
Abstract
Alzheimer’s disease (AD) is characterized by memory and cognitive deficits that in part are related to a diminished ability to activity-dependent synaptic plasticity. In AD, an attenuated long-term potentiation has been correlated with a deficit of synaptic plasticity-relevant proteins and protein turnover. The ubiquitin-proteasome system (UPS) critically regulates the protein turnover and contributes to dynamic changes of the protein milieu within synapses. In AD, UPS aberration has been implicated in inadequate proteostasis and synaptic malfunction. However, here we show that the inhibition of proteasome-mediated protein degradation by MG132 or lactacystin restored an impaired activity-dependent synaptic plasticity in an AD-like mouse model. In this whole-cell voltage-clamp study, we provided evidence that an amelioration of long-term plasticity by modulating UPS activity in pyramidal neurons.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) is proposed as the new epidemic of the 21st century [1]. It needs an urgent and effective therapeutic intervention. Emerging evidence suggests that AD begins with the subtle failure of hippocampal synaptic efficacy before neuronal degeneration [2]. In the hippocampus, the ubiquitin-proteasome system (UPS) mediated protein quality control system regulates the turnover of key regulatory proteins including those critical for synaptic plasticity that are dysregulated in AD [3]. Earlier we and others reported a triadic relationship between UPS, long-term plasticity, and amyloid-β (Aβ), and suggested that protein degradation through the UPS is a critical regulator of neuroplasticity [4]. The mounting evidence has demonstrated that at the early phase of AD, UPS malfunction, consequent lack of plasticity-related proteins (PRPs) and signaling pathways have contributed to synaptic failure [5, 6]. Furthermore, with evidence from biochemical, physiological, and behavioral experiments, we have established that levels of early induction phase PRPs can be restored by the inhibition of protein degradation using UPS inhibitors [4].
In this study, we have investigated how UPS regulation affects plasticity at CA1 pyramidal neurons that are particularly vulnerable and dysregulated in AD. Due to the cellular specificity of neurodegeneration in AD, the examination of long-term potentiation (LTP) at specific CA1 pyramidal neuron subpopulations by whole-cell voltage-clamp electrophysiology has become an exceptional tool for identifying synaptic mechanisms underlying LTP. Considering the disparate and varying roles of UPS in different neuronal subtypes, compartments, physiological and disease conditions, the examination of single-neuron physiology is crucial. Therefore, this functional analysis makes it a useful physiological readout for synaptic changes caused by Aβ oligomers and for the study of therapeutic interventions [3]. Recently, the proteasome inhibitors have been reported to alleviate various neurodegenerative disorders (ND) and brain-related disorders such as AD, Parkinson’s disease, stroke, ischemic brain injury, and central nervous system malignancies through their neuroprotective, anti-inflammatory, and pro-survival effects [6–9]. Thus, a paradigm shift in the ubiquitin-proteasome research has already repositioned the ubiquitin-proteasome from being a prime suspect in the pathology of ND to an attractive therapeutic target [10].
RESULTS
In this study, we have evaluated LTP in pyramidal neurons from the CA1 Schaffer-collateral pathway of the hippocampus (see the Supplementary Material for Methods). First, in the control study, we observed that the induction of synaptic potentiation by the pairing of Schaffer-collateral activation and postsynaptic depolarization 5 min after cell-break-in evoked a robust excitatory postsynaptic current (EPSC) potentiation (LTP) that lasted for at least 50 min in the wild-type mice (Fig. 1B, n = 8). However, the same induction protocol was insufficient to evoke a 50 min potentiation of EPSCs in CA1 neurons of APP/PS1 mice that declined within 20 min back to baseline (Fig. 1C, n = 7). These data are in line with previous field electrophysiology studies, suggesting that dysfunction in the synaptic signaling pathways contributed to AD [4, 11]. The oligomeric Aβ induces widespread plasticity proteome impairment in neurons [3]. In AD, the plasticity protein turnover and proteostasis mediated by the ubiquitin-proteasome system has also been implicated in memory deficit as a result of Aβ toxicity [12, 13]. Intriguingly, the application of the 5μM of proteasome inhibitor MG132 or lactacystin (See the Supplementary materials for pharmacology) ranged from 15 min before to 30 min after high-frequency stimulation restored the synaptic plasticity within the 50 min time window of recording (Fig. 1D, E) without affecting basal EPSC (Fig. 1F, G). This result supports other findings in various other ND that inhibition of the ubiquitin-proteasome system offers neuroprotective, anti-inflammatory, and pro-survival effects, in particular restoration of plasticity players and related mechanisms in AD. However, the co-application of protein synthesis inhibitor, emetine (20μM) for 25 min (Fig. 1H, I) together with MG132 or lactacystin proved that restoration of plasticity by proteasome inhibitors is based on the modulation of the protein turnover balance (protein synthesis and protein degradation) in favor of upregulation of plasticity proteins. On the contrary, the application of MG132 or lactacystin in wild-type slices attenuated late-LTP (Fig. 1J-K). This suggests that the regulation of the UPS with proteasome inhibitor altered the protein turnover balance in favor of synaptic potentiation in APP/PS1 mice. For complete statistical comparison, refer to Supplementary Table 1.
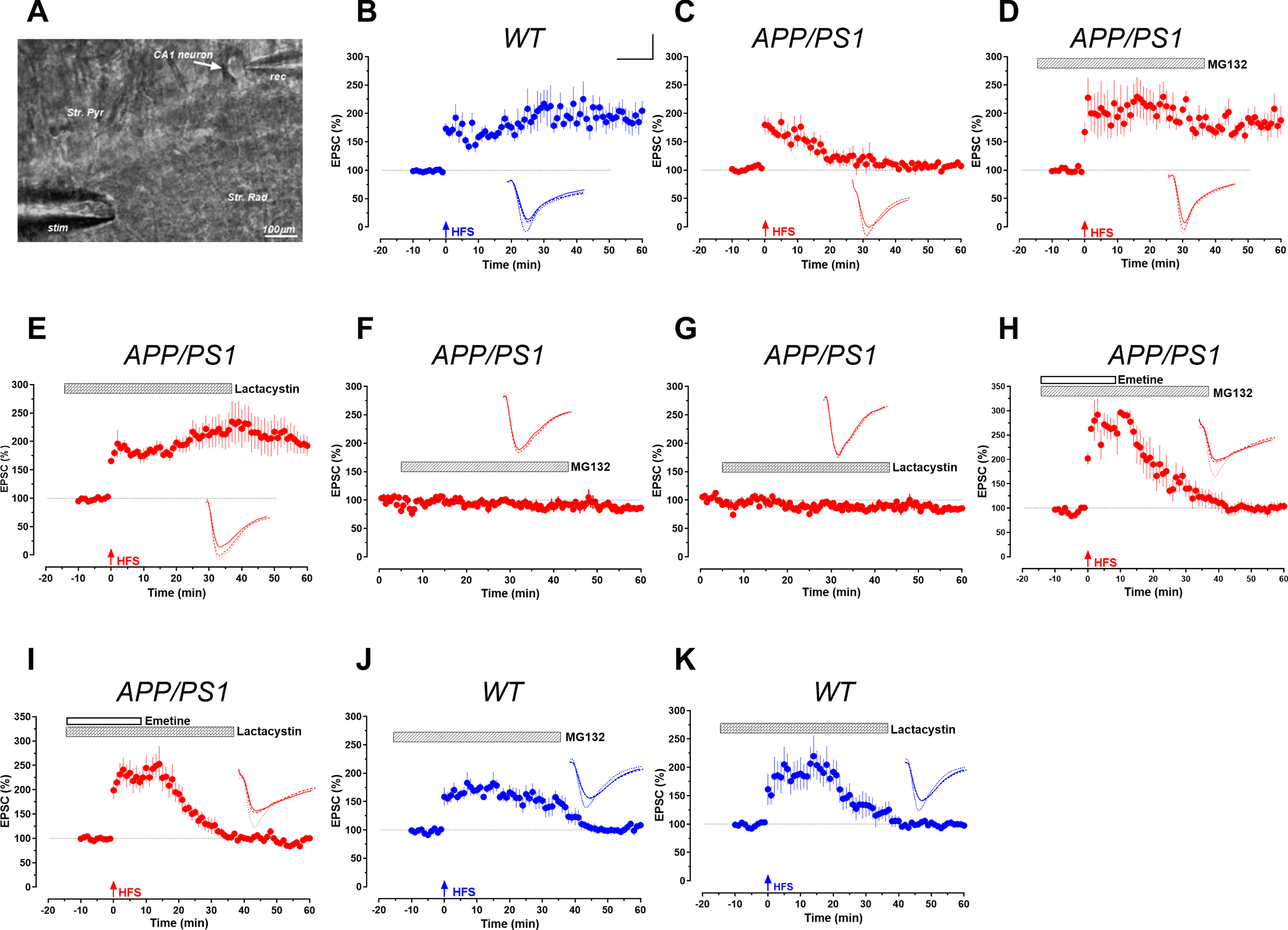
Proteasomal regulation of LTP by whole-cell recording. A) Hippocampal slice and the relative positions of the recording (rec) and stimulating (stim) electrodes in the stratum pyramidale (str. pyr.) and stratum radiatum (str. rad.), respectively. B) LTP induction by HFS in single pyramidal CA1 neuron from wild-type mice showed significant potentiation (n = 6). C) LTP is attenuated in APP/PS1 mice (n = 8). D, E) Attenuated LTP was restored by treatment with proteasome inhibitors MG132 (n = 8) and Lactacystin (n = 8). F.G) Basal EPSC activity remains stable for 60 min when treated with MG132 (n = 5) and Lactacystin (n = 5) respectively. H, I) Restoration of LTP is protein synthesis-dependent (n = 7). J, K) LTP was affected by MG132 and Lactacystin treatment in wild-type animals (n = 6). Inset traces represent typical EPSC recorded before and after HFS for each condition at time points -5, 20, and 50 min. Scale bars for analogs are 10 ms/50 pA.
DISCUSSION
AD and synaptopathy
In our study, the long-term plasticity was impaired already in 3-month-old AD-like mice. We have observed that long-term synaptic plasticity is attenuated in individual pyramidal neurons of AD-like mice (Fig. 1). This is in compliance with multiple lines of evidence suggesting the pre-plaque phase of memory deficits in AD preceding plaques and neurofibrillary tangles [4, 14] and as a result AD mediated memory decline has been linked to synaptopathy [4, 14]. Experimental evidence from human patients and animals present early onset of AD as early as 30 years of age [15, 16]. In the early phase of AD, there is a 25–35% decrease in synapses in rodents and a 45% decrease in boutons and spines in patients, which precedes the damage of the neurons [17, 18]. In vivo and in vitro studies in AD models established that the soluble Aβ blocks L-LTP [4, 19]. Both strengthening of functional neuronal connections (functional plasticity) and structural rearrangement of neurons (structural plasticity) that underlies long-term memory are impaired by amyloid toxicity in AD. AD downregulated important plasticity players like GluR1, PSD 95, CaMKII, and PKMzeta. We have demonstrated that the synaptic dysfunction in AD is particularly mediated by a deficit in the new plasticity-related protiens during the early induction phase of L-LTP [4]. Furthermore, we have reported that the impaired mTOR pathway is the critical culprit behind the deficit in plasticity proteins and synaptic dysfunction in AD conditions [4, 20].
Ubiquitin proteasome regulation has disparate roles
On the other hand in line with previous studies, our whole-cell voltage-clamp study also confirms that proteasomal inhibition in wild-type control animals can attenuate LTP in the Schaffer-collateral pathway (Fig. 1). This is attributed to an accumulation of negative regulators of synaptic plasticity such as transcription repressors, particularly during the long-term memory maintenance phase [21–23]. In control animals, the inhibition of the UPS blocked the transcription of BDNF and CREB through repressors which led to the cessation of plasticity protein synthesis for LTP maintenance. However, contrary to AD and proteasome inhibited control conditions, UPS inhibition increased the accumulation of early induction proteins in an activity-dependent manner during the early phase of LTP [4, 20]. UPS inhibition prevented the degradation of early initiation proteins of the long-term plasticity in AD. Thus, we propose that UPS performs disparate proteostatic functions not only in different parts of the neuron but asymmetrically in physiological and pathological conditions [3, 21]. With this conceptual foundation, we hypothesized that the amelioration of synaptic dysfunction caused by lack of plasticity proteins by blocking the degradation.
Proteasomal regulation facilitates L-LTP by balancing plasticity proteins availability
We discovered the re-establishment of long-term synaptic plasticity in the pyramidal neurons of AD models by UPS inhibition. Our study has found that proteasomal inhibition using MG132 and lactacystin could effectively alleviate the attenuation of LTP (Fig. 1). MG132 and lactacystin are two 26S proteasome-specific protease inhibitors, MG132 is a peptide aldehyde that blocks the proteolytic activity of the 26S proteasome complex by binding reversibly to the N-terminal Thr residue of the β1 subunit. Lactacystin is a specific, potent, irreversible and non-peptidic proteasome inhibitor. It binds covalently and inhibits specific catalytic subunits of the proteasome [20]. The UPS inhibition is known to have an antagonistic effect on the pathophysiology of AD at the functional and structural level of plasticity (i.e., upregulation of critical molecular players of synaptic plasticity), thus leading to amelioration of both structural and functional plasticity. A growing body of literature indicates that the following set of downstream synaptic proteins have contrasting occurrence in AD and during UPS inhibition (upregulated during proteasome inhibition and downregulated in AD): GluR1, aurora kinase, CaMKII-α, CPEB dependent synaptic proteins, PSD, Shank, GKAP, and AKAP79/150, Homer 1a, and DUNC-13 [4, 24–29]. In other words, most synaptic and plasticity players that are downregulated in AD are restored by blocking the degradation of those critical players of plasticity by proteasome inhibition. Similar experience and identical trend with plasticity relevant proteins such as BDNF [11, 31], CREB [32], CAMKII, PKMζ [4], and mTOR signaling [4, 20] has been observed by others and us. In particular, the mTOR pathway-mediated translation of ‘cognitive kinases’ (such as classical PKC, PKA, and CaMKIIα) that plays a key role in the consolidation of L-LTP, has been rescued by proteasome inhibition [4]. PKMζ upregulates synaptic potentiation and LTP maintenance by decreasing postsynaptic AMPAR endocytosis and doubling the number of functional AMPAR channels. Recently we have reported upregulation of p70S6K and PKMζ, the mTOR downstream plasticity-related proteins during UPS inhibition [4]. This way, UPS regulation guides the balance of newly synthesized plasticity proteins and counters the effect Aβ which significantly reduces plasticity relevant protein synthesis during the early phase [20]. Our study highlights that UPS inhibition balances between degradation and synthesis towards a net increase in protein synthesis and positive effector synaptic proteins by blocking the degradation of positive effector proteins.
The translational and therapeutic value of UPS regulation
Taking into consideration that proteasome inhibitors Bortezomib (Velcade®), Carfilzomib, and Ixazomib are already approved by FDA and achieved great success for other diseases, our findings warrant further mechanistic and translational exploration against ND. New proteasome inhibitor drugs are in animal testing for various ND and PS-519, in particular, have completed phase I clinical trials for ischemic stroke [33–35]. Besides, the presently approved treatments for AD provide only symptomatic relief and are ineffective in preventing the progression because synaptic dysfunction occurs decades earlier and becomes irreversible by the time the clinical symptoms appear. Our results demonstrate high efficacy for UPS inhibitors in restoring long-term plasticity and provide a strong case for potential therapeutic intervention at the early initiation stage of AD. Furthermore, our recent findings support that irrespective of the etiology of the disease, the idea of targeting memory amelioration mechanisms can be a valid candidate for therapeutic intervention. In this study, we preferred the low cost MG132 and lactacycystin, the derivatives of FDA approved drugs over the commercially available FDA drugs due to the availability of literature for plasticity related mechanisms from us and others [4, 20].
Footnotes
ACKNOWLEDGMENTS
We thank Ang Ruixia Sheila for critical review of the manuscript.
This work was supported by Ministry of Education Academic Research Fund Tier 3 (MOE2017-T3-1-002), NUSMED-FOS Joint Research Programme, (NUHSRO/2018/075/NUSMed-FoS/01) grants and NUHS seed fund (NUHSRO/2020/145/RO5+6/Seed-Sep/05) and Ministry of Health (MOH-000641-00). T.B. was supported by the Natural Science Foundation of China (31871076) and Shanghai Municipal Science and Technology Major Project (No. 2018SHZDZX01), ZJLab, and Shanghai Center for Brain Science and Brain-Inspired Technology.
DECLARATIONS
This study was performed in accordance with the ethical review and received approval from the Institutional Animal Care and Use Committee (IACUC) of the National University of Singapore (Approval numbers R17-1008 and R13-5711).