stress-inducible HSPA1A (Hsp70–1). Neurons in the adult human brain exhibit high levels of HSPA8 relative to non-neuronal tissues which we have suggested could provide neuronal pre-protection without the time lag needed to upregulate inducible Hsps following proteotoxic stress [51, 52]. We have previously reported that variable levels of HSPA8 in different neuronal populations correlate with the frequency of neurodegenerative diseases in the human population [51]. For example, cortical neurons, associated with AD and high disease frequency, exhibit low levels of cortical HSPA8, and hence reduced buffering capacity against protein misfolding and aggregation. Whereas motor neurons, associated with amyotrophic lateral sclerosis and low disease frequency, demonstrate high levels of constitutively expressed HSPA8, and hence increased buffering capacity against protein misfolding. The present report demonstrates that constitutively expressed HSPA8 localizes to the periphery of cytoplasmic protein aggregates following treatment of differentiated human neuronal cells with the proteotoxic agent MG132. This suggests that HSPA8 could provide a rapid response to proteotoxic stress in neuronal cells, circumventing the time lag required to upregulate inducible Hsps. Interestingly, it has recently been reported that constitutively expressed HSPA8 acts more efficiently than stress-inducible HSPA1A as a key component of the mammalian protein disaggregation/refolding machine [47, 53].
We have previously identified nuclear speckles and the granular component layer of the nucleolus as heat-sensitive sites in differentiated human SH-SY5Y neuronal cells that are targeted by HSPA1A and HSPA8 proteins during recovery [28, 54]. Unlike MG132-induced HSPA6 and HSPA1A that are committed to cytoplasmic stress recovery mechanisms during proteotoxic stress in neuronal cells, HSPA8 is also available to translocate to the neuronal nucleus during subsequent exposure to heat stress. Following stress-induced upregulation of Hsps, levels of constitutively expressed HSPA8 in human cells exceed the level of stress-inducible HSPA (Hsp70) proteins [55, 56]. Thus limited levels of inducible Hsps may be titrated away by an initial stress while the relative abundance of HSPA8 provides a buffer that can be mobilized in response to an additional stress. This is supported by the observation that in differentiated human neuronal cells under proteotoxic stress, HSPA8 is retained at the periphery of cytoplasmic aggregates upon subsequent exposure to heat shock; however, HSPA8 was also available to translocate to heat-sensitive nuclear structures, unlike the inducible isoforms. This observation underscores the importance of high levels of HSPA8 for neuronal protection from multiple stressors. In aging neurons that experience chronic proteotoxicity as a result of neurodegenerative disease-associated protein misfolding and aggregation, high levels of HSPA8 may be important to protect from added stressors that would otherwise exacerbate protein aggregation and accelerate disease progression.
During aging there is a decline in the efficacy of protein quality control in neuronal cells which is thought to contribute to increasing levels of proteotoxicity that could lead to the development of neurodegenerative diseases [7, 12]. This occurs when protein misfolding and aggregation overwhelm protein quality control mechanisms [7–9, 12]. The present data advance knowledge of HSPA6 that is present in the human genome and not found in mouse and rat and hence is lacking in current animal models of neurodegenerative diseases. HSPA6 has recently been reported to be upregulated in the brains of patients with AD, Parkinson’s disease, and dementia with Lewy bodies [20–22] which likely reflects a defense attempt to maintain protein homeostasis. Therapeutic enhancement of HSPA6 expression could potentiate buffering capacity against protein misfolding and aggregation.