
Abstract
We report the case of a woman firstly referred to our Memory Clinic at the age of 61, following the development of cognitive complaints and difficulties in sustained attention. The investigation that was performed showed: predominant executive dysfunctions at the neuropsychological evaluation, with mild, partial and stable involvement of the memory domain; cortical and subcortical atrophy with well-preserved hippocampal structures at MRI; marked fronto-temporal and moderate parietal hypometabolism from 18F-FDG PET study with a sparing of the posterior cingulate and precuneus; positivity of amyloid-β at 18F-Flutemetamol PET; an hexanucleotide intermediate repeats expansion of C9ORF72 gene (12//38 repeats) and ApoE genotype ɛ4/ɛ4. The patient was diagnosed with probable early onset frontal variant of Alzheimer’s disease (AD), presenting with a major executive function impairment. The lack of specific areas of brain atrophy, as well as the failure to meet the clinical criteria for any frontotemporal dementia, drove us to perform the aforementioned investigations, which yielded our final diagnosis. The present case highlights the need to take into consideration a diagnosis of frontal variant of AD when the metabolic and the clinical picture are somehow dissonant.
INTRODUCTION
Alzheimer’s disease (AD) is the most common form of progressive neurodegenerative syndrome, typically occurring in middle or late life [1]. The classical clinical phenotype comprises of episodic memory impairment together with deficits in other cognitive domains. Based on the evidence of peculiar presentations of AD, the term “atypical AD” has been postulated, indicating less usual but well described phenotypes that occur with AD pathology hallmarks [2,3, 2,3]. According to the most recent criteria, these atypical phenotypic presentations incorporate: primary progressive aphasia, logopenic aphasia, frontal variant of AD (fvAD), and posterior cortical atrophy [2]. Considering the clinical overlap that can be observed in the atypical presentations of AD, differential diagnosis in a clinical setting may sometimes be challenging. In particular, atypical frontal phenotypes of AD might mimic frontotemporal dementia (FTD), where patients present with initial and prevalent executive dysfunctions [3].
Despite the extensive use of neuroimaging and metabolic investigation, a recent study found that up to 40% of patients clinically diagnosed with FTD are found to have AD at postmortem examination. Correspondingly, many patients with pathologically confirmed FTD were previously diagnosed with AD [4]. Since the presence of amyloid-β is considered to be a diagnostic milestone in the construct of biological AD [5,6, 5,6], amyloid-β PET can be useful to identify the presence of AD pathology in vivo. Yet, it is not able to distinguish among the several AD subtypes.
Moreover, a genetic background can have an impact on AD development, especially in early onset AD (EOAD), where almost 60% of cases are hereditary [7]. Three genes are mainly recognized in autosomal cases of AD: amyloid precursor protein (APP), presenilin-1 (PSEN1), and presenilin-2 (PSEN2) [7]. The role of C9ORF72 hexanucleotide repeat expansions (HREs), a known causal mutation for FTD and amyotrophic sclerosis, is yet to be deeply investigated in the context of AD and available data are still controversial [8, 42].
Here, we report clinical, neuropsychological, neuroimaging, and genetic data suggesting a case of frontal variant of EOAD.
Case presentation
A 61-year-old right-handed woman was admitted to our Memory Clinic in 2017. Her family history was positive for dementia since her father was diagnosed with late onset AD. The patient is a married English teacher mother of two and she was referred to us because, throughout the last two years, she has had difficulty in: finding words, writing, focusing when trying to make simple calculus, correcting students’ tests, and in shifting from a paper format to a newly introduced technological tool, indispensable to her job position. However, no concerns were raised by family members regarding her episodic memory or behavior. Insight of her deficits was maintained and mild depressive symptoms were present. Upon admission the patient was independent on her activities of daily living, alert, and oriented in time and space. At neurologic examination, eye movements, cranial nerves, gait, and postural reflexes were normal. No focal motor or sensibility deficits were detected. Tendon hyperreflexia at lower limbs and Babinski’s sign were present bilaterally.
The clinical investigation at first admission included a structural magnetic resonance imaging (MRI) and a neuropsychological evaluation. Subsequently, few months later, the patient underwent a 18F-FDG PET and an amyloid-β PET. After one year from the first neuropsychological battery, at the age of 62, the patient was re-evaluated to explore the evolution of her cognitive deficits. Finally, the most common genetic mutations causing dementia were analyzed.
The clinical, neuropsychological, metabolic, and genetic evidence suggested a neurodegenerative disease of Alzheimer type. However, the executive dysfunctions were prominent at onset and deteriorated overtime, while the mild memory deficit, that was initially detected, remained substantially stable over the two evaluations. The patient retired prematurely from her job and is currently supported in instrumental activities of daily living. Moreover, she is now under treatment with memantine and rivastigmine.
MATERIALS AND METHODS
Neuropsychological evaluation
Two neuropsychological evaluations were performed: one at first admission in April 2017 and the other at the latest follow-up in March 2018. Representative psychometric test results, from both evaluations, are included in Table 1 (raw scores, corrected scores, and cut-offs). The first evaluation included the following tests: Clinical Dementia Rating (CDR) [9], Mini-Mental State Examination (MMSE) [10], Digit Span [11], Visuospatial Span [11], Short-Story Memory Test [11], Attentive Matrices [12], Phonemic and Semantic Verbal Fluencies [11,12, 11,12], Token Test [11], Design Copy Test [12], Apraxia Test [13], Clock Drawing Test [13], Cognitive Estimation Test [13], Rey Auditory Verbal Learning Test (RAVLT) [14], Trail Making Test (TMT) A and B [15], and the Neuropsychiatric Inventory (NPI) [16]. The second evaluation included all the previous tests in addition to other tests to better explore our patient’s cognition: Interference Memory Test [13], Free and Cued Selective Reminding Test (FCSRT) [17], Stroop Test [19], Visual Words Comprehension [18], Auditory Sentence Comprehension [18], Visual Sentence Comprehension [18], and Calculation Processing [18].
First (2017) and second (2018) neuropsychological evaluations. The table shows the performances on tests for memory, attention, language, constructional praxia, executive functions, and the comprehensive neuropsychiatric profile (raw, corrected scores, and cut-off values are reported)
MMSE, Mini-Mental State Examination; RAVLT, Rey Auditory Verbal Learning Test; IR = immediate recall; DR = delayed recall; NPI, Neuropsychiatric Inventory; FCSRT, Free and Cued Selective Reminding Test; N.A., not applicable; N.E., not executable; IFR, immediate free recall; ITR, immediate total recall; DFR, delayed free recall; DTR, delayed total recall; ISC, Index of sensitivity of cueing; scores below normal range are
Structural and nuclear medicine imaging
Structural MRI was performed with 1.5-T Magnetom 15 mT of gradients with four channels (Siemens Magnetom Avanto). We used T1 and T2 and T2 FLAIR weighted sequences.
The patient underwent two PET brain scans, first with 18F-FDG and then with 18F-Flutemetamol (Vizamil®), using a PET tomograph Discovery 710 (General Electric Healthcare). The CT scan was used for attenuation and scatter correction with set voltage tuned to 120 kV. The FDG scan was obtained over 15 min starting 60 min after i.v. of 227 MBq. The 18F-flutemetamol study (Amyloid- β PET) was obtained over 20 min starting 90 min after i.v. injection of 198 MBq. The images of the two scans were visually assessed only.
Genetic analysis
Considering the clinical phenotype and age as well as the penetrance of mutations responsible for genetic FTD, initially we analyzed the C9ORF72 gene and evaluated progranulin plasma levels. We extracted genomic DNA from peripheral blood samples using standard procedures (Flexi Gene DNA Kit, Qiagen). PCR reagents were optimized for the amplification of the C9ORF72 hexanucleotide repeats (AmplideX®PCR/CE C9ORF72 Kit, Asuragen, Inc.). Amplicons were sized using capillary electrophoresis (CE) on a 3100 Genetic Analyzer (Thermo Fisher). Progranulin plasma levels were determined by using commercial ELISA kit according to the procedure of the manufacturer (AdipoGen, Korea). To investigate the presence of mutations associated with AD, including APP, PSEN1, and PSEN2, and other rare dementias, we took advantage of a next generation sequencing technique [20]. ApoE was determined by allelic discrimination [21].
RESULTS
Neuropsychological evaluation
First evaluation (April 2017)
The patient was oriented in time and space and collaborative, even though she was not able to maintain adequate attention throughout the whole execution. The extent of the cognitive impairment was assessed with CDR [9] scale that resulted in 0.5, meaning mild cognitive impairment. At the MMSE (raw score = 24; corrected for age and education = 22.5) [10], the patient showed dysgraphia, with substitutions of letters in single words and difficulties in making backwards calculation. Overall, the evaluation revealed a multi-domain cognitive deficit: partial and mild memory deficit (Visuo-Spatial Span [11] and Short-Story Memory Test [11]) and attention deficit (Attentive Matrices [12]); apraxia (Design Copy Test [13], Apraxia Test [13]). Regarding the executive dysfunction, the Clock Drawing Test [13] and the Cognitive Estimation Test [13] were under the expected value considering the age and education of the patient. While during the TMT B [15], the patient could not implement the rule of the task, presenting a difficulty in alternating letters and numbers. The TMT B test was, therefore, not concluded. The NPI [16] did not reveal any behavioral or psychotic disorder.
Second evaluation (March 2018)
At follow-up, the patient was oriented in time and space, collaborative and, again, she showed a deficit in sustained attention. The overall cognitive impairment was, again, assessed with the CDR that resulted in a score of 1, meaning mild initial dementia [9]. Overall, the evaluation revealed a multi-domain cognitive impairment with mild and stable memory deficit (impaired: Visuo-Spatial Span [11], Short Story Memory test [11]; spared: interference memory test [13], RAVLT [14], and FCSRT [17]). The second evaluation showed also stable attention deficit (impaired: attentive matrices [11]), partial language deficit (impaired: Fluencies tests [12] and Token test [11]; spared: Visual Words Comprehension [18], Auditory Sentence Comprehension [18] and Visual Sentence Comprehension [18]) and apraxia (impaired: Design Copy Test [12], Apraxia test [13]).
Regarding the executive dysfunction, the Clock Drawing test [13], Cognitive Estimation test [13], and the Stroop test [19] were moderately impaired. The TMT B [15] was, again, not concluded and the Calculation Test [18] was not executable due to a severe deficit in calculus abilities. Moreover, the NPI [16] did not show any behavioral or psychotic disorder. Overall, the assessment documented a persistent impairment in executive functions, in attention, in language skills with a mild and stable memory deficit.
Structural and nuclear medicine imaging
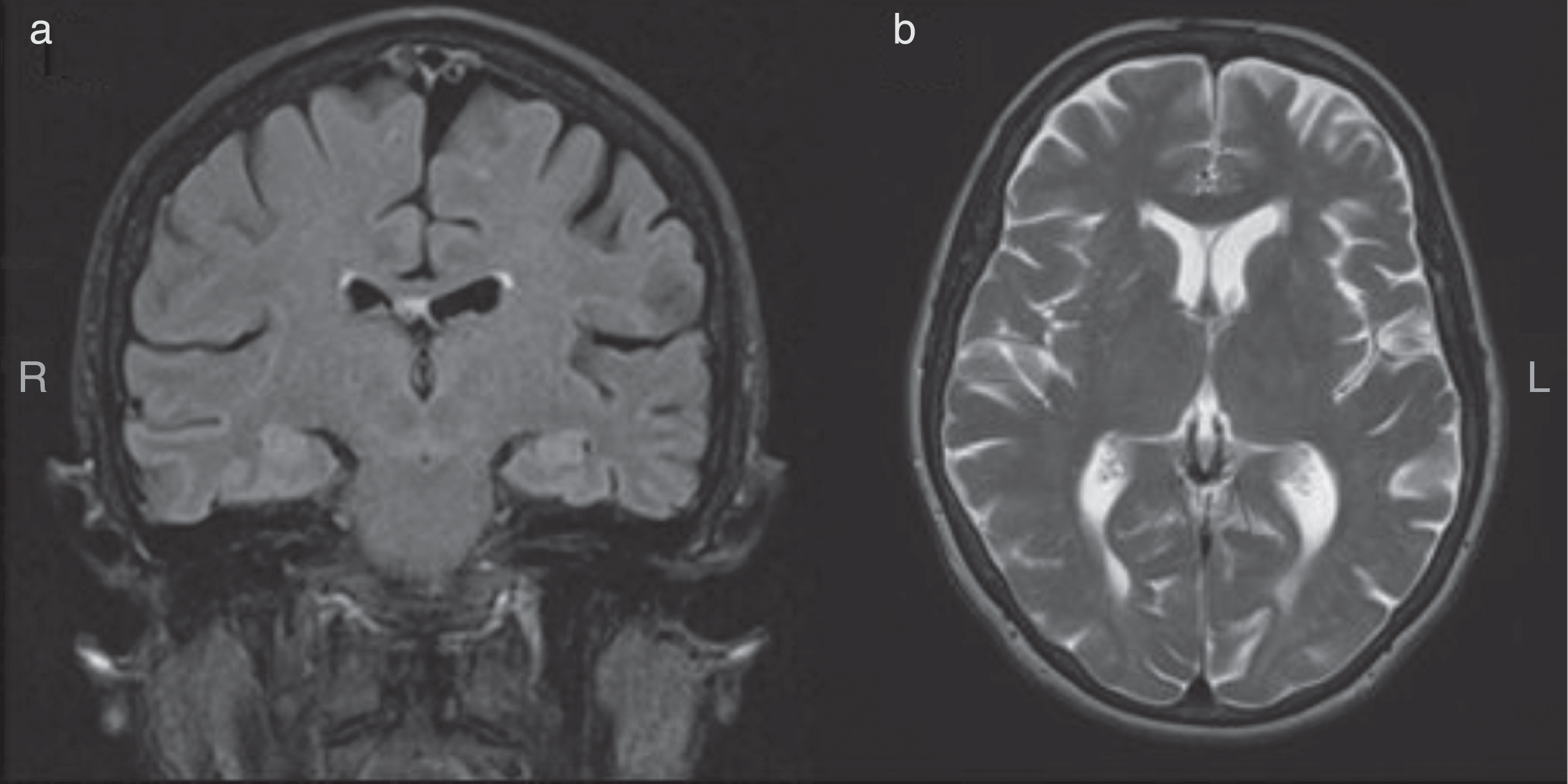
Structural MRI showed mild generalized cortical atrophy and subcortical atrophy with enlargement of the lateral ventricles (Fig. 1a, b). Hippocampal structures appeared substantially preserved at coronal FLAIR (Fig. 1a).
The 18F-FDG scan showed discrete-to-marked bilateral frontal and temporal hypometabolism, particularly on the left side (Fig. 2a-c) and moderate left parietal hypometabolism (Fig. 2d, e), with a sparing of the posterior cingulate and the precuneus.
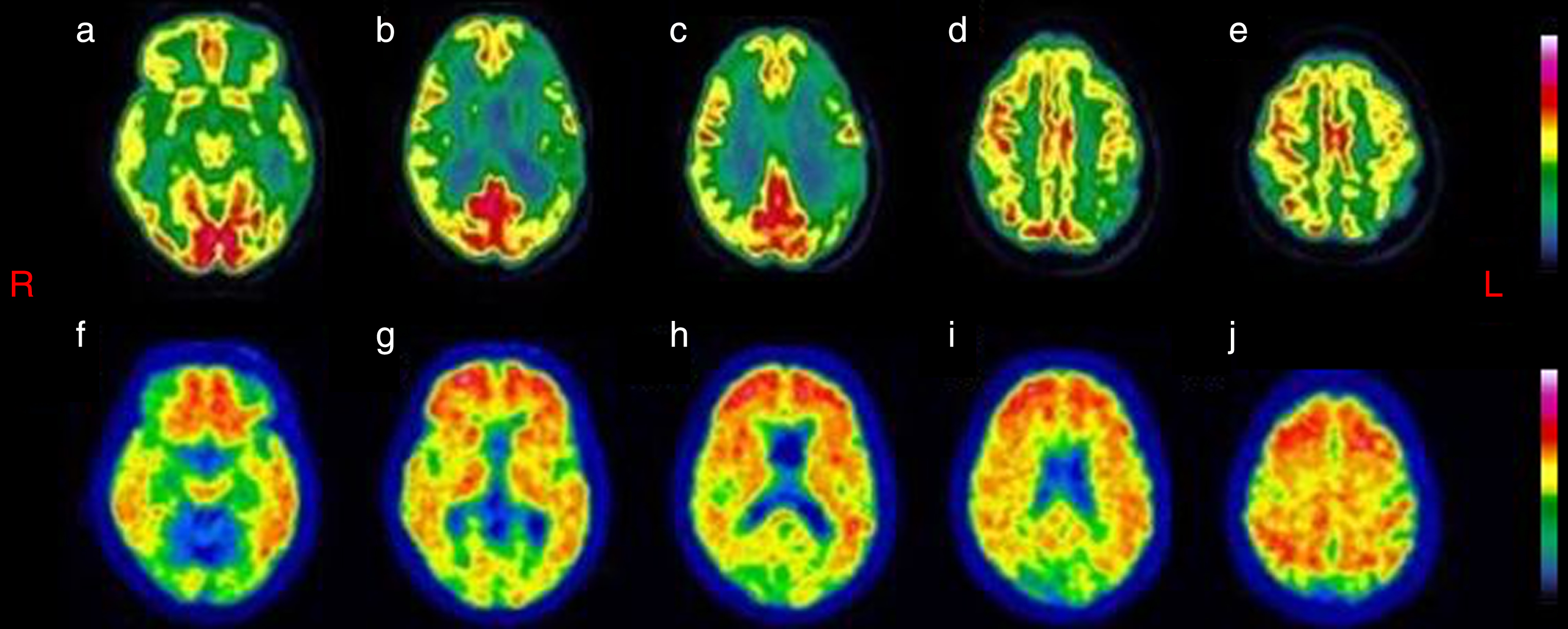
(a-e) 18F-FDG was visually inspected and shows discrete-to-marked hypometabolism in bilateral lateral-frontal and temporal cortex, more evident on the left side, and moderate hypometabolism of the left parietal cortex. Fig. 2f-j The 18F-Flutemetamol scan (amyloid tracer) showing a diffused uptake of the amyloid tracer at the level of cerebral cortex.
The 18F-Flutemetamol scan showed a diffused uptake of the amyloid tracer at the level of cerebral cortex (Fig. 2f-j).
Genetic analysis
C9ORF72 analysis disclosed the presence of an expanded allele (genotype:12//38 repeats). Progranulin plasma levels were found to be within normal range (147 pg/mL; NV > 61 pg/mL), suggesting the absence of haploinsufficiency mutations. Considering mutations associated with AD (APP, PSEN1, and PSEN2), no mutations were found and the ApoE genotype was ɛ4/ɛ4.
DISCUSSION
Our patient presented with an overall multi-domain, prominently dysexecutive, cognitive impairment. Also, she presented with a metabolic pattern at 18F-FDG PET showing a marked hypometabolism in bilateral lateral-frontal, left temporal areas (Fig. 2a-c), and a moderate hypometabolism in the left parietal region (Fig. 2d, e). The selective sparing of the posterior cingulate and precuneus (Fig. 2b-d) and the prominent frontotemporal hypometabolism (Fig. 2a-c) with less marked hypometabolism of the parietal lobes (Fig. 2d, e), directs toward an anterior form, more than a posterior form of AD. The amyloid-β PET revealed a diffuse amyloid-β presence (Fig. 2f-i, l). Besides, the genetic analysis showed C9ORF72 HREs with intermediate repeat expansion and an ApoE genotype ɛ4/ɛ4.
Taking all the evidence together, we oriented toward a diagnosis of fvAD, described in research studies and case reports [3,22-28, 3,22-28].
Regarding our patient’s performance on the neuropsychological examinations, a deficit in executive functions was predominant already from the first assessment in 2017 and deteriorated even more over the following year (TMT B [15], Clock Drawing Test [13], and Cognitive Estimation Test [13] of 2017 and 2018). On the other hand, the mild memory deficit remained stable overtime and was not the predominant aspect, as observed in typical AD. For example, RAVLT [14], a sensitive exam that suggests the presence of typical “hippocampal” AD, was found to be within a normal range in both evaluations. Moreover, during the second assessment, the patient underwent additional memory tests (Interference Memory Test [13] and FCSRT [17]) and she performed within a normality range. Indeed, the performance on the FCSRT [17] demonstrated that patient’s memory is not the leading deficit, as in typical AD presentation. Besides, a mild memory deficit has been reported in patients with fvAD, as also described by a recent study that included 29 autopsy/biomarker-confirmed dysexecutive fvAD patients. Indeed, they found that the onset of cognitive complaints at baseline in the fvAD group consisted of memory impairments (38%), executive dysfunctions (10%), both (38%) or neither (14%) [22]. Moreover, an attention deficit was detected (Attentive Matrices), and we consider it to be substantially stable over the one-year follow-up. The same pattern has been reported in another single case presentation of a patient with fvAD, where the patient presented with deficit on attention tests that remained stable over the first follow up [29]. Additionally, the patient presented with early visuoconstructural deficits (Design Copy and Apraxia Test), probably related to the left fronto-parietal dysfunction, as detected at the 18F-FDG PET (Fig. 2d, e).
Our patient’s cognitive profile is in line with a group study [30], which found that fvAD patients display a distinctive clinical course compared to the “classical” AD and FTD patients. Accordingly, a retrospective study based on neuropathological findings reported that, at onset, both fvAD with executive dysfunctions and behavioral-AD had lower executive functions score than typical AD [31]. Interestingly, they found that both frontal variants of AD are not characterized by frontal grey matter atrophy, as opposed to classical behavioral frontotemporal dementia (bvFTD) [31]. Notably, in our patient, structural MRI showed mild and diffused cortical atrophy, not suggestive for any frontotemporal lobar degenerations [32,33, 32,33].
Our patient’s 18F-FDG PET findings could have misled us toward a diagnosis of initial FTD. However, the neuropsychological profile and structural MRI pattern did not match with that hypothesis.
On the other hand, the amyloid-β PET showed a diffused uptake of the amyloid-β tracer. This data was crucial to endorse the final diagnosis, as it revealed the presence of the AD pathology in vivo. As a matter of fact, an inclusion criterion for any AD subtype is the presence of AD pathology, investigated either in vivo (amyloid-β PET, CSF markers) or postmortem [2]. The investigation using amyloid-β PET has been found useful to discriminate among AD and FTD. Indeed, a study found that 7 out of 7 AD patients had a positive scan and 4 out of 12 patients, that had previously received a FTD diagnosis, had positive scans as well [34]. One of the 4 patients had a clinical framework similar to our patient, presenting marked executive dysfunctions, yielding a diagnosis of fvAD [34]. However, according to recent literature, it is not possible to differentiate typical AD pathology with atypical phenotypic presentation of AD with amyloid- β PET. Indeed, multimodal imaging studies that combined amyloid-β PET and 18F-FDG PET did not find significant correlation between the patterns of hypometabolism and amyloid-β deposition: the latter was diffused in the neocortex in all AD syndromes equally [35,36, 35,36].
Moreover, a case of a 55-year-old man with behavioral disturbances, initially misdiagnosed with bvFTD, was then diagnosed with fvAD following amyloid-β PET [37]. This case and ours have similar patterns of onset and imaging framework. However, the predominant symptom of our case was an overall cognitive impairment with a more striking executive dysfunction.
Given that clinical phenotype in AD is closely linked to patterns of glucose hypometabolism (but not amyloid-β deposition), we can speculate that the severe hypometabolism of the dorsolateral frontal cortex, appreciated from the 18F-FDG PET study, could explain the executive dysfunctions. At the same time, the relative sparing of the mesial frontal cortex could be related to the absence of behavioral symptoms [38].
Regarding the genetic analysis, an expansion of 12//38 repeats in C9ORF72 was detected. The cut-off to discriminate between normal repeat alleles and pathogenic expanded repeats has not been unequivocally established. Although most authors use an arbitrary cut-off of 30 or 60 units [39], some affected individuals were reported to have smaller repeats [40]. Repeats alleles spanning the range between 20 and 22 have already been observed in FTD patients and their affected siblings [41], as well as in AD families [42]. However, it remains unclear whether the smaller repeat expansions observed in our patient could cause the specific disease phenotype. Besides, largest repeat expansions have been observed to affect the anticipation of the disease [43]. While C9ORF72 HREs has been widely documented in FTD, there is very little evidence regarding the involvement of the aforementioned HREs in patients with AD. In a study carried out on a sample of 342 families with members affected by AD, they found that the C9ORF72 HREs were present in 3 families (<1%) apparently affected by AD (6 out of 771 patients with probable AD) [44]. Following a postmortem analysis, 2 out of the 6 patients carrying the HREs showed neuropathological findings consistent with a primary diagnosis of FTD and not AD pathology [44].
In conclusion, although we cannot demonstrate that the C9ORF72 expansion plays a role in the clinical presentation as no functional studies have been carried out, recent literature supports the hypothesis that expansions of >30 are associated with a wide spectrum of clinical presentations, including memory impairment and executive dysfunction. Nevertheless, epigenetic modifications such as methylation has not been detected in alleles <43 repeats, thus questioning the cut-off of 30 repeats for pathogenicity [45]. Moreover, the genotype of the ApoE gene was ɛ4/ɛ4. Therefore, the patient has the highest risk for AD known so far. However, the present literature suggests that the ApoE ɛ4/ɛ4 allele is more frequent in the amnesic form of AD compared to the executive predominant group [23,24, 23,24] and studies on the possible gene-gene interaction between C9ORF72 and ApoE ɛ4/ɛ4 are still lacking. As discussed above, since the literature on the role of intermediate C9ORF72 alleles is controversial, the ApoE gene is more likely to be associated with the development of AD, rather than the C9ORF72.
Overall, here we describe the case of an EOAD patient presenting with predominantly frontal/dysexecutive clinical features and marked frontotemporal hypometabolism, with less marked parietal involvement, from the 18F-FDG PET study. Although atypical, the occurrence of an FTD-like metabolic pattern in AD is not a very uncommon phenomenon, particularly in patients with early AD onset. The lack of specific areas of brain atrophy, as well as the failure to meet clinical criteria for bvFTD and primary progressive aphasia, drove us to perform further investigations, which finally yielded to a diagnosis of AD. The present case highlights the need to take into consideration a diagnosis of fvAD when the metabolic and the clinical pictures are somehow dissonant. However, more data will be required to establish the possible pathological role played by HREs of the C9ORF72 gene in these cases of AD.
Finally, it will be important to follow-up the patient longitudinally to see if, as reported in many subjects with intermediate repeats of C9ORF72, she will experience behavioral disturbances, or if the patient will advance with a typical progression of the dysexecutive form of AD.
DISCLOSURE STATEMENT
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/18-0715r2).