
Abstract
Alzheimer’s disease (AD) is one of the most prevalent neurodegenerative diseases that is characterized by progressive memory loss and two main pathological hallmarks, including the extracellular amyloid plaques and intracellular neurofibrillary tangles. The microtubule-related protein tau is involved in the pathogenesis of many neurological diseases commonly known as tauopathies and is found to be abnormally hyperphosphorylated in AD and accumulated in neurons. Besides hyperphosphorylation, tau also undergoes abnormal glycosylation, ubiquitination, glycation, and other posttranslational modifications. These abnormalities lead to the aberrant aggregation of tau in the synaptic loci in AD. In this review, we highlighted the most recent studies about how tau is abnormally regulated and how those abnormalities play important roles in the pathogenesis of AD.
INTRODUCTION
Alzheimer’s disease (AD) is a progressive neurodegenerative disease in aged people. The etiology of AD is not clear yet although the first case had been reported for over 100 years. Currently, the known therapeutic strategies for AD can only partially retard the progression of some clinical manifestation. Pathologically, AD is characterized by the senile plaques that are constituted of aggregation of amyloid-β (Aβ) peptides and the neurofibrillary tangles (NFTs) which are composed of the abnormally hyperphosphorylated microtubule-associated protein tau [1, 2]. Two main forms of AD exist, the familial or early onset AD representing less than 1% of all cases and caused by mutations in multiple risk genes, including presenilins (PS) 1 and 2, amyloid precursor protein (APP), and other genes identified by genome-wide association studies, such as TREM2, PICALM, PLD3, UNC5C, AKAP9, and ADAM10 [3–5]. The other form is late-onset AD (LOAD), which is the most common (>95%). The etiology of LOAD is not clear and all of the genetic and environmental factors are thought to be crucial. Moreover, in primary neuronal culture, loss of tau also causes iron retention, by lessening surface trafficking of amyloid-β protein precursor (AβPP). These studies suggest that the loss of soluble tau could contribute to toxic neuronal iron accumulation in AD [6]. Furthermore, those findings have shown the suitability of lithium as a potential treatment for disorders where brain iron is elevated as the case in AD [7].
Tau is one of the microtubules associated proteins (MAP) that is predominantly localized in the axons in the developed neurons and is thought to play important role in the maintenance of microtubule stability [8–10]. In the human brain, there are six isoforms of tau [11]. These isoforms are coded by a gene on chromosome 17 and are produced by alternative splicing of its pre-mRNA [12, 13]. In many neurological disorders, tau is dissociated from microtubules and accumulated in the cytosol [1]. Numerous phosphorylation sites (>40) under the physiological and pathological conditions have been mapped. Specifically, many serine-proline or threonine-proline epitopes phosphorylations have been seen in AD [8].
Abnormal aggregation of tau is related to many neurodegenerative diseases including AD [1]. In AD, tau protein is abnormally hyperphosphorylated and accumulated into packs of fibers [12], and abnormally collapsed and hyperphosphorylated tau (p-tau) gathers in axons, dendrites, and somas [9, 15]. Tau abnormalities are implicated in many tauopathies, including AD and some frontotemporal dementias (FTD) [3]. In this review, we first consider the normal structure and physiological function of tau, then we summarized data on tau abnormalities and its deregulation in AD.
STRUCTURE AND FUNCTION OF TAU PROTEIN
The 4R2N tau is the longest human tau isoform with a total of 441 amino acids (tau441) long [12]. The tau protein is encoded by the MAPT gene situated on chromosome 17 [16, 17]. In the adult human brain, mRNA alternative splicing of exons 2, 3, and 10 yields six tau isoforms. The six isoforms of tau protein vary from 352 to 441 amino acids residues and differ as per the contents of three (3R) or four (4R) repeats tubulin binding spaces of 31 or 32 amino acids in the C-terminal piece of tau protein and two (2N), one (1N), or no insert of 29 amino acids each in the N-terminal portion of the molecule. These isoforms, that differ in size from 352 to 441 amino acid remains, are identified to the existence or absence of sequences encoded by exons 2, 3, or 10. Consideration of the exon 10 prompts to the expression of tau holding four microtubule-binding (4R tau: 0N4R, 1N4R, 2N4R), while avoidance of exon 10 results in splicing produces having tau with three MTBRs (3R tau: 0N3R, 1N3R, 2N3R) (Fig. 1). Apart from the six main tau isoforms in the brain and CNS, in the peripheral nervous system, there are additional tau isoforms containing an additional exon, exon 4a [18]. However, the longest human CNS tau isoform with 441 residues has usually been selected as a model to exemplify tau primary structure [11]. In the normal human brain, the ratio of 4R to 3R tau is about one; however, in various tauopathies, this ratio is altered [3, 19]. Beside adjusting microtubule dynamics, tau may regulate axonal transport through various mechanisms. A small amount of tau is also discovered in dendrites but the function of tau here is vague. Tau is also found in the nucleus, where it might play a part in maintaining the safety of genomic DNA [20].
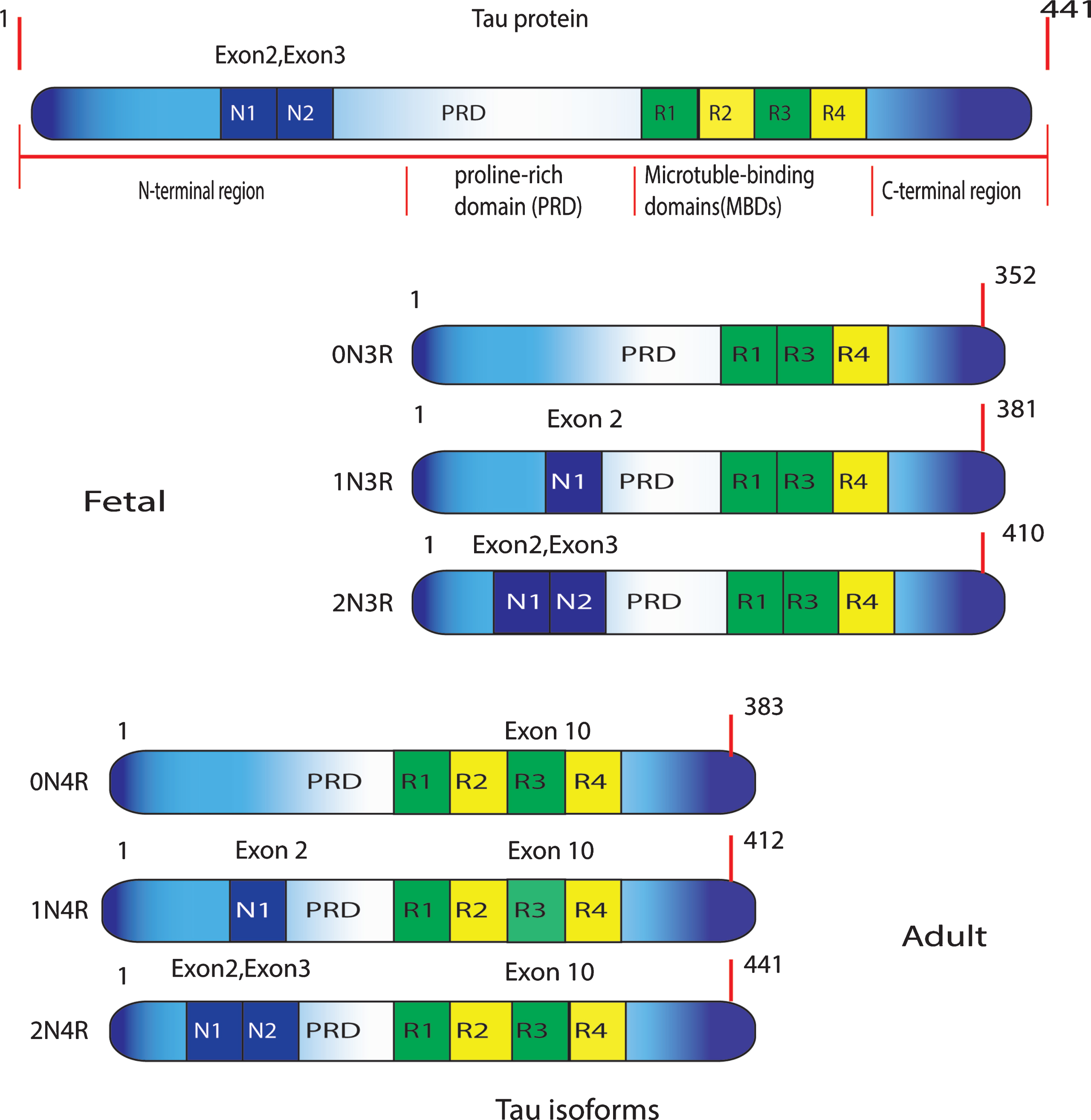
Structure of the six human tau isoforms. Tau has an overwhelmingly acidic N-terminal region, a proline-rich domain (PRD), and a generally neutral C-terminal region. The six human brain isoforms of tau are represented. They differ by the inclusion or exclusion of exons 2 (1N), exon 3 (2N), and exon 10 (R1–R4).
Tau has several physiological functions. In normal, healthy neurons, it is fundamentally distributed in axons and plays an important role in stabilizing microtubules. Tau protein binds microtubules through some repeated areas (R1-R4) (encoded by exons 9–12) situated at the C-terminus of the molecule (Fig. 2) [21]. Each repeat comprises extensions of a highly monitored 18 remains that are iteratated three times in the fetal tau protein and four times in the adult form [21]. The repeats are isolated from each other by 13- or 14-residues spacer areas. The main function of tau as a promoter of tubulin polymerization depends mostly on the MTBR [20, 21]. Lowering tau protein might affect its physiological role prompting unfavorable impacts. In additional to its role in the brain, a previous study showed the presence of tau protein in cardiac tissue. That study bolsters a functional role of tau in the heart and loss of this protein leads to a weakening in cardiovascular performance which worsens with age [22]. Another report highlights the involvement of tau in mediating stroke-induced iron accumulation, as well as the therapeutic potential of ferroptosis inhibition in ferroptotic injury [23].
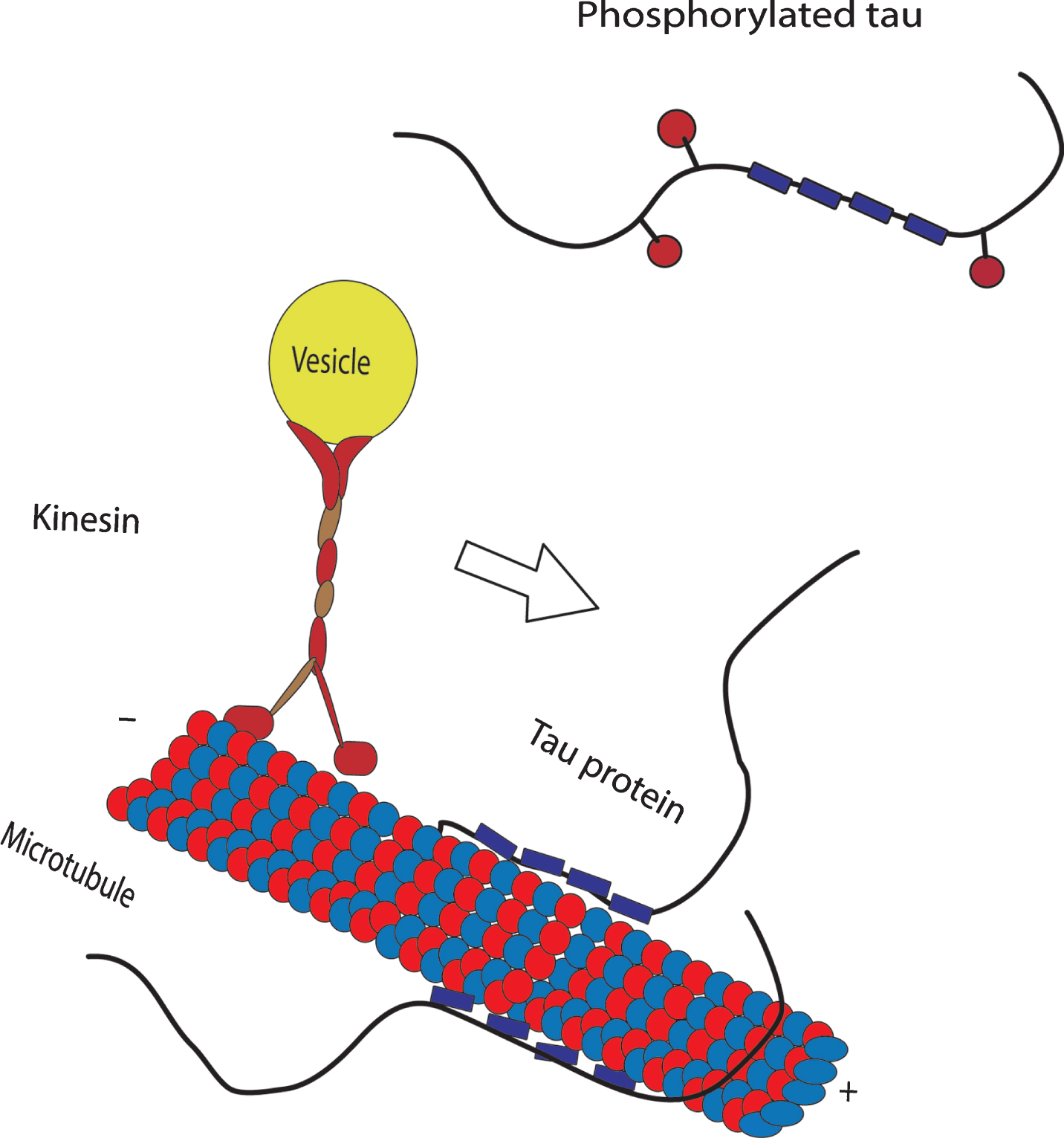
The function of human tau in the stabilization of microtubules. This is achieved through four tubulin binding domains (blue boxes). Phosphorylation of tau (red balls) can directly affect its ability to bind to microtubules and its role in axonal transport. Tau protein may repress addition to end-coordinated transport of vesicles along microtubules by kinesin.
PATHOPHYSIOLOGY OF TAU PROTEIN
The pathological significance of tau protein was confirmed by the revelation of acquired mutations in the MAPT gene, in contrast to the obvious redundancy of the physiological function of this protein, as exhibited in tau-deficient mice [24]. These genes are intimately connected to the disease in families with autosomal dominant frontotemporal dementia with Parkinsonism linked to chromosome 17 (FTDP-17) [25–28]. Again, genetic discoveries set up tau protein as a key agent in the pathogenesis of a subgroup of FTD, a heterogeneous form of tauopathies portrayed by dementia and movement disorders [29].
Tau has multiple physiological functions. In normal healthy neurons, it is mainly distributed in axons and plays an important role in stabilizing microtubules. Besides regulating microtubule dynamics, tau may adjust axonal transport through various mechanisms. Pathological tau cannot enter the nucleus that may result in DNA damage due to the loss of the DNA-protective function of tau [30]. Finally, tau might form aggregates, leading to a deterioration of neuronal function. Furthermore, tau aggregates may be released into extracellular space and be taken up again by other neurons, leading to the expansion of tau pathology [31]. Hyperphosphorylation of tau prompts conformational transformations that notably disable its capability to tie to microtubules. Tau hyperphosphorylation is a result of deregulation in the equilibrium between kinases and phosphatases [32]. More than thirty kinases have been depicted directing tau phosphorylation in vitro and at the same time, just a couple were affirmed in vivo [32]. Among them, GSK3β may be a paramount kinase which phosphorylates tau with respect to more than 30 sites, what appears to play a critical part in NFT advancement. A change in the activity from one or numerous kinases or phosphatases is mostly recognized to launch the pathological phosphorylation of tau seen in AD [32].
TAU PATHOLOGY
In AD, the normal part of tau protein is insufficient to keep the cytoskeleton efficient in the axonal transport because this protein loses its ability to bind to microtubules. This strange conduct is because of conformational changes and misfolding in the primary structure of tau [20, 33–35] that prompts its abnormal aggregation to fibrillary assemblies inside the neurons of demented individuals [20, 37]. In this manner, the greater part of the changed pools of tau protein in the disease is redistributed and aggregated in both the somatodendritic compartment and axons of affected neurons. The mechanisms by which tau protein turns into a nonfunctional substance are still unclear; however, abnormal posttranslational alterations are proposed to be the primary cause of this failure [38, 39]. In such a manner, anomalous phosphorylation (hyperphosphorylation), ubiquitination, acetylation, nitration, glycation, proteolytic cleavage (truncation), conformational changes, and other alterations [20, 40–42] have been suggested to cause the loss of the ability of tau to exert its normal function and therefore leading to tau pathology [20].
An abnormal tau 3R/4R ratio, either increased or decreased, is thought to alter the capacity of tau in the microtubule to get together for stabilization and normal axonal transport. Inherited mutations at that point are ventured to incite comparable problems, while mutant tau, as a rule, is more inclined to phosphorylation and polymerization [29, 43–45]. Eventually, the role played by anomalous structural highlights of tau protein in human tauopathies should be dependent on the evidence of its normal cellular functions [29]. In contrast to normal tau, anomalous tau does not induce the in vitro assembly of microtubules [46, 47]. Enzymatic dephosphorylation, but not deglycosylation, of tau protein can reestablish the microtubule assembly inducing movement of both paired helical filament (PHF)-tau and the AD P-tau [40, 49]. Previous studies [46, 50] highlighted that the abnormally phosphorylated tau is not just latent in inducing microtubule assembly but also causes dismantling of microtubules association with tau, MAP1, and MAP2. Therefore, neurofibrillary degeneration may be avoided by restraining abnormal hyperphosphorylation of tau [40].
Lack of pathology in some neurons in AD, as cerebellar neurons which are impervious to degeneration [15, 52] has been reported, while aggregates in neurons (e.g., Purkinje cells) are not found. This might be because of the low amount of tau in these neurons [15, 53], although in diseases like Niemann-Pick type C disorder, cerebellar tau might be hyperphosphorylated [53]. In PNS neurons, no aberrant tau aggregates have been found. This may mirror the presence of the extra exon 4A in the tau molecule that prevents tau self-assembly [15, 54].
STAGING OF TAU PATHOLOGY
During the 1990s, the importance of tau pathology for neurodegenerative diseases, specifically for AD, stayed in the shadow of the amyloid hypothesis as the distribution and general quantity of Aβ ended up being of constrained essentialness for pathological staging of AD progression and an indication of seriousness. After more advanced investigations in the development and dissemination of NFTs demonstrating a relationship with the level of cognitive decline and memory impairment in AD, Braak and Braak proposed a neuropathological staging of the continuous affidavit of abnormal tau inside vulnerable neurons in brain areas as either NFT or neuropil threads [55]. In the first place, they utilized traditional silver staining [55, 56] and later immunohistochemical staining for hyperphosphorylated tau utilizing AT8 antibody [55, 57]. They found that NFT furnish a superior relationship with cognitive impairment and was confirmed by different researchers [55, 59], supporting a significant role of tau pathology in the disease. The Braak arranging system classified the topographic progression of AD neurofibrillary degeneration into six phases, spreading from the transentorhinal area to the hippocampal formation (beginning stages I and II, which clinically correspond with subjective or objective impairment of memory for late events and mild spatial disorientation, however with safeguarding of general subjective working with or without least minimum impairment of activities of daily living), then to the temporal, frontal, and parietal neocortex (intermediate stages III and IV, which correlate with impaired recall, delayed word recall, and word revenues difficulties, disorientation in time and space, and impaired concentration, comprehension and conceptualization among other symptoms of dementia), and eventually to unimodal and essential sensory and motor areas of the neocortex (late stages V and VI, which almost relate with disorder in object recognition, and other perceptual and motor skills) [55].
TAU GENETIC MUTATIONS
The effect of tau gene mutations in familial frontotemporal dementia and Parkinsonism linked to chromosome 17 (FTDP-17) on different tauopathies has been demonstrated. The current recognizable proof of the significant protein segment of the microtubule-associated protein tau constitutes one of the characterizing neuropathological elements of AD. FTDP-17 alludes to the aggregation of inherited neurodegenerative disorders portrayed by early behavioral changes later taken after by psychological and motor deficits [60, 61]. Pathologically, FTDP-17 is described by degeneration of the frontal and temporal lobes with nerve cell death, astrocytic gliosis, and microvacuolation of the neuropil mostly located in the shallow cortical layers. The basal ganglia and substantia nigra are among the most affected subcortical structures [60, 61].
Variable pathologies among FTDP-17 families are probably depending on the area of the different mutations as of late portrayed in the tau gene [28, 63]. The most widely recognizable mutations happen in an anticipated stem-loop structure in the 5∞-splice site of exon 10. The 5∞-splice site mutations are thought to destabilize the intron 10 stem-loop structure and in this manner, offer ascent to an increased splicing in of exon 10 which builds generations of the four repeat isoforms [27, 61]. Four repeat isoforms in this manner prevail in both dissolvable tau and filamentous tau conformations in brains from individuals. Besides, recent reviews have revealed that mutations in FTDP-17 do not just influence the ratio of four to three repeat isoforms; however, some exonic mutations (in exons 10, 12 and 13) do likewise modify the biochemical properties of tau. These exonic mutations appear to diminish the microtubule-binding ability of tau to induce microtubule assembly [27, 64].
Tau mutations in the intron taking place after exon 10 or in exon 10 itself lead to a neuronal and glial pathology, though mutations situated outside exon 10 command to a predominantly neuronal tau pathology [65]. In the normally grown-up human brain, tau is predominantly found in nerve cells, with little low levels in glial cells [64]. The incomplete loss-of-function due to the lessened capacity of mutated tau to induce microtubule assembly is presumably the essential impact of the tau mutations. It might represent a fundamental stride for allowing the consequent hyperphosphorylation of tau which, in conjunction with different elements, is said to lead to assembly into fibers. Whether these mutations affect tau phosphorylation and filament assembly remains to be elucidated [64, 66]. Intronic tau mutations that lead to a considerable expansion of exon 10 have been related to a number of neuronal and glial pathology, with the glial part being more conspicuous than that created by the P301L and P301S changes [67].
ALTERNATIVE SPLICING OF TAU
Alternative splicing is of real significance in producing proteomic differing qualities, and in adjusting protein activities in a timely and spatial way [68]. Alternative splicing produces a great part of the gigantic assorted qualities required in the proteins required in framing particular synaptic associations and in interceding synaptic transmission [68]. Alternative splicing of exons 2, 3, and 10 manufacture tau proteins comprising 3 or 4 microtubules binding repeats, tau3R or tau4R. In the young human brain, the proportion of 3R and 4R tau transcripts is roughly one [67]. Alternative splicing on the tau gene is also regulated through development. Splicing mutations can happen in the coding region or intronic area. A few mutations can influence both the peptide succession and alternative splicing of the tau gene (e.g., N279K). A couple of single nucleotide changes that do not influence tau peptide assembly (silent mutations [e.g., L284L, N296N, and S305S]) influence alternative splicing of the tau gene [67, 69]. Exon 10 alternative splicing can be regulated by many splicing factors together with their kinases and phosphatases suggesting that the dysregulation of the alternative splicing of tau exon 10 results might in alterations of the ratio of 3R-tau, and 4R-tau, that have been seen in numerous tauopathies [70]. Transactive response DNA binding protein of 43 kDa (TDP-43) is associated with the processing of RNA, including splicing, indicating that dysregulation from tau exon 10 splicing is enough to induce neurodegeneration [70].
ABERRANT TAU POST-TRANSCRIPTIONAL REGULATION AND MODIFICATION
Tau expression is transcriptionally organized and tissue-specific. Besides, gel electrophoresis analysis shows that transcriptional regulators SP-1 and AP-2 are essential for basal expression but not required for neuron-specific expression of the tau transcript [71]. Numerous tau modifications have not yet been studied as broadly as phosphorylation, including glycosylation, nitration, ubiquitination, deamidation, oxidation, cross-linking, or glycation, methylation, and acetylation [69]. However, in pathological situations, tau may experience modifications, predominantly through phosphorylation, that can bring about conditions that are harmful to neurons. In pathological conditions, tau may undergo post-translational modifications, for instance, hyperphosphorylation, acetylation, ubiquitination or truncation that could lead to the detachment of tau from microtubules, resulting in microtubule disassembly in axons (Fig. 3). Additionally, tau protein comprises multiple isoforms that are regulated along with the development and are mostly formed by alternative mRNA splicing. MicroRNAs (miRNAs) which are small noncoding RNAs that control gene regulation at the posttranscriptional level by attaching to the 3’ untranslated locale (3’UTR) of target mRNAs causing their translational suppression or sometimes degradation. Several miRNAs are particularly synthesized or found in high amount in the brain [72–75], among which some were linked to neuronal differentiation, synaptic plasticity, and memory formation [76]. miRNAs were also reported to be associated with aging and life expectancy control in adult worms [77, 78].

Schematic illustration of the mechanism of the post-translational modifications of tau under normal and pathological conditions.
TAU PHOSPHORYLATION
In the 1980s, tau was characterized as a phosphoprotein in a wide range of studies. While these experiments focused on the serine/threonine phosphorylation of the tau protein, lately some consideration has been paid to its phosphorylation on tyrosine [15]. Tau contains a strangely high number of phosphorylation sites (45 serines, 35 threonines, and four tyrosines), and for a couple of those, fastidious antibodies have been developed. Under physiological conditions, there are on the average 2-3 moles of phosphate for each mole of tau protein, while, under pathological conditions, this quantitative relationship is brought up to 7-8 moles [20, 79]. This has been termed ‘hyperphosphorylation’ [79, 80]: some sites are phosphorylated to a higher degree in the diseased than in the healthy brain; others are newly phosphorylated. Hyperphosphorylation is essential for tau to disengage from microtubules and is supposed to be a prerequisite for its aggregation [15, 79]. As cytoplasmic tau levels enlarge further, tau aggregates and insoluble fibrillary structure formation also increases. With expanding tau levels in the cell, tau further abnormally interacts with the host cell proteins, altering their normal physiological functions [79]. Despite the repeated interest in tau phosphorylation, function, and dysfunction, an appropriate animal model that carefully recapitulates the tau pathology and neuronal degeneration found in human tauopathies has not been established. While the models that have been produced may not be completely precise in their impersonation of human disease states, they do supply worthy insights into how tau functions in normal and pathological states and the part that phosphorylation play in modulating tau function [81]. In general, an increase in tau phosphorylation decreases its binding ability to MT and thereby regulates the traditional biological activity of tau association to microtubule assembly and stability. It should be noted that phosphorylation of tau protein is part of the traditional physiological regulatory mechanism and not pathogenic in itself [29, 83].
A decrease in the tau phosphatase activity in AD brains has been reported, indicating that the tau is abnormally phosphorylated in AD brains, likely because of a disruption in the protein phosphorylation/dephosphorylation framework [40]. A major wanting link in the understanding of the role of the abnormal phosphorylation of tau in neurofibrillary degeneration has been the nature of the participation of the HMW-MAPs [15]. At a cellular level, abnormal phosphorylation of tau presents modifications in several procedures which are specifically controlled by the appropriate organization of the microtubule network. In a typically developed neuron, tubulin is available over tenfold than tau, and thus practically all tau protein is microtubule limited in the cell [20, 85]. In AD neurons, abnormally phosphorylated cytosolic tau (AD P-tau) neither binds to tubulin nor prompt microtubule assembly [20, 86]. Rather, this protein prevents the assembly and damage the microtubule organization [46]. Furthermore, it was reported that abnormally phosphorylated tau protein disassembles ordinary tau from microtubules into the cytosolic compartment [46]. As much as 40% of the abnormally hyperphosphorylated tau in the brain of AD patients is available in the cytosol and not polymerized to PHFs or forming NFT [20, 87].
Tau phosphorylation is extraordinarily increased in response to various stressors. Phosphorylation, despite the fact that to a lesser level than in AD brain, gives off an impression of being assembled by neurons to regulate the activity of tau transiently and reversibly as required. A prime example is a hibernation which is a versatile process that represents a powerful physiological procedure to withstand periods of limited energy supply. Hibernation is a hypometabolic state with declining body temperature, intermittently hindered by brief unconstrained periods of rewarming to center temperatures, whereby particular articulation and control of kinases and phosphatases is a versatile reaction for long-haul survival [29, 88]. Highly phosphorylated tau protein promptly gathers and even seems to accumulate in the brain of sleeping animals. Strikingly the whole procedure is completely switched when animals arouse, restoring typically temperature and metabolism without harming neurons or networks [29, 89–91]. Tau phosphorylation was also found to be affected by miRNAs. In primary neurons, overexpression of miR-125b was found to induce tau hyperphosphorylation and an upregulation of p35, cdk5, and p44/42-MAPK signaling. It has recently been discovered that some miRNAs are modified in the brains of AD patients [92–96]. miRNAs typically focus on numerous mRNAs and can, subsequently, modify a few cellular pathways in parallel [97]. As such, deregulation of miR-29a/b-1 and miR-101 in AD may induce Aβ production by targeting amyloid-β protein protein (AβPP) and/or β-site APP cleaving enzyme (BACE1) [77, 98].
PHOSPHORYLATION BEFORE OR AFTER PHF ASSEMBLY
A further question that emerges from the conceivable connection between tau phosphorylation and aggregation is whether tau phosphorylation happens before or after PHF assembly. If tau phosphorylation happens in the first place, and it happens in areas that will be covered after PHF assembly, the site will not be available for phosphatases following assembly, and we can theorize that such a site was phosphorylated before tau assembly [46, 99]. This proposes tau phosphorylation happens before its assembly, a hypothesis that is bolstered by different studies [46, 100]. Indeed, a solid indication exists that the phosphorylation of tau induces its self-aggregation [46, 101].
TAU PHOSPHORYLATION AND DEVELOPMENT
The phosphorylation of tau is developmentally organized; it is higher in fetal neurons and decrease with age. Moreover, a gigantic increment in the phosphorylation of tau emerges in neurotic circumstances (tauopathies) [40, 102–104]. Strangely, phosphorylation at various sites could happen in various tau isoforms [15, 99]. This could be due to the distinctive cellular localization or subcellular compartmentalization of the varied tau isoforms, or the way that different kinases or phosphatases can adjust tau phosphorylation in an unexpected way. The likelihood of distinguishing the phosphorylation of particular tau isoforms by analyzing the electrophoretic versatility of these isoforms in the presence or absence of phosphatases has been demonstrated [46, 102]. In any case, it must be conceived as a top priority that the binding of tau isoforms to different proteins (microtubules) or membrane may cover these sites, preserving their phosphorylation in nonpathological conditions, or like tau itself, in pathological conditions. Then again, hyperphosphorylated tau has been portrayed in extracts of both typical and AD tissues [46, 87]. Numerous tau isoforms are phosphorylated at numerous sites under physiological situations by similar kinase (e.g., glycogen synthase kinase 3). This could be because of the fact that such kinases can alter both primed (those sites that can be phosphorylated after a past change at an adjacent site by other kinases) and unprimed sites. Though, in pathological conditions, similar to AD, a solitary tau isoform can be altered at an expanded number of sites.
The protein tau is modified by many post-translational processes which in normal conditions equilibrate each other. In pathological conditions, a downregulation of glycosylation facilitates abnormal phosphorylation of tau and cause an imbalance between tau phosphorylation and dephosphorylation procedures foremost to tau hyperphosphorylation and microtubule destabilization [105]. Other post-translational modifications, such as acetylation, oxidation, isomerization, glycation, nitration, ubiquitination, sumoylation, polyamination, and proteolytic cleavage of tau interact with each other and are involved in the formation of tau pathological aggregates (NFT).
TAU GLYCOSYLATION
Glycosylation is a standout among the most well-known, and the most intricate, types of post-translational modification of proteins. However, protein glycosylation in AD is a subject that has not been completely researched [106]. Both N-and O-glycosylation of tau have been reported, with N-glycosylation happening in hyperphosphorylated tau, though unmodified tau can be O-glycosylated. Three potential tau N-glycosylation sites have been reported, and the presence of N-linked glycans on tau has been confirmed by a combination of lectin recoloring monosaccharide structure examination, and N-glycosidase F treatments [106–108]. The connection between phosphorylation and O-glycosylation of tau proteins may have a role in the nuclear localization of tau [15]. As several studies have suggested that protein glycosylation is adjusted in AD, this comparatively unexplored subject merits more consideration, and could conceivably be profoundly critical for the advancement of enhanced biomarkers and treatment technique for AD. As opposed to N-glycosylation, O-glycosylation happens just after protein translation [106]. An experiment by Lefebvre et al. [109] incited the hyperphosphorylation of tau by repressing phosphatase action and found that the incorporation of O-GlcNAc into tau has diminished, as did the translocation of tau to the cell nucleus [15]. The finding that O-GlcNAc modification occurs in tau, and its effect on tau phosphorylation has attracted recent research attention in O-GlcNAc studies in AD field. Modification of proteins by O-GlcNAc happens widely in the brain. The level and activities of the enzymes catalyzing O-GlcNAc cycling are significantly higher in the brain than in the peripheral tissues. The levels of O-GlcNAcylation of brain proteins inclusive tau are reduced in AD brain, possibly due to diminished brain glucose metabolism. This decrease in brain O-GlcNAcylation seems to mediate the molecular mechanism by which diminished brain glucose metabolism leads to neurodegeneration [110]. Many other studies on the impact of N-glycosylation on the phosphorylation of tau have similarly been done. One of those studies proposed that the glycosylation of tau is an early abnormality from the norm that can lead to the hyperphosphorylation of tau in AD brain [108]. Another study suggested that deglycosylation of the distortedly glycosylated tau diminished the resulting phosphorylation of tau at Ser214, Ser262, and Ser356 in vitro by protein kinase A [111]. Additionally, it has been shown that atypical glycosylation of tau in the AD may lead to neurofibrillary degeneration by inducing anomalous hyperphosphorylation by cdk5 and GSK-3β [112].
TAU UBIQUITINATION
Ubiquitin is a 76-amino acid stress protein involved in the ATP-dependent degradation of short-lived proteins or the expulsion of abnormal or harmed proteins [113]. Ubiquitination is a particular posttranslational modification on proteins that signal for their degradation in the cytosol by the UPS (ubiquitin-proteasome system). In non-pathological conditions, tau appeared to be ubiquitinated and proteolytically prepared by UPS [97, 98]. Tau can be ubiquitinated, and in support of this, ubiquitinated tau has essentially been found in aberrant aggregates, for example, incorporation bodies found in Pick’s or Parkinson’s diseases or in a few sorts of PHF found in AD [15]. Elevated amounts of ubiquitinated tau proteins were found in PHF and cerebrospinal fluid of AD patients. The occurrence of the conjugated type of ubiquitin in PHF has been studied by the marking of PHF with antibodies particularly for conjugated ubiquitin [114, 115]. It has been demonstrated that the ubiquitinated proteins in PHF are tau proteins and that the conjugation sites, including Lys254, 257, 311, and 317, are restricted in the microtubule-binding area. It is in all likelihood that abnormally phosphorylated full-length tau aggregates as PHF-tau, which is then stepped by step proteolyzed in its N-terminal part is thereafter ubiquitinated in its C-terminal domain [116]. Tau ubiquitination can happen in its C-terminus, at K254, K311, and K353, and was also found in the microtubule-binding domain [39].
TAU GLYCATION
Proteins with moderate turnover rates can be changed at lysine residues by non-enzymatic reactions including the addition of a sugar aldehyde or ketone assemble with the € -NH2-groups of the lysine [15]. The results of this reaction can experience irreversible changes to shape the propelled glycation end results that can bring about the cross-linking of the modified proteins [117]. Tau secluded from PHF is glycated [118], and this glycation may induce the collection of PHF into more complex aggregates NFTs [119]. In addition, it has been discovered that the presence of glycated tau into cultured cells might produce oxygen free radicals capable of exasperating neuronal functional capacity [15, 120].
TAU OXIDATION
Tau protein can be oxidized, prompting in vitro and in vivo PHF aggregation [39, 121]. Chronic oxidative stress has been said to promote tau phosphorylation in vitro [39, 122]. The presence of one or two cysteines at the tau isoforms missing or containing exon 10 has elevated the likelihood of tau forming dimers through the arrangement of intermolecular S-S bonds. In this condition, the oxidation of tau could bring about its variant aggregation [121]. In any case, within the sight of exon 10, the likelihood of intramolecular S-S bonds shaping exists. As of late, and in connection with tau oxidation, tyrosine nitration has been depicted in a tau particle or the arrangement of dityrosine cross-linking [15, 123]. Oxidation happens at C322 which is limited in the R3 area of MBD region of the tau molecule [39, 121]. To date, few studies have investigated this tau posttranslational alteration, and it is still uncertain whether tau oxidation at C322 is involved in tau lesions [39].
TAU ACETYLATION
The progress in the pathology of neurodegenerative tauopathies, for example, AD, Pick’s disease, FTD, and dynamic supranuclear palsy, basically gathered around phosphorylation and hyperphosphorylation of the tau protein. Tau acetylation is widely observed in human brains at the early and middle Braak stages, until before the formation and aggregation of NFTs, in this way supporting tau acetylation as an early occasion in disease pathology [124]. Tau acetylation at lysine residue 280 increases tau fibrillization and reduces tau-microtubule assembly in vitro. Tau acetylation was additionally observed to be strongly connected with tau hyperphosphorylation in AD, corticobasal degeneration, advanced supranuclear palsy, and in tau transgenic mouse models of tauopathies [125–127]. Recently it has been reported that acetylation of tau protein at lysine 280 may be a critical step in the molecular pathology of these neurodegenerative diseases preceding tau hyperphosphorylation [128], and tau acetylation has been opened to both disassemble tau of the microtubule and furthermore encourage tau aggregation [129]. Therefore, preventing tau acetylation could possibly serve as a novel target for stopping neurodegeneration before it fully starts. Salsalate and methylene blue have both been reported to diminish tau acetylation in pre-clinical models [130]. But still, the mechanism is not clear and therefore requires further studies. Acetylation of tau plays an important role in the process of neurodegeneration, and was found to be increased in AD brain [128]. It has recently been demonstrated that acetylation of tau is related to tauopathy and significantly influences tau function in neurons [131]. Acetylation inhibits tau degradation [42], prevents tau microtubule-binding [126, 132], and favors tau aggregation [126, 133]. Acetylated tau could repress the activity-dependent induction of postsynaptic AMPA-type glutamate receptors required for plasticity by meddling with the postsynaptic localization of KIBRA. Tau acetylation was additionally increased in cultured neurons treated with Aβ oligomers [42]. The acetylation of tau may promote toxicity by upgrading tau oligomerization. Tau oligomers are upregulated in AD brain and may assume a role in the primary stages of pathogenesis [134, 135]. Acetylation of tau could instigate pathogenesis by promoting the accumulation of phosphorylated or truncated species of tau that are known to be toxic [136].
TAU TRUNCATION
Truncation is another post-translational alteration that might have an etiological part in tau pathology. Many cell-based studies demonstrated that the truncation of either the C-terminal or both the N-and C-termini of tau impact its biochemical and functional properties and triggered its toxic transformation [137–141]. N-terminal truncation may likewise impact other tau properties, for example, polymerization [142] and cell localization [137]. Tau truncation in AD may alter glycation [118] and ubiquitination [39, 144]. Abnormal post-translational modifications, like truncation, are probably implicated in the pathological process [137]. Tau truncation has been characterized, for example, the cleavage of tau at the glutamic acid residue 391 [15, 145]. This modification could favor abnormal tau aggregation [145]. Aggregation of truncated tau protein at E391 and D421 have been recognized in AD brains and not in control brains [146]. In addition, this type of tau and tau truncated at D13 have been found in AD brains and corresponded with the disease advancement [39, 148]. These truncations increase the ability of tau to aggregate and participate in the execution of neuronal apoptosis [39, 149–151]. These truncated structures are particularly found in PHFs stipulating that tau truncation may participate in tau aggregation in AD brains [39, 152]. Tau cleavage is related to its dephosphorylation in cerebellar, hippocampal, and cortical neurons [153–155]. Numerous studies have confirmed that tau hyperphosphorylation happens before its cleavage [156, 157] and that tau cleavage happens before NFT formation [39]. The deamidation of tau at asparagine or glutamine sites have been depicted [158] and could likewise have a part to play in tau accumulation [15, 159]. A noteworthy advance onward in understanding the role of tau truncation is the identification of the exact cleavage sites of the few truncated tau pieces that are seen in AD brains, particularly those truncated at the N-terminus, which are fewer portrayed than those truncated at the C-terminus [137].
OTHER MODIFICATIONS OF TAU
It has been suggested that tau may develop cross-linkage through the enzymatic response tweaked by transglutaminase. Besides, modifications including a conformational modification of tau protein that could contain proline cis-trans isomerization likewise occur [15]. Tau is known to experience a few PTMs including ubiquitination, glycation, glycosylation, methylation, polyamination, prolyl-isomerization, acetylation, nitration, and truncation [125]. Tau prolyl-isomerization is completed by peptidyl-prolyl isomerase Pin1 (peptidyl-prolyl cis/trans isomerase NIMA reactivating-1; NIMA: not ever in mitosis gene A) [39]. Pin1 perceives phosphorylated serine or threonine took after by proline [160]. Tau nitration is the increase of nitrogen dioxide in tyrosine from an organic molecule [39]. Like hyperphosphorylation, tau nitration was proposed to be linked with tau accumulation [161]. Nitration incidentally happens on both soluble tau and insoluble [39, 163]. The response of tau polyamination by transglutaminases includes a glutamine (Q) as an acyl donor and a lysine (K) as an acyl acceptor [39]. It produces a g-glutamyl-e-lysine isopeptide bond and induces protein cross-linking [39, 164].
TAU AGGREGATIONS
It has been demonstrated already, in cell culture experiments, that extracellular tau aggregates could influence the transmission of tau to misfold from outside to within the cell where these aggregates might persuade fibrils formation of recombinant tau monomer [165]. Besides, a similar report displayed that tau aggregates were exchanged between co-cultured cells [166]. Another recent study depicted that brain extracts from NFT-bearing mouse brain infused in wild-type tau carrying mice induces seeding of tau fibrils in neurons [167]. In a mouse model with embryonic neurofibrillary pathology, the pool of free hyperphosphorylated tau was converted into tau aggregates that collect after some time [29, 168]. Tau protein forms unsolvable filamentous inclusions that are closely connected with nerve cell death in numerous neurodegenerative diseases. How neurons die in these tauopathies remains to be elucidated. Previous studies found an increased expression of MFGE8 in the frontal cortex of FTD cases for MAPT mutations (P301L and FTDP+3) or sporadic Pick’s disease but not in the cerebellum of P301L cases, demonstrating that the expression level of MFGE8 relies upon the presence of tau aggregates. Conservation of neurons, albeit with tau inclusions, protects a working network and gives a substrate for potential treatments, which by adjusting aggregation and toxicity may well have the capacity to prevent neuronal loss. Consequently, MFGE8 expressing is elevated in transgenic P301S-tau mouse brains for tau inclusions and in tau inclusion-rich brain areas of several human tauopathies, indicating shared mechanisms of the disease [169]. Preventing phagocytosis of living neurons will maintain them for treatments that inhibit tau aggregation and toxicity.
THE AGGREGATION OF TAU IN VITRO
The molecule of tau has long extensions of positively and negatively charged areas that are not favorable for intermolecular hydrophobic affiliation [84, 170]. The β-structure in monomeric tau is focused just on R2 (exon 10) and R3 (exon 11), which can self-aggregate into fibers and co-aggregate with heparin as a synthesized inducer [20, 171]. Evidence from in vitro studies has shown that self-aggregation of tau into fibers is limited to the N-terminal and the C-terminal, which rest over the MTBR and prevent the association between these sticky areas [20]. Abnormal phosphorylation of the N-terminal and the C-terminal extended areas may lead to a relaxed conformational change in the tau molecule that unclips the two extremes from the MTBR area leading to the self-association between these sticky areas resulting in the development of PHF/SF (Fig. 3) [20, 101].
Although in vitro tau polymers have been shown by laser scattering, spectroscopy, and electron microscopy [172–175], recent studies show that prefibrillar tau oligomers can occur in vitro by light-stimulated binding of tau together with benzophenone-4-maleimide (B4M) [20, 175]. These oligomers of tau were likewise seen in situ at an AD early stage when a monoclonal and specific antibody to these oligomeric elements of tau were evaluated in the brain of AD patients [20, 175]. Oligomeric types of tau protein are shown to have high cytotoxicity over soluble and high-level fibrillary aggregates, like NFTs [20, 176–178]. In transgenic mice that overexpress tau, the vast majority of the reported behavioral deficits arose at phases of the significant appearance of multimer tau aggregates before the NFTs development [20, 178].
TAU AGGREGATION INHIBITORS
To isolate potential tau aggregation inhibitors, a research utilized the tau develop K19 which comprises the three-repeat of the fetal human isoform htau23 for the high throughput test (Fig. 4). This protein aggregates overnight to PHFs with high reproducibility, which makes it suitable for a mechanized screening framework. The increase of three-repeat tau correlates strongly with the PHF structure and contains the hexapeptide VQIVYK, the transformation of which to a beta-sheet structure facilitates aggregation [179, 180].

Diagram of the full-length tau isoform htau40 and the repeat domain construct used in the PHF inhibition assay (construct K19, repeat domain with 3 repeats, R2 absent).
The propensity of phosphorylated tau to separate from microtubules and accumulate into fibrils rely upon its phosphorylation state [179, 181]. However, the screening was complete with unphosphorylated recombinant tau, since this protein polymerizes well into PHFs. The similarity of these filaments to those obtained from AFr brain tissues has been evaluated by electron microscopy and spectroscopic techniques, and the result revealed that the in vitro aggregation is a solid PHF [179, 182]. Beginning from 200,000 compounds, 77 of them were distinguished that could repress tau aggregation and to break up preformed PHFs. The chemical class of rhodanines was recognized as one of the dynamic hits. Utilizing 21 of the 77 compounds, an in-silico screen was performed from which the phenyl-thiazolyl-hydrazides were first recognized as a hit [179, 183].
TAU DISAGGREGATING AGENTS
The aggregation of neurofibrillary pathology in neurons is related to cognitive decrease and neurodegeneration [184]. A few classes of compounds, including those reported in recent screens [185–188], have now been recognized to hinder tau aggregation as well as dismantle existing fibrils in vitro. These include a few chemical classes such as phenothiazines, cyanine colors, amino thieno pyridazines, anthraquinones, phenyl-thiazolyl-hydrazides, and rhodanines [3].
MicroRNAs AND TAUOPATHIES
There are several tau protein isoforms that are developmentally regulated and these are the result of mRNA alternative splicing [21]. MicroRNAs (miRNAs) are shown to be strong regulators of neuronal gene expression that participate in both normal physiological and pathological processes [97]. Many folds studies have proved that miRNAs are implicated in various brain functions, inclusive development, cognition, and synaptic plasticity [189] whereby, from a pathologic point of view, alteration of miRNAs expression has been related to a few pathological processes, including neurodegeneration [190], thus indicating that stabled levels of miRNA are pivotal for appropriate tau functioning and neuronal survival. However, how aberrant miRNA levels might contribute to the improvement of neurodegenerative diseases remains to be clarified [191]. More interesting miRNAs have an incredible potential as biomarkers in the analysis, monitoring of disease progression and therapeutic reaction in AD and in other neurodegenerative diseases [190]. Some of them have been reliably recognized as AD-specific miRNAs and their targets likewise seem to be implicated in the etiopathogenesis of AD [190]. As the general posttranslational regulator of gene expression, miRNAs are possible modulators of kinase, phosphatase, and/or splicing factor expression [192]. Therefore, taken together all these make mRNA a potential target in therapy for tauopathies. Interestingly, the difference in splicing of the tau transcript has been reported in AD [193, 194]. Additionally, studies have showed that genetic background may affect the extent of cognitive weakness in tau mice. Therefore, moderate lowering of tau should be evaluated in therapeutic strategies for AD [195].
ABNORMAL TAU LOCALIZATION AND SYNAPTIC DISORDER IN AD
Protein tau is a major reason for synaptic and neuronal degeneration in AD. In its earliest clinical stage, AD characteristically produces a remarkably unadulterated impairment of memory. Substantial evidence revealed that this disorder starts with subtle alterations of hippocampal synaptic viability preceding the actual neuronal degeneration and that the synaptic dysfunction is caused by the aggregation of Aβ diffusible oligomer [196]. Normal tau is existent at both presynaptic and postsynaptic terminals of control human brains. In AD, tau progresses toward becoming hyperphosphorylated and misfolded at both presynaptic and postsynaptic terminals, and this abnormally post-translationally modified tau is abundantly present in synaptoneurosomal subfractions. Synaptic tau seems to be hyperphosphorylated and ubiquitinated and forms stable oligomers that resist SDS denaturation. These synaptic aggregated hyperphosphorylated tau oligomers of AD patients was found to be related to increased ubiquitinated substrates and increased proteasome components and consistent with the dysfunction of the ubiquitin-proteasome system. So, synaptic hyperphosphorylated tau oligomers might be an essential mediator of the proteotoxicity that altered synaptic structure and function in AD [9]. In AD, aggregates of neurofibrillary structures and lack of synapses in the neocortex and limbic system all significantly correlate with cognitive impairment [9]. Tangles are composed of misfolded hyperphosphorylated tau proteins, but it is still not clear how tau abnormality affects synaptic function. Tau dissociates from its normal axonal microtubule location and translocates into pre-and postsynaptic terminals. AD patients’ present the devastating signs of early synaptic degeneration before tau build up; however, the basic mechanism is still unclear [197]. Unbound tau may mislocalize into presynaptic terminals and induce synaptic dysfunction, causing a decrease in the number of synaptic vesicles in presynaptic terminals and neural connection loss. Likewise, tau may enter dendrites and postsynaptic compartments, and along these lines leads to postsynaptic dysfunction and promoting synaptic degeneration [111].
Recent studies using brains of transgenic models to summarize the role of both plaques and tangles have led to new insights into their role in degenerative processes and their effects on synaptic function and plasticity. Mapping the brain areas occupied by both plaques and tangles in AD brain could enable one to establish their relationship with important neurologic processes like learning and memory, synaptic plasticity, and brain inflammation [198]. The finding that intraneuronal Aβ is the trigger of cognitive alterations is consistent with the fact that it also induces synaptic dysfunction. Both cognitive and synaptic plasticity deficits manifest before the aggregation of these lesions and correlate best with intraneuronal Aβ [199]. However, it is not yet evident whether the buildup happens in pre- or post-synaptic cells. In general, these findings highlighted that both cognitive and synaptic impairments are early events in the AD pathogenesis. These results are in line with findings from another transgenic model where synaptic deficits were reported to occur independently of plaque accumulation [200].
INSOLUBLE TAU AGGREGATES AND SYNAPTIC FUNCTION
Synaptic plasticity is believed to be the pathway to learning and the acquisition of new memories. In AD, synaptic loss and the underlying cognitive deficits seem to be related to neurodegenerative processes triggered by both Aβ and tau. After death, human brain samples have been found to present signs of gliosis and oxidative stress in the region of amyloid plaques and NFTs that may further aggravate the synaptic changes [201, 202]. The overexpressing of Aβ in mice uncovered neurite degeneration after plaque formation [202, 203]. Furthermore, restorative therapeutic in AD mouse models suggested that plaques are dormant and an expansion in this metastable aggregate is not related to neurological deficiencies [204], but instead is beneficial since subjective capacity was enhanced in mice [202, 205]. Neurites encompassing plaques regularly contain phosphorylated tau aggregates [206]. Throughout AD, tau is hyperphosphorylated and assembled into fibrillary aggregates in the somatodendritic compartment [202, 207].
PROTEOLYSIS OF ABNORMALLY HYPERPHOSPHORYLATED TAU
In situ hybridization experiments have uncovered no critical changes in the expression of tau mRNA in AD brain [208]. Therefore, the increase in tau protein level seen in AD brain is presumably generally because of a decrease in its turnover caused by the hyperphosphorylation [48, 210]. The AD unusual hyperphosphorylation of tau makes tau reluctant to both calcium-activated dependent proteases, calpains, and its degradation through the ubiquitin-proteasome pathway. Not at all like typical tau, the AD hyperphosphorylated tau is resistant to proteolysis by calpains [48]. The ubiquitination of the abnormally hyperphosphorylated tau in NFTs evidently does not cause its clearance by absorption in the proteasome. This could somewhat be owing to a quicker rate of accumulation of the ubiquitinated phospho-tau than the capacity of the proteasomes of the degenerating neurons to process it [12]. Suppression of the proteasome by its inhibitor, lactacystin, increase the accumulation of both typical and hyperphosphorylated tau in rats [211].
CONCLUSIONS
Although the pathogenic idea of each species of protein accumulation has been a dubious issue for a long time, it is presently progressively acknowledged that abnormal types of tau protein are specifically associated with the onset of the neurodegenerative process. Several pathogenic events may contribute, either specifically or by implication, to tau hyperphosphorylation and aggregation. However, it is clear that many pathogenic events might directly or indirectly contribute to tau hyperphosphorylation and/or aggregation and has shed light on potential mechanisms of tau-induced neuronal cell death and dysfunction. There has been considerable knowledge of the sequence of events that lead to the abnormal phosphorylation and accumulation of tau in AD and other tau-related diseases. In pathological circumstances, tau might be hyperphosphorylated and accumulated in a distorted way. The outcome of these modifications is neuronal damage leading neurological disorders, with dementias, which are commonly known as tauopathies. Brains specimens from AD patients show markers whose position and components are reflective of the disease. A standout amongst the most established markers is phosphorylated and aggregated tau protein. This type of tau can fill in as a manual for the beginnings of the pathology. The advancement of aggregation inhibitors will be linked to the comprehension of their coupling mode at the molecular level, and the capacity to incorporate vital components of pharmacology at an early stage might attenuate the in vivo toxicity, brain permeability, and plasma stability. We believe that solving the genesis from conformational changes of tau protein confirmed by these posttranslational modifications and its role in fibrillization in disease are leading achievements in tau-directed therapies. Moreover, careful determination of modified tau protein on the cerebrospinal fluid and other body fluids may supply a better expectation to foresee the onset and evolution of dementia.
Footnotes
ACKNOWLEDGMENTS
This work was supported in part by the National Natural Science Foundation of China (81771150, 81761138043, 91632114, and 31571039 to L.-Q.Z.), and National Program for Support of Top-Notch Young Professionals, and Academic Frontier Youth Team of HUST to L.-Q.Z. and Program for Changjiang Scholars and Innovative Research Team in University (IRT13016).