
Abstract
Alzheimer’s disease (AD) is increasingly viewed as a neurological disease accompanied by a systemic disorder. Accumulating evidence supports that angiotensin II and angiotensin 1-7 exert opposite effects on various organs including the brain. However, the interaction between angiotensin II and angiotensin 1-7 in AD remains to be defined. The present study was undertaken to examine the interaction between these peptides in AD. 5XFAD mice, a useful model of AD, were separated into three groups: 1) saline-infused, 2) angiotensin II-infused, and 3) angiotensin II-infused and angiotensin 1-7-co-infused. These peptides were systemically given to 5XFAD mice via osmotic minipump for 4 weeks. Systemic angiotensin II infusion for 4 weeks induced significant hypertension in both wild-type and 5XFAD mice. Angiotensin II induced cognitive abnormality in 5XFAD mice as estimated by the Morris water maze test and the nest building test, and this effect was associated with cerebral blood flow reduction, cortical arterial amyloid-β deposition, hippocampal inflammation, and neuron loss in 5XFAD mice. In addition, angiotensin II infusion led to gastrocnemius muscle atrophy in 5XFAD mice. Co-infusion of angiotensin 1-7 prevented the above mentioned detrimental effects of angiotensin II in the brain and gastrocnemius muscle in 5XFAD mice, without significant influence on blood pressure. The left ventricular hypertrophic response to angiotensin II was attenuated in 5XFAD mice compared with wild-type mice, which was not significantly altered by co-administration of angiotensin 1-7. Our results show that angiotensin 1-7 counteracts angiotensin II-induced cognitive impairment, brain injury, and skeletal muscle injury in AD mice.
Keywords
INTRODUCTION
Alzheimer’s disease (AD), the leading cause of dementia, has traditionally been considered a neurodegenerative disease, and is characterized by intraneuronal tau hyperphosphorylation and extracellular amyloid-β (Aβ) depositions [1]. Contrary to this view, previous studies have shown that AD patients also display a peripheral organ pathology characterized by cardiac dysfunction, low body weight in later life, sensory-motor impairment, and skeletal muscle atrophy [2–5]. Hence, AD is increasingly viewed as a systemic disease [6]. This might be explained by the ubiquitous expression of Aβ and its role in various cellular pathogenic processes [2, 7–9].
There is a general understanding that AD develops slowly from a preclinical stage to the prodromal stage and finally the dementia stage [10, 11]. The identification of patients at early stages provides the opportunity of medical treatment and lifestyle modification, and thus to delay or even prevent cognitive decline [12]. Although brain pathology remains in the focus of AD research, the systemic abnormalities in AD remain largely undefined, especially in the early stage.
The renin-angiotensin system (RAS) is critical for the maintenance of blood pressure and vascular homeostasis, mainly through angiotensin (Ang) II, III, and 1-7 [13]. Its effects have been well characterized in both the brain and the periphery [14]. Emerging evidence indicates the involvement of the RAS in AD [13, 15]. Angiotensin-converting enzyme (ACE)/Ang II/Ang II type 1 receptor axis is linked to the pathogenesis of AD. We and other group of investigators have previously reported that intracerebroventricular (ICV) Ang II infusion in five familial Alzheimer’s disease mutations (5XFAD mice) induces cerebrovascular Aβ deposition and cognitive impairment [16], ICV Ang II infusion also causes cerebral tau hyperphosphorylation, and cognitive impairment in normal rat [17]. Clinical and epidemiological studies supported that patients treated with ACE inhibitors or angiotensin receptor blockers had lower risk of developing cognitive impairment [18–21]. Further, the ACE2/Ang 1-7/Mas axis, which counteracts the effects of Ang II, is suggested to be involved in the improvement of cognitive function. ICV administration of Ang 1-7 ameliorates cognitive impairment in rats with chronic cerebral hypoperfusion [22] and rats with streptozotocin-induced diabetes [23]. Furthermore, we have recently shown that ICV administration of Ang 1-7 improved cognitive impairment in 17-month-old 5XFAD mice, a useful model of AD [24], thereby suggesting the critical role of brain Ang 1-7 in AD pathology. However, it is unclear whether Ang 1-7 can counteract Ang II-induced cognitive impairment, brain injury, and peripheral organ injuries such as the heart and skeletal muscle in AD.
In the current study, using 6-month-old 5XFAD mice, we investigated the potential role of systemic Ang 1-7 in Ang II-induced brain AD pathology and cardiac and skeletal muscle injuries in AD mice.
MATERIALS AND METHODS
Animals
All experimental procedures were approved by the Kumamoto University Animal Care and Use Committee. The generation of 5XFAD mice has been described previously [25]. 5XFAD mice harbor human amyloid precursor protein (APP695) (Swedish: K670 N, M671 L; Florida: I716 V; London: V717I) and human presenilin-1 (M146 L and L286 V) mutations, with neural-specific elements of the mouse Thy-1 promoter [26]. The 5XFAD strain (C57BL/6J genetic background) was maintained by crossing heterozygous transgenic mice with C57BL/6J mice (SLC Japan, Shizuoka, Japan). Mice were housed in a temperature-controlled (20±2°C) and humidity-controlled (60%) room under a 12-h light/dark cycle (8 : 00/20 : 00), with free access to food and water.
Experimental model and drug administration
Drugs treatment was started at the age of 6 months and was continued for 4 weeks. Six-month-old male wild-type (WT) mice were randomly assigned to two groups: 1) control group receiving 0.9% NaCl (WT-C, n = 7) and 2) group receiving Ang II (WT-AII, n = 8). The 5XFAD mice with the same age were randomly assigned to three groups: 1) control group receiving 0.9% NaCl (5X-C, n = 8), 2) group receiving Ang II (5X-AII, n = 8), and 3) group receiving Ang II and Ang 1-7 (5X-AII + A(1-7), n = 8). Ang II and Ang 1-7 were systemically administered via subcutaneously implanted osmotic pump (Alzet 1004, Durect Corp., Cupertino, CA, USA) for 4 weeks. The dose of Ang II was 1,000 ng/kg/min because this dose of Ang II has been most generally used to investigate the detrimental effects of Ang II in vivo and is established to significantly cause hypertension and organ injury [27–32]. The dose of Ang 1-7 was 400 ng/kg/min, since such a dose of Ang 1-7 is also generally used dose according to previous reports [29, 32–34]. Human Ang II (Code: 4001) and Ang 1-7 (Code: 4332) were purchased from Peptide Institute Inc. (Osaka, Japan). After priming for 24 h at 37°C, osmotic pumps were subcutaneously implanted in the intrascapular region in a sterile manner and the incision was closed using sutures. For analgesia, meloxicam (1 mg/kg weight) was subcutaneously administered immediately after the surgical procedure.
Measurement of body weight and blood pressure
Body weight was measured every week, and blood pressure was recorded by a noninvasive tail-cuff system (Softron BP-98A, Tokyo, Japan) at 0, 2, and 4 weeks after pump implantation, as described previously [16].
Rotarod test
The rotarod test was applied to evaluate motor function and sensorimotor coordination [16]. On the testing days, mice were first trained to walk at a rate of 4 rotations per minute (rpm) for 1 min on the horizontal drum (MK-630B, Muromachi Kikai Co., LTD, Tokyo, Japan). Then, the mice were placed to walk on the accelerating spindle (4 to 40 rpm) for 5 min, and the latency to fall was recorded as the time mice gripped around for two successive revolutions or fell off the spindle. The latency of three trails of the test was averaged for each mouse.
Nest building behavior
Nest building was used to evaluate general home cage behavior, according to a previous method [35] with a slight modification. Briefly, mice were housed individually and were supplied pressed cotton squares (2 pieces, total 3.0 g per cage) at 7 pm, then nest building results were scored at 9 am on the next day as following: 1 = cotton was not noticeably touched (≥90% intact); 2 = cotton was partially torn (50% –90% remaining intact); 3 = most cotton was torn (10% –50% remaining intact) but a nest was not obviously made; 3.5 = most cotton was torn (10% –50% remaining intact) and identifiable but flat nest was made; 4 = most of all cotton was torn (≤10% remaining intact) with flat nest; 4.5 = most cotton was torn (10% –50% remaining intact) and bowl nest was made; 5 = most of all cotton was torn (≤10% remaining intact) with bowl nest.
Morris water maze test
Morris water maze test was performed to assess spatial learning and memory, as described [16]. The movement of mice was video-recorded and was analyzed by the software (Muromachi Kikai, Tokyo, Japan). After training, hidden test was performed for 4 days (day 1 to day 4), with 4 sessions per day. And the time to find the platform was recorded as the escape latency. The submerged platform was removed on day 5, and memory retrieval was examined by probe test for 100 s. The number of times the mice crossed the platform location was measured.
Measurement of cerebral blood flow
Cerebral blood flow (CBF) was measured with a laser speckle blood flow imager (Omega Zone, Omegawave, Tokyo, Japan), as described [16]. Briefly, mice were thermostatically controlled at 37°C by a warming pad. Then the skull was exposed by a midline scalp incision under 2% isoflurane. The bilateral cortical surface was diffusely illuminated by 780 nm semiconductor laser light. The results of 10 measurements in each mouse were averaged in a blinded manner.
Histopathology and immunohistochemistry
At the end of the experiments, all mice were sacrificed and tissue harvesting was performed as described previously [36]. Briefly, mice were weighed and placed under anesthesia. Blood was collected by right ventricular puncture, and serum was separated and stored at –80°C. The heart was perfused first with sterile cold phosphate buffered saline, and rapidly dissected, fixed in 4% paraformaldehyde and embedded in paraffin. Brains were extracted and separated at the point of bregma, and the caudal side of each brain was embedded into Tissue-Tek O.C.T. compound (Sakura Finetek, Tokyo, Japan) and immediately frozen on dry ice, then stored at –80°C. An 8-μm-thick section was made at 1.43 to 2.43 mm caudally from the bregma for histological evaluations. Further, the gastrocnemius muscle of each mouse was weighed and frozen in Tissue-Tek O.C.T. compound or fixed in 4% paraformaldehyde and embedded in paraffin for the histological evaluations.
Measurement of tissue inflammation
To evaluate brain and gastrocnemius muscle macrophage infiltration, 8-μm-thick frozen sections from those organs were incubated overnight with CD68 antibody (1 : 500, rat anti-mouse CD68; AbD Serotec, Oxford, UK) followed by anti-rat secondary antibody (Biosource, Camarillo, CA, USA), as described [16]. To evaluate cardiac interstitial macrophage infiltration, 4-μm-thick paraffin sections from left ventricle (LV) of each mouse were incubated overnight with rabbit polyclonal anti-ionized calcium-binding adaptor molecular 1 (Iba1) antibody (1 : 200, Catalog No. 019-19741; Wako Pure Chemical Ind., Ltd, Osaka, Japan) followed by horseradish peroxidase labelled polymer anti-rabbit secondary antibody, and visualized with 3, 3-diaminobenzidine (Dako Cytomation, Glostrup, Denmark). To quantitate brain macrophage/microglia, the percentage of CD68-positive cell area was evaluated in bilateral hippocampal CA1 regions for each mouse at×200 magnification. In LV, 10 fields per section were taken at×200 magnification, and Iba1-positive cell number per field area (mm2) was counted. In gastrocnemius muscle, 3 fields per section were randomly taken at×200 magnification, and CD68 positive cell number per field area (mm2) was counted in each mouse.
Nissl staining
For the hippocampal neuron counting, frozen brain sections were subjected to Nissl staining. The number of neurons in bilateral hippocampal CA1 regions in each slice at×200 magnification was counted and compared between each group [37].
Immunohistochemical analysis of brain Aβ deposition and cortical cerebral amyloid angiopathy
To measure Aβ1-40 and Aβ1-42 levels, brain sections (8-μm thickness) were incubated for 5 min in 90% formic acid followed by 30 min in 0.3% H2O2/methanol, and then incubated overnight with Aβ1-40 or Aβ1-42 antibody (1 : 200, Code No. 18580 and Code No. 18582, respectively; Immuno-Biological Laboratories Co., Ltd, Gunma, Japan) at 4°C. Then the tissues were incubated for 1 h with horseradish peroxidase-conjugated anti-rabbit IgG secondary antibody at room temperature, visualizing using 3, 3′-diaminobenzidine (Dako Cytomation, Glostrup, Denmark), as described [16]. The accumulation of Aβ1-40 and Aβ1-42 in bilateral somatosensory cortex and hippocampal CA1 regions at×200 magnification were evaluated by WinRoof version 5.8 (Mitani Corporation, Fukui, Japan) and were expressed as the mean percentage of the positive area per interest region.
To measure cerebral amyloid angiopathy (CAA), brain slices were incubated with an antibody for the basement membrane marker collagen IV (1 : 200, rabbit; Abcam, Cambridge, MA, USA) with Alexa Fluro 568 goat anti-rabbit secondary IgG antibody (1 : 200; Invitrogen, Carlsbad, CA, USA), immersed in a thioflavin S solution (0.05% in 50% ethanol) (Sigma-Aldrich, St. Louis, MO) [16]. To quantify the thioflavin S-positive cortical middle cerebral arteries, photos were taken on bilateral cortical surface (total of 4 cortical arteries) at×400 magnification. The percentage of thioflavin S co-localized with collagen IV was measured by WinRoof version 5.8 (Mitani Corporation, Fukui, Japan).
Measurement of cardiac cell size and fibrosis
To measure cardiomyocyte size, 4-μm-thick paraffin sections from LV were prepared, and the cardiomyocyte cell membranes were stained with fluorescein-tagged wheat germ agglutinin (FITC-WGA, Sigma-Aldrich, St. Louis, MO, USA) and the nuclei with 4′,6′-diamidino-2-phenylindole (DAPI, Vector Laboratories, Burlingame, CA, USA) [38]. The cell size of cardiomyocytes in situ was determined by Image J (National Institutes of Health, Bethesda, MD, USA). The minimal Feret’s diameter of more than 100 cardiomyocytes was measured [39].
To measure LV interstitial fibrosis, Sirius Red F3BA staining (0.5% wt/vol in saturated aqueous picric acid; Aldrich Chemical Company, St. Louis, MO, USA) was performed. Cardiac fibrosis was quantified by examining 10 fields per section by WinRoof version 5.8 (Mitani Corporation, Fukui, Japan), as described [16].
LV Aβ enzyme-linked immunosorbent assay
The levels of LV Aβ were measured using human Aβ1-40 and Aβ1-42 enzyme-linked immunosorbent assay (ELISA) kits (Code No. 27713 and Code No. 27711, respectively; Immuno-Biological Laboratories Co., Ltd, Gunma, Japan), as described previously [24]. Briefly, fresh frozen hearts were homogenized in 10 volumes (w/v) of 1% Triton X-100 in Tris-buffered saline (25 mM Tris and 137 mM NaCl, pH 7.6) with protease inhibitors (protease inhibitor cocktail, 1 tablet in 50 ml solution; Roche), and then centrifuged at 100,000 g for 60 min at 4°C. The insoluble fractions of Aβ peptides were collected and stored at –80°C as described previously [40]. The insoluble fraction samples in the heart were diluted in the ratio of 1 : 4 in the ELISA diluent buffer. The amounts of Aβ1-40 and Aβ1-42 were measured in duplicate following the manufacturer’s protocol and were normalized for the weight of the heart.
Measurement of gastrocnemius muscle fiber size
Four-μm-thick gastrocnemius muscle paraffin slices were stained with hematoxylin-eosin. The minimal Feret’s diameter was used for assessment of muscle fiber size [39]. In individual mice, at least 100 gastrocnemius muscle fibers per section were measured by Image J (National Institutes of Health, Bethesda, MD, USA).
Statistical analysis
Data are presented as mean±standard error (SE). Graphpad Prism version 7.00 (Graphpad Software Inc., San Diego, CA, USA) and Statcel (OMS Publication, Saitama, Japan) were used for statistical analysis. One-way or two-way ANOVA followed by Tukey post hoc test and the Kruskal-Wallis test followed by Steel-Dwass post-hoc test were applied as appropriate. A two-sided p-value <0.05 was considered significant. The detail of statistical analysis was described in each figure and table legends.
RESULTS
Blood pressure and body weight
Two mice in the WT-AII group, one mouse in 5X-C group and one mouse in 5X-AII + A(1-7) group died before sacrifice, and were thus excluded from the analysis. At the beginning of the study, there were no significant differences in systolic blood pressure (SBP) and body weight between groups. After 4 weeks of treatment, SBP increased significantly in Ang II-infused groups (165.5±4.12 mmHg in WT-AII mice, p < 0.05; 161.0±7.88 mmHg in 5X-AII mice, p < 0.05; and 174.5±5.03 mmHg in 5X-AII + A(1-7) mice, p < 0.05) in comparison with the respective control group in both strain (Table 1). Ang II-induced hypertension was not altered by Ang 1-7 infusion after 4 weeks of treatment. Blood pressure remained at its higher level in both genotypes until the end of experiment. After treatment, body weight was lower in 5X-AII (p < 0.01) and 5X-AII + A(1-7) (p < 0.01) groups compared with WT-C groups, as shown in Table 1. In addition, there were no significant differences in heart rate and tibia length (TL) between groups (Table 1).
Heart rate, blood pressure, body weight, and tibia length in wild-type and 5XFAD mice
WT-C, wild-type mice with 0.9% NaCl infusion; WT-AII, wild-type mice with angiotensin II infusion; 5X-C, 5XFAD mice with 0.9% NaCl infusion; 5X-AII, 5XFAD mice with angiotensin II infusion; 5X-AII + A(1-7), 5XFAD mice with angiotensin II + angiotensin 1-7 infusion. Data are presented as mean±standard error. Statistical analysis was performed by the one-way ANOVA followed by Tukey’s multiple comparisons or the Kruskal-Wallis test followed by Steel-Dwass post-hoc test for multiple comparisons. †p < 0.05, *p < 0.01 compared with the WT-C group; §p < 0.05 compared with 5X-C group.
Morris water maze test, nest building test, and cerebral blood flow
As shown by the hidden test in Figure 1A, the escape latency was not significantly different between WT-C and WT-AII groups. In contrast, 5X-AII group exhibited longer escape latency in the hidden platform test compared with WT-C, WT-AII and 5X-C groups (p < 0.01 for all). Such cognitive impairment shown by Ang II-infusion in 5XFAD group was confirmed by the probe test. In this phase, a significant difference in the number of platform crossings was found between 5X-AII and WT-C groups (p < 0.05) (Fig. 1B). No significant differences were obtained for the comparison of WT-C and WT-AII groups (Fig. 1B).
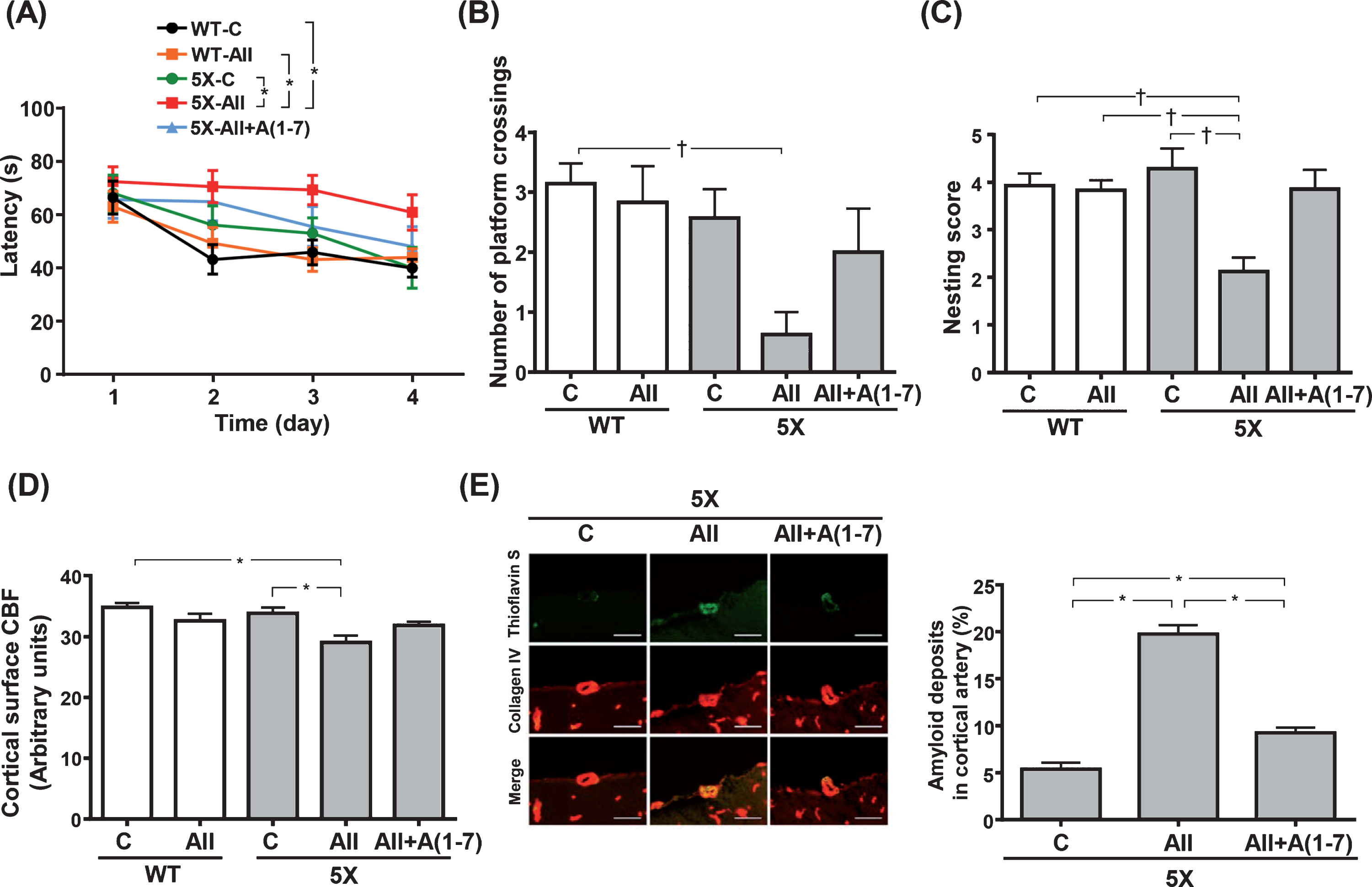
Escape latency of the hidden platform test (A), number of platform crossings of the probe test (B), nest building score (C), resting cerebral blood flow (D) and quantification of cortical artery amyloid deposition (E). E) Left panels show representative images of thioflavin S (green) and collage IV (red) double immunostaining in the cortical branch of middle cerebral artery and right panel shows quantification of percent area of thioflavin S colocalized with collage IV. Data are presented as mean±standard error. n = 7 in WT-C, n = 6 in WT-AII, n = 7 in 5X-C, n = 8 in 5X-AII and n = 7 in 5X-AII + A(1-7). In A, statistical analysis was performed by two-way ANOVA followed by Tukey post hoc test between each group. In B and C, statistical analysis was conducted by the Kruskal-Wallis test followed by Steel-Dwass post-hoc test between each group. In D and E, statistical analysis was done by the one-way ANOVA followed by Tukey’s multiple comparison post hoc test. †p < 0.05, *p < 0.01. Scale bar = 50μm. WT-C, wild-type mice with 0.9% NaCl infusion; WT-AII, wild-type mice with angiotensin II infusion; 5X-C, 5XFAD mice with 0.9% NaCl infusion; 5X-AII, 5XFAD mice with angiotensin II infusion; 5X-AII + A(1-7), 5XFAD mice with angiotensin II + angiotensin 1-7 infusion; CBF, cerebral blood flow.
We also tested the performance of the mice in the nest building test. We found no differences in the nesting score between WT-C and WT-AII groups (Fig. 1 C). However, 5X-AII group had a lower nesting score compared with WT-C, WT-AII and 5X-C groups (p < 0.05 for all) (Fig. 1C).
Moreover, no significant differences were found for cortical surface CBF between WT-C and WT-AII groups (Fig. 1D). On the other hand, 5X-AII group showed significantly lower CBF compared with both WT-C and 5X-C groups (p < 0.01 for both) (Fig. 1D).
Cerebrovascular and cerebral parenchymal amyloid deposition in 5XFAD mice
As shown by collagen IV and thioflavin S double immunostaining, the degree of cerebrovascular Aβ deposition was significantly greater in both 5X-AII and 5X-AII + A(1-7) groups compared with 5X-C group (p < 0.01 for both) (Fig. 1E). However, Ang 1-7 coinfusion significantly alleviated Ang II-induced increases in cerebrovascular Aβ deposition in 5XFAD mice (p < 0.01) (Fig. 1E).
The detection of Aβ1-40 and Aβ1-42 deposition in both cortical and hippocampal area by immunostaining was also conducted. As shown in Figure 2, quantitative analysis demonstrated that there were no significant differences among groups for percentage area of Aβ1-40 or Aβ1-42 deposits in hippocampus (Fig. 2B, C) or cortex (Fig. 2D, E).
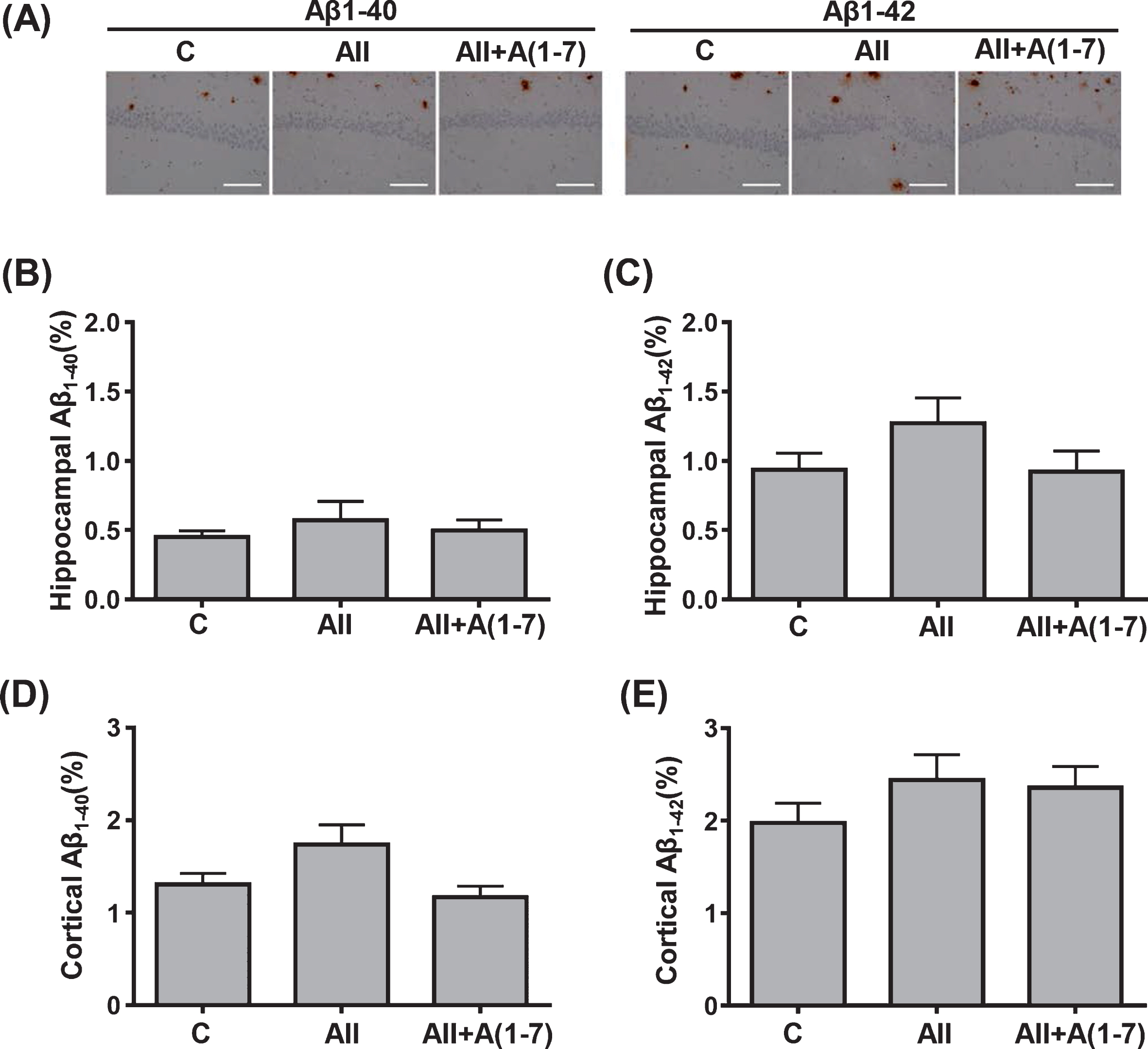
Quantification of cerebral parenchymal Aβ1-40 and Aβ1-42 deposition in hippocampus (B and C) and cortex (D and E) in transgenic 5XFAD mice. A) Representative images of hippocampal CA1 sections with Aβ1-40 and Aβ1-42 immunostaining. Data are presented as mean±standard error. n = 7 in 5X-C, n = 8 in 5X-AII and n = 7 in 5X-AII + A(1-7). In B, D and E, statistical analysis was conducted by the Kruskal-Wallis test followed by Steel-Dwass post-hoc test between each group. In C, statistical analysis was done by the one-way ANOVA followed by Tukey’s multiple comparison post hoc test. Scale bar = 100μm. C, 5XFAD mice with 0.9% NaCl infusion; AII, 5XFAD mice with angiotensin II infusion; AII + A(1-7), 5XFAD mice with angiotensin II + angiotensin 1-7 infusion; Aβ, amyloid-β.
Hippocampal macrophage/microglia and neuronal cell
The percentage of hippocampal CD68-positive cell area was significantly greater in 5X-AII group, compared with WT-C and 5X-C groups (p < 0.05 for both) (Fig. 3A). However, no significant difference was noted between 5X-AII+A1-7 group and 5X-C group. There was also a significant decrease in neuronal cell number in hippocampal CA1 region in 5X-AII group than in WT-C (p < 0.01) and 5X-C groups (p < 0.05), while neuronal cell numbers did not differ between 5X-C and 5X-AII+A(1-7) groups (Fig. 3B).

Hippocampal CA1 region inflammation (percentage CD68-postitive cell area) (A) and neuronal cell number (B). A) Upper panels show representative images of CD68-stained hippocampal sections. B) Upper panels show representative images of Nissl-stained hippocampal sections. Data are presented as mean±standard error. n = 7 in WT-C, n = 6 in WT-AII, n = 7 in 5X-C, n = 8 in 5X-AII and n = 7 in 5X-AII + A(1-7). In A, statistical analysis was conducted by the Kruskal-Wallis test followed by Steel-Dwass post-hoc test between each group. In B, statistical analysis was done by the one-way ANOVA followed by Tukey’s multiple comparison post hoc test. †p < 0.05, *p < 0.01. Scale bar = 100μm. Abbreviations used are the same as in Fig. 1.
Cerebral IgG extravasation
As shown in Supplementary Figure 1, the degree of cerebral IgG extravasation in WT and 5XFAD mice tended to be greater in respective Ang II group, but the differences did not reach statistical significance.
LV hypertrophy and remodeling
As shown in Figure 4A and 4B, Ang II infusion for 4 weeks significantly induced LV hypertrophy in WT group as assessed by the calculated LV/TL (P < 0.05) (Fig. 4A) and cardiomyocyte minimal Feret’s diameter (p < 0.05) (Fig. 4B). In contrast, the LV hypertrophic response to Ang II infusion was significantly attenuated in 5XFAD mice (Fig. 4A, B).
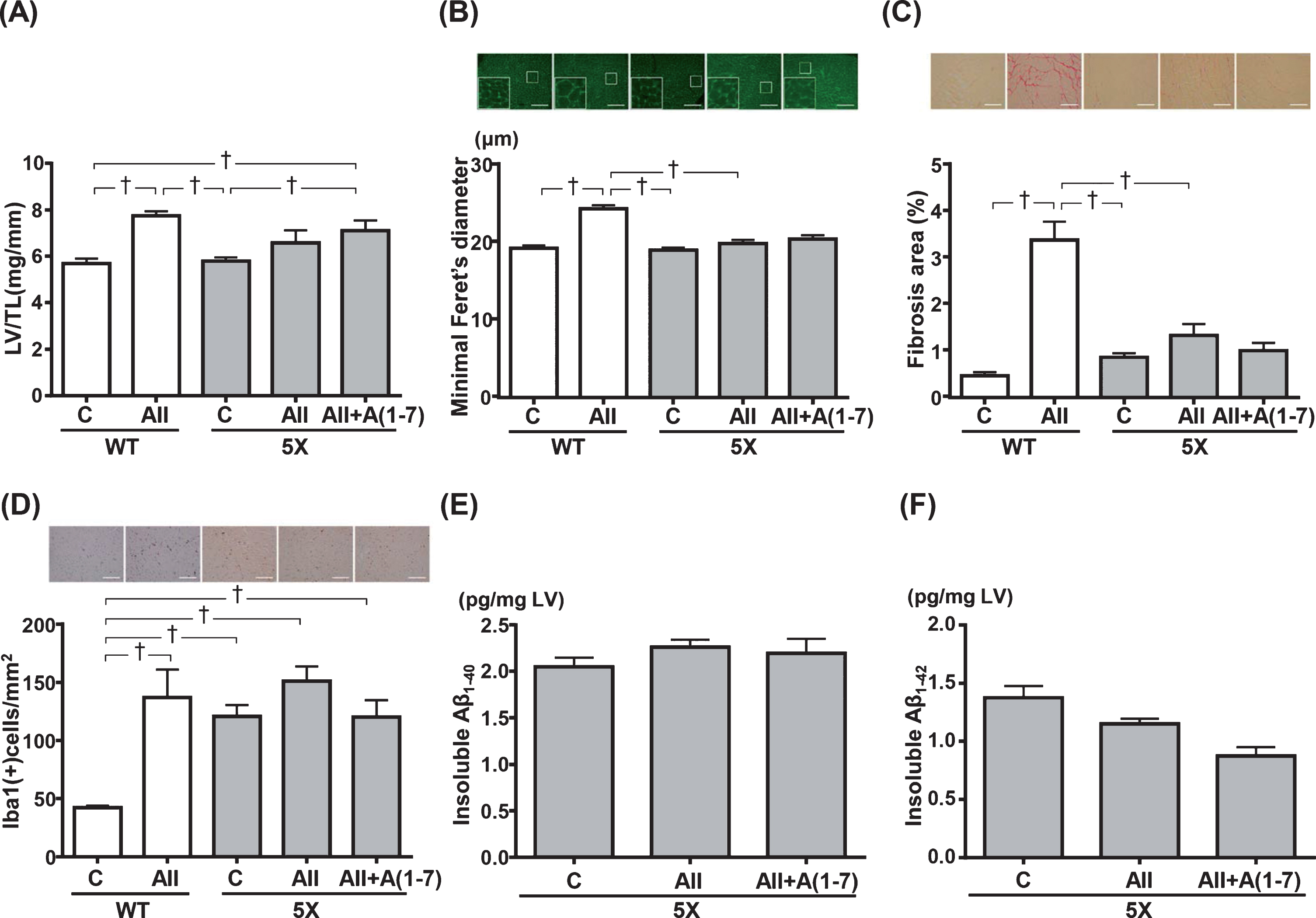
Left ventricular (LV) weight (A), LV minimal Feret’s diameter of cardiomyocyte cell size (B), LV fibrosis (C), LV Iba-1-positive cell numbers (D), quantification of LV insoluble Aβ1-40 (E) and Aβ1-42 (F) levels. B) Upper panels show representative immunofluorescence-stained images. C) Upper panels show representative Sirius red-stained images. D) Upper panels show representative Iba-1 staining images. Data are presented as mean±standard error. n = 7 in WT-C, n = 6 in WT-AII, n = 7 in 5X-C, n = 8 in 5X-AII and n = 7 in 5X-AII + A(1-7). In A, B, C, D, and F, statistical analysis was conducted by the Kruskal-Wallis test followed by Steel-Dwass post-hoc test between each group. In E, statistical analysis was done by the one-way ANOVA followed by Tukey’s multiple comparison post hoc test. †p < 0.05. Scale bar = 100μm. LV/TL, left ventricular/tibia length; Aβ, amyloid-β. Other abbreviations used are the same as in Fig.1.
Sirius Red-stained LV sections were examined to assess fibrosis. As shown in Figure 4 C, WT-AII group exhibited greater LV interstitial fibrosis than WT-C, 5X-C and 5X-AII groups (p < 0.05 for all). LV Iba1-positive cell numbers, as marker of inflammation, was quantified. As shown in Figure 4D, chronic Ang II infusion triggered a significant increase in the number of Iba1 positive cells in WT-AII group compared with WT-C group (p < 0.05). Further, 5X-C, 5X-AII and 5X-AII + A(1-7) groups showed significant increases in the number of Iba1 positive cells compared with WT-C group (p < 0.05 for all) (Fig. 4D). However, there was no significant difference in LV Iba1-positive cell numbers among the 3 groups of 5XFAD mice (Fig. 4D).
Cardiac Aβ deposition in 5XFAD mice
In our preliminary study, we found that it is difficult to identify and quantify intracellular or extracellular Aβ deposits by traditional immunohistochemical staining in both the LV and skeletal muscle of WT and 5XFAD mice (data not shown). Thus, ELISA study was conducted. As shown in Figure 4E and 4F, the presence of both insoluble Aβ1-40 and Aβ1-42 in the LV of 5XFAD mice were confirmed and quantified. The insoluble Aβ1-40 and Aβ1-42 levels in 5XFAD mice were not significantly changed by Ang II or Ang II + Ang 1-7 administration.
Rotarod test, gastrocnemius muscle weight, muscle minimal Feret’s diameter, and inflammation
As shown by rotarod test in Fig. 5A, there were no significant differences in latency to fall between WT-C and WT-AII groups. On the other hand, 5X-AII group showed a significant decrease in latency to fall compared with WT-C (p < 0.01) and 5X-C groups (p < 0.05), while latency to fall did not differ between 5X-C and 5X-AII+A(1-7) group (Fig. 5A). Gastrocnemius muscle weight corrected for TL was significantly decreased in 5X-AII group compared with WT-C (p < 0.01) and 5X-C groups (p < 0.05), but its weight was similar between 5X-C and 5X-AII+A(1-7) groups (Fig. 5B). On the other hand, there were no significant differences in muscle minimal Feret’s diameter between groups (Fig. 5 C). In addition, Ang II increased the number of CD68-positive cells in both WT (p < 0.05) and 5X group (p < 0.01) in comparison with the respective control groups in both strains (Fig. 5D). Coinfusion with Ang 1-7 ameliorated Ang II-induced increase in CD68-positive cell numbers in 5XFAD group (p < 0.05) (Fig. 5D).
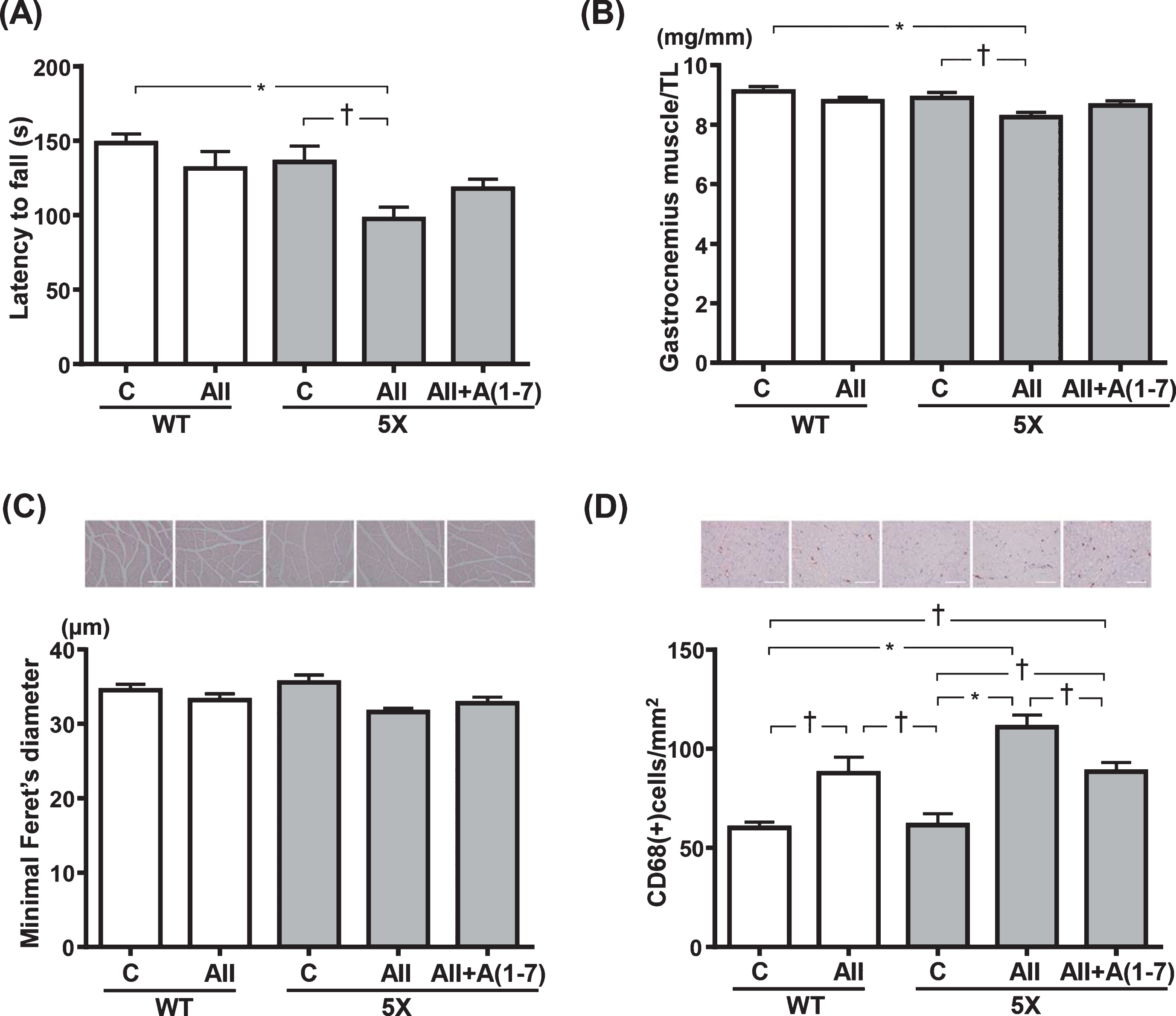
Latency to fall in rotarod test (A), gastrocnemius muscle weight (B), minimal Feret’s diameter of gastrocnemius muscle cell size (C), and CD68-positive cell numbers (D). C) Upper panels show representative images of hematoxylin-eosin stained of gastrocnemius muscle sections. D) Upper panels show representative images of CD68-stained gastrocnemius muscle sections. Data are presented as mean±standard error. n = 7 in WT-C, n = 6 in WT-AII, n = 7 in 5X-C, n = 8 in 5X-AII and n = 7 in 5X-AII + A(1-7). In C, statistical analysis was conducted by the Kruskal-Wallis test followed by Steel-Dwass post-hoc test between each group. In A, B, and D, statistical analysis was done by the one-way ANOVA followed by Tukey’s multiple comparison post hoc test. †p < 0.05, *p < 0.01. Scale bar = 100μm. TL, tibia length. Other abbreviations used are the same as in Fig. 1.
Effect of isoproterenol infusion on LV hypertrophy of 5XFAD mice compared with WT mice
As shown in Supplementary Figure 2, chronic isoproterenol infusion in WT mice significantly increased LV/TL (p < 0.01) (Supplementary Figure 2A) and cardiomyocyte minimal Feret’s diameter (p < 0.05) (Supplementary Figure 2B). However, isoproterenol-induced cardiac hypertrophic response was less in 5XFAD than in WT mice (Supplementary Figure 2).
DISCUSSION
The major findings of the current study were that 1) chronic Ang 1-7 infusion protected against Ang II-induced cognitive dysfunction and skeletal muscle atrophy in 5XFAD mice, a mouse model of AD; 2) 5XFAD mice exhibited Ang II-induced LV hypertrophy and remodeling to a lesser extent than control mice in spite of similar hypertension by Ang II. Therefore, our present study provided the experimental evidence that Ang 1-7 counteracts Ang II-induced brain and skeletal muscle injuries in AD mice and AD mice exhibit the impairment of cardiac hypertrophic response.
Previous reports by us [16] and other groups [27, 41] suggest that AD mice (including 5XFAD mice) are more vulnerable to Ang II-induced cognitive abnormality and AD neuropathology. Furthermore, brain Ang 1-7 is reduced in AD mice [42]. We have previously demonstrated the neuroprotective effect of centrally administered Ang 1-7 in 17-month-old 5XFAD mice [24]. However, it is unclear whether Ang 1-7 can protect against Ang II-induced cognitive impairment and tissue injuries including brain in AD mice. In the present study, as estimated by Morris water maze test and the nest building test, we found that 6-month-old 5XFAD mice were more vulnerable to Ang II-triggered cognitive dysfunction than control mice, and this finding is similar to our previous findings on older (12-month-old) 5XFAD mice subjected to central Ang II infusion [16]. Ang II-induced cognitive dysfunction in 5XFAD mice in the present study was associated with the decreased CBF, the increased hippocampal inflammation, and hippocampal neuronal loss, thereby suggesting the possible involvement of CBF disturbance and cerebral inflammation in cognitive dysfunction by Ang II in 5XFAD mice. Furthermore, cortical arterial Aβ deposition in 5XFAD mice was significantly increased by Ang II infusion, thereby suggesting the important role of cerebrovascular Aβ deposition in the vulnerability of 5XFAD mice to Ang II-triggered cognitive dysfunction. Notably, we found that Ang 1-7 ameliorated Ang II-mediated cognitive dysfunction in 5XFAD mice, without significant effect on Ang II-induced hypertension. The protective effect of Ang 1-7 against cognitive impairment was associated with the decrease in cortical arterial Aβ deposition, the improvement of CBF, and the amelioration of hippocampal inflammation and of neuronal loss. Our results provided the evidence indicating that Ang 1-7 can counteract Ang II-induced cognitive impairment and brain injury in AD mice independently of blood pressure.
We have recently reported that centrally infused Ang II induces less cardiac hypertrophic response in 12-month-old 5XFAD mice than in the same-aged WT-type mice [16]. In the present study, we examined the effect of systemic infusion of Ang II and Ang 1-7 in 6-month-old 5XFAD mice. We found that cardiac hypertrophy, inflammation, and fibrosis by Ang II were significantly less in 5XFAD mice than control mice. Thus, our present findings, taken together with our previous report [16], confirm that 5XFAD mice exhibit impaired cardiac hypertrophic response to Ang II. To examine whether the impaired cardiac hypertrophic response in 5XFAD mice is specific for Ang II, we examined the effect of isoproterenol (β adrenergic agonist) on the heart of 5XFAD mice, and found that isoproterenol-induced cardiac hypertrophic response was also less in 5XFAD mice than in WT-mice. Thus, 5XFAD mice seem to exhibit the abnormality of cardiac hypertrophic response. However, further study is needed to define the significance of impaired cardiac hypertrophic response in 5XFAD mice.
Skeletal muscle atrophy and injury is another feature of peripheral pathophysiological change in AD [4, 16]. Recent studies highlight the role of RAS in skeletal muscle [43–45]. We have previously reported that 12-month-old 5XFAD mice were more vulnerable to centrally administered Ang II-induced skeletal muscle atrophy and injury [16]. In addition, in normal mice, Ang 1-7, via Mas receptor, is reported to counteract Ang II-triggered skeletal muscle atrophy and injury [46, 47]. However, the role of Ang 1-7 in skeletal muscle pathology in AD mice remains to be determined. Therefore, in the present study, we examined the systemic effect of Ang 1-7 on Ang II-induced skeletal muscle injury of 5XFAD mice. The present results showed that 6-month-old 5XFAD mice was susceptible to skeletal muscle injury by systemic Ang II infusion compared with control mice and coinfusion of Ang 1-7 attenuated Ang II-induced skeletal muscle atrophy and injury in 5XFAD mice. Thus, our work suggests that Ang 1-7 might play a protective role in skeletal muscle injury in AD.
There are several study limitations in the present work. First, the present study did not allow us to elucidate the potential role of cardiac Aβ in the impairment of cardiac hypertrophic response in 5XFAD mice, and cardiac Aβ levels were not significantly affected by Ang II or Ang 1-7. A growing body of evidence suggests the potential link between AD and cardiac diseases. Previous studies have demonstrated the presence of Aβ in the myocardium of AD patients with cardiac dysfunction [2] or heart failure patients [48]. In vitro study indicates that Aβ is toxic to human cardiomyocytes and causes cell death or apoptosis [48]. Therefore, it cannot be excluded that cardiac Aβ might be responsible for the impairment of cardiac hypertrophic response in AD mice. Further study is required to determine the precise mechanism of impaired cardiac hypertrophic response in AD mice. Second, the potential role of Aβ in skeletal muscle injury in 5XFAD mice was not examined in the present study. It has been suggested that the accumulation of Aβ is associated with increased oxidative stress, inflammation, cell apoptosis and death in skeletal muscle [49, 50]. Therefore, it cannot be ruled out that Aβ might participate in Ang 1-7-mediated protection against skeletal muscle injury in 5XFAD mice. Third, the dose of Ang 1-7 in the present study was generally used dose for pharmacological experiment [29, 32–34]. However, further study using different doses of Ang 1-7 is needed to elucidate the optimal dose for preventing Ang II-induced cognitive impairment and skeletal muscle injury. Finally, in the present study, the blood-brain barrier was not significantly disrupted by systemic Ang II infusion in 5XFAD mice. However, we have previously reported the significant disruption of the blood-brain barrier by central Ang II infusion in 5 XFAD mice [16]. This discrepancy between our present work and our previous report [16] seems to be explained by the different experimental conditions between the two studies such as different age of mice (6-month-old versus 12-month old), different administration route of Ang II (systemic versus central), and different dose of Ang II infusion, etc. However, further study is needed to define whether the blood–brain barrier might be involved in Ang 1-7-mediated brain protective effects or not.
In conclusion, we provided the experimental evidence indicating that Ang 1-7 counteracted Ang II-induced cognitive impairment, brain injury, and skeletal muscle injury in young 5XFAD mice and cardiac hypertrophic response is impaired in 5XFAD mice. However, further study is required to define the underlying mechanisms and the significance of Ang 1-7-induced protective effects in AD mice.