
Abstract
Alzheimers’ disease (AD) is the most common cause of dementia, with an estimated 5 million new cases occurring annually. Among the elderly, AD shortens life expectancy, results in disability, decreases quality of life, and ultimately, leads to institutionalization. Despite extensive research in the last few decades, its heterogeneous pathophysiology and etiopathogenesis have made it difficult to develop an effective treatment and prevention strategy. Aging is the biggest risk factor for AD and evidence suggest that the total number of older people in the population is going to increase astronomically in the next decades. Also, there is evidence that air pollution and increasing income inequality may result in higher incidence and prevalence of AD. This makes the need for a comprehensive understanding of the etiopathogenesis and pathophysiology of the disease extremely critical. In this paper, a quintuple framework of thyroid dysfunction, vitamin D deficiency, sex hormones, and mitochondria dysfunction and oxidative stress are used to provide a comprehensive description of AD etiopathogenesis and pathophysiology. The individual role of each factor, their synergistic and genetic interactions, as well as the limitations of the framework are discussed.
Keywords
INTRODUCTION
Dementia can be defined as a clinical syndrome characterized by cognitive impairment that leads to complete dependence for basic functions of daily life [1]. Alzheimers’ disease (AD) is the most common cause of dementia, making up almost 80% of all dementia cases [1]. It is estimated that over 25 million people worldwide are affected by dementia, most suffering from AD, with an estimated 5 million new cases reported annually [2, 3]. The economic, health, and social burden of AD is staggering. Currently, AD ranks number six in the cause of death in America and number five in the cause of death in elderly Americans (65 years or older) [4]. Among the elderly, AD shortens life expectancy, results in poor quality of life, disability, and being committed to an institution [1]. In the United States (US), after cancer and coronary heart disease, AD is the third most costly disorder exceeding $US100 billion annually [4]. The cost of AD was approximately $US216 billion in 2012 in the US, and it is expected to surpass $203 billion in 2013 [4]. Also, AD has a tremendous impact on the sufferer’s family members and caregivers. Caregivers of AD patients suffer greatly from the long time spent caring for AD patients and many experience higher levels of mental illnesses [5].
Furthermore, the world faces an unprecedented global epidemic of AD as the number of elderly people in the population increases [6]. Currently, someone in America develops AD every 68 seconds, but by 2050, that is expected to drop to as low as every 33 seconds, or nearly a million new cases annually [7]. Globally, the incidence of AD is expected to rise dramatically from the current 36 million sufferers to as much as 115 million by 2050 [8]. Despite extensive research on dementia and AD in the last few decades, its heterogeneous etiopathogenesis and pathophysiology hinder the development of effective treatment or an outright cure for the disease. The projected spike in AD prevalence and the associated health, social, and economic burden that accompanies it makes the need to better understand the etiopathogenetic and neuropathophysiological basis of the disease extremely critical as such understanding is crucial to reducing its burden and arresting its projected spike.
The objective of this paper is to reduce the complexity around the etiopathogenesis and pathophysiology of AD and improve understanding of the disease. This could help improve treatment strategies and slow the progression of the disease.
PATHOGENESIS AND PATHOPHYSIOLOGY OF AD
The factors recognized to play a role in AD etiopathogenesis, and pathophysiology are numerous. The main pathological changes observed in AD brain tissue are amyloid-β (Aβ) peptide, a product of the sequential cleavage of amyloid-β protein precursor (AβPP), and hyperphosphorylated tau protein, a major component of neurofibrillary tangles [9, 10]. Multiple lines of evidence have also implicated α-synuclein in the pathogenesis AD [11–13]. The main species of Aβ are Aβ40 and Aβ42 peptides, and Aβ42 has an affinity to aggregate, creating the toxic amyloid fibrils observed in AD [14]. AD is classified into early-onset AD (onset <65 years) or familial AD which accounts for about 1–5% of all cases, and late-onset AD (onset ≥65 years) or sporadic which accounts for>95% of AD sufferers. Familial AD is caused by mutations in any of these three genes: APP, Presenilin-1 (PS1), and Presenilin-2 (PS2) and these mutations are connected to the excessive production of Aβ1 - 42 [9].
Furthermore, factors such as alcohol consumption, obesity, smoking, blood pressure, type 2 diabetes, traumatic brain injury, obesity, cerebral and cardiovascular diseases, and diet have been associated with increased risk of AD [1, 9]. Research indicates that cholinergic dysfunction [15, 16], as well as cortisol dysregulation, are associated with AD pathophysiology and pathogenesis [17, 18]. An active socially integrated late lifestyle and high levels of social engagement have been found to reduce the risk of AD [19]. Also, depression has been recognized to confer an increased risk for later developing AD [20], and several lines of evidence point to the role of aluminum and metals in the etiopathogenesis of AD [21–23]. Moreover, cytokines and neuroinflammation have been implicated in AD [24, 25], and excitatory glutamatergic neurotransmission due to excessive N-methyl-d-aspartate receptor (NMDAR) activity has been associated with AD pathogenesis and pathophysiology [26].
Also, degenerative changes in selected brain regions have been implicated in AD pathophysiology [27]. Multiple lines of evidence indicate that there is a significant cognitive dysfunction (impairment) linked to hippocampal-amygdala atrophy in AD patients [28–30]. Furthermore, circadian abnormalities and sleep disorders have been implicated in the risk of AD [31]. Evidence from several lines of investigation has implicated sleep-related disorders and deprivation with a negative effect on Aβ load in the human brain [32, 33]. Obstructive sleep apnea is very common in AD patients and has been suggested to contribute to AD cognitive decline progression [34]. Also, metabolic abnormalities of red blood cells have been proposed as a risk factor for AD [35] as well as monoaminergic dysfunction [36].
These numerous factors make AD very complex and add to the difficulty to develop adequate clinical/treatment strategies to fight the disease development and progression. Therefore, the need for a framework that integrates all these different factors to better understand the disease pathogenesis and pathophysiology cannot be overemphasized.
SEARCH STRATEGY
Search was done by accessing PubMed/Medline, EBSCO, and PsycINFO databases. The search string used was “(dementia* OR Alzheimer’s) AND (pathophysiology* OR pathogenesis)”. New key terms were identified (new term included: “vitamin D, thyroid hormone, mitochondria dysfunction, oxidative stress, testosterone, estrogen, melatonin, progesterone, luteinizing hormone, Aβ, and hyperphosphorylated tau”). The electronic databases were searched for titles or abstracts containing these terms in all published articles between January 1, 1965, and June 31, 2018. The search was limited to studies published in English and other languages involving both animal and human subjects. Articles selected included randomized clinical trials (RCTs), observational studies, meta-analyses, and systemic reviews providing primary quantitative data with a measure of AD or AD symptoms as an outcome. Exclusion criteria were case reports and unpublished data of any form including conference proceedings and dissertations. The articles cited were mostly observational with sample sizes of less than 1,000 subjects. There were very few large double-blind RCTs particularly on thyroid dysfunction and AD.
NEW HYPOTHESIS
The etiopathogenesis and pathophysiology of AD are multifactorial and complex. However, a comprehensive framework that captures all recognized risk factors can be derived using five fundamental factors: mitochondria dysfunction and oxidative stress (MtD and OS), vitamin D (VD), sex hormones, melatonin, and thyroid hormones. Each factor has an independent and direct etiopathogenetic, pathophysiological, and genetic interactive effect on AD and acts as the secondary pathogenic mechanism for other recognized AD risk factors.
Vitamin D (VD)
Several lines of investigation have found an association between VD deficiency and risk of AD [37–39]. Evidence suggests that VD and its receptors (VDR) regulate the AβPP processing pathway, are co-localized with AβPP in neurons, and play an essential role in Aβ homeostasis and the prevention of Aβ toxicity [39–42]. Also, evidence suggests that VD inhibits α-synuclein aggregation [43].
Findings from research indicate that VD affects many processes in the developing brain which are associated with AD including calcium homeostasis, cytokine regulation, regulation of nerve growth factor, neuronal differentiation, and metabolism [44, 45]. VD deficiency has been implicated in the hastening of aging and age-related diseases because it regulates many processes related to aging including oxidative stress, inflammation, epigenetic changes, autophagy, mitochondrial dysfunction, and DNA disorders [46]. Furthermore, several studies have observed an association between cognitive impairment and VD hypovitaminosis [37, 47]. Evidence suggests that VD improves cognitive function in AD patients and promotes hippocampus neurogenesis and hippocampal synaptic function [48–50]. Moreover, research has found a downregulation in VDR mRNA levels in hippocampus of AD patients [51], strengthening the argument that VD plays an essential role in the hippocampal cognitive decline observed in AD patients.
Also, evidence suggests that VD may play an essential role in sleep quality because its receptors are present in brain areas associated with the initiation and maintenance of sleep [52]. Chronic sleep problems negatively affect immune function and elevate proinflammatory mediators such as cytokines [53, 54]. Cytokines are involved in the modulation of sleep [54–56], and evidence suggests that they play an essential role in the pathogenesis of AD [24, 25]. Research indicates the VD plays a critical role in the immunoregulation of cytokines and the downregulation of proinflammatory cytokine response [57, 58]. VD has been found to significantly reduce retinal inflammation and levels of Aβ accumulation, which is a hallmark of aging and AD [59].
Furthermore, VD deficiency is linked to depression and evidence suggests it plays a role in the regulation of monoamines [60–63]. Additionally, findings from research indicate that VD is involved in the glutamate-GABAergic system. Evidence suggests that the tilt toward glutamate excitotoxicity in AD is connected to a failure to maintain calcium homeostasis in the cell [64]. VD maintains low intracellular Ca2 + levels, but in VD deficiency, Ca2 + balance is disrupted [41]. Moreover, cholesterol is associated with increased risk of AD and evidence suggests that VD deficiency is associated with lower high-density lipoprotein cholesterol dyslipidemia [65].
In summary, evidence suggests that VD plays a key role in Aβ deposition, tau hyperphosphorylation, α-synuclein anti-aggregation, brain function, brain areas involved in AD, glutamate excitotoxicity system, neuropsychiatric disorders, aging, antioxidant system, cytokines regulation, circadian abnormalities and sleep-related disorders, and the immune system.
Mitochondrial dysfunction (MtD) and oxidative stress (OS)
Numerous lines of evidence support a role for MtD in the pathogenesis and pathophysiology of AD [66, 67]. Aβ has been observed accumulating in mitochondria postmortem AD brains and in the brains of AD living patients [68]. Aβ has been observed in mitochondria prior to amyloid plaque deposition, indicating that MtD plays an essential role in the etiopathogenesis of AD [68, 69]. Also, research indicates that MtD plays an important role in promoting tau pathology in AD [70].
Mitochondria are one of the main sources of reactive oxygen species and nitrogen reactive species, and MtD could result in overproduction of these species and/or failure of the antioxidant defense leading to oxidative/nitrative stress, a condition associated with AD [71, 72]. Both MtD and OS have been implicated in AD etiopathogenesis [73, 74]. Aβ, neurofibrillary tangles, and MtD have been shown to promote OS, and multiple lines of evidence indicate that OS precedes and contributes to the progression of Aβ pathology [75–78]. OS is involved in the promotion of Aβ deposition, tau hyperphosphorylation, and the loss of synapses and neurons [79]. Elevated levels of OS have been found to accelerate tau phosphorylation through the activation of glycogen synthase kinase 3 (GSK-3β) [80], and influence the alteration of tau protein which affects the tau fibrils formation [74]. Also, research indicates that both MtD and OS can induce α-synuclein oligomerization and aggregation [81, 82].
Furthermore, OS plays a role in sleep disorders. Evidence suggests that Aβ increases during wakefulness but sleep helps to reduce their accumulation, and any disruption to the sleep-wake cycle increases OS and slows the clearance of Aβ [83, 84]. Moreover, evidence suggests that OS contributes to glutamatergic excitotoxicity in AD by altering critical components of the glutamate system, and Aβ plays an essential role in this process [85, 86]. Research indicates that OS mediated via NMDAR plays a critical role in tau hyperphosphorylation and synapse dysfunction in AD [87].
Also, OS plays a role in brain areas affected in AD. Evidence suggests that age-related memory impairment is a consequence of the decline in brain and plasma antioxidants defense system [88, 89]. Damage from elevated levels of OS has been observed in the postmortem frontal cortex of AD patients, indicating that it plays a role in executive dysfunction and cognitive decline in AD [90–92].
Many recognized risk factors of AD are well documented to increase OS, indicating that it may be their link to the disease. As an example, cholesterol, a risk factor of AD, has been found to induce OS [93, 94]. Neuroinflammation, which is implicated in AD pathology, leads to increased OS which causes subsequent inflammation [95], and antioxidants protect neurons by reducing OS and chronic inflammation in AD [96]. Furthermore, research indicates that OS is central to the pathogenesis of both depression, an AD risk factor, and AD [97], and evidence suggests that depression is a state elevated OS [98]. Chronic stress is considered a risk factor for AD and evidence suggest that OS and MtD underlie its pathophysiology in AD [99]. Hypothalamic dysfunction has been implicated in AD pathology [100], and evidence suggests that OS may be the underlying culprit in hypothalamic-pituitary-adrenal (HPA) axis dysfunction as part of the aging process [101]. Also, several lines of evidence indicate that obesity and obesity-related comorbidities (e.g., insulin resistance, leptin dysfunction, hyperglycemia, and type 2 diabetes) increase the risk of cognitive impairment and AD [102–104]. Research indicates that diabetes, obesity, and leptin dysfunction (both hyperleptinemia and leptin resistance) are states of elevated OS [105, 106]. Smoking has been implicated in the risk of AD and evidence suggest that OS may underlie its pathophysiology in AD [107]. Homocysteine has been associated with increased risk of AD [108], and evidence suggests that altered levels of homocysteine promote OS and the pathogenesis of AD [109, 110]. Moreover, cholinergic dysfunction is associated with AD and several lines of evidence suggest that OS is the cause of cholinergic dysfunction [111–113]. Nerve growth factor has been associated with AD [114], and multiple lines of evidence indicate that it is a potent antioxidant that attenuates OS [115, 116]. Insulin-like growth factor-1 has been associated with AD pathogenesis [117], and evidence indicates that it is a powerful antioxidant [118], Similarly, selenium deficiency has been implicated in AD [119], and multiple lines of evidence indicate that it is a powerful antioxidant that can reduce AD pathology [120, 121]. Furthermore, research indicates that in AD, there is increased accumulation of metals in the brain (Fe, Al, and Hg), and evidence suggests that OS underlies their role in AD pathogenesis and pathophysiology [122, 123]. Also, metabolic abnormalities of erythrocytes due to poor antioxidant defense have been proposed as a risk factor of AD, indicating that OS may be the primary cause of the glucose metabolism disturbance which results in neurobiological changes observed in the AD brains [35]. Cognitive activity has been found to reduce the risk of dementia and AD [124, 125], and evidence suggests that it reduces stress [126], which is associated with lower levels of cortisol or HPA activity and thus OS.
In summary, evidence suggests that MtD and OS play a prominent role in Aβ deposition, tau hyperphosphorylation, α-synuclein aggregation and oligomerization, loss of synapses and neurons, cognitive decline, glutamate excitotoxicity, neuropsychiatric disorders, aging, cytokines regulation, circadian abnormalities and sleep-related disorders, HPA axis, and many factors that are involved in the pathogenesis and pathophysiology of AD.
Melatonin
Several lines of investigation have implicated melatonin in AD pathophysiology [127, 128]. Evidence from several studies indicates that melatonin levels are significantly lower in the serum and cerebrospinal fluid (CSF) of AD patients [129–131]. Indeed, evidence suggests that melatonin is practically non-existent in AD and its levels in CSF diminishes with the progression of the disease [130]. Research indicates that melatonin confers a neuroprotective effect against AD via different mechanisms including, inhibition of Aβ deposition, Aβ fiber formation, tau protein hyperphosphorylation, and prevention of cognitive impairment and decline [132]. Numerous lines of evidence suggest that melatonin’s role in AD involves its anti-apoptotic, anti-amyloidogenic, and antioxidant properties [133, 134].
Melatonin has been found to promote nonamyloidogenic processing, suppress the amyloidogenic processing of AβPP [135], and counteract the hyperphosphorylation of tau protein [132, 136]. Furthermore, evidence suggests that melatonin alters the secondary structure of the Aβ peptide and hinders the formation of beta-sheets and amyloid fibrils [137]. Melatonin affects the secretion of soluble derivatives of AβPP by disrupting its maturation [138] and protects against Aβ1 - 42-induced neurotoxicity by reducing the overexpression of caspase-9, caspase-3, and PARP-1 level [139]. Melatonin uses its antioxidant properties to counteract neuronal and astrocytic death triggered by Aβ toxicity [140]. Melatonin has been found to counteract Aβ-induced neurotoxicity by increasing the survival of glial cells [141], reducing the negative effects of OS and maintaining intracellular Ca2 + levels [142, 143]. Also, evidence suggests that melatonin counteracts α-synuclein aggregation, cytotoxicity, and oligomerization and suppresses its fibril formation [144, 145].
Furthermore, elevated levels of melatonin have been found to be associated with higher cognitive function in the elderly [146, 147]. Research indicates that melatonin can modulate plasticity in hippocampal pyramidal neurons [147], and evidence suggests that melatonin plays a critical role in hippocampal neurogenesis [148, 149]. Also, evidence suggests that melatonin slows hippocampal pathological progression and improves amygdala-dependent emotional memory in AD [150]. Fear conditioning is impaired in AD [151] and melatonin has been found to restore this impairment by facilitating the extinction of conditional cued fear without disturbing its acquisition or expression [152].
Also, melatonin plays a major role in sleep regulation. Research indicates that melatonin counteracts Aβ induced circadian alterations [153], and its supplementation ameliorates sundowning, as well as improve sleep quality in AD patients [147, 154].
Moreover, melatonin affects the glutamate-GABAergic system. It regulates the NMDA receptor [155] and controls intracellular free Ca2 + by binding to calmodulin [156]. Melatonin has been found to counteract Aβ neurotoxicity by suppressing glutamate excitatory tonus, and the activation of GABA receptors [157].
In addition, melatonin influences many AD risk factors. As an example, cholesterol is a risk factor for AD and evidence suggests that melatonin has a hypocholesterolemic effect [158, 159]. HPA axis dysregulation has been implicated in AD and evidence suggests that melatonin modulates it [160]. Cholinergic dysfunction has been implicated in AD and several lines of evidence suggest that melatonin facilitates choline transport and could restore the cholinergic system [136, 161]. Leptin dysfunction has been associated with AD and evidence suggests that melatonin interacts with leptin and prevents leptin resistance [162]. Furthermore, multiple lines of investigation indicate that melatonin has strong anti-inflammatory properties [163, 164] and modulates the expression of different inflammatory mediators including both pro-and anti-inflammatory cytokines [165, 166].
In summary, evidence suggests that melatonin plays a prominent role in Aβ deposition, tau hyperphosphorylation, α-synuclein aggregation and cytotoxicity, AD cognitive decline, glutamate excitotoxicity, neuropsychiatric disorders, antioxidant system, cytokines regulation, circadian abnormalities and sleep-related disorders, HPA axis, and many factors that increase the risk of AD.
Thyroid hormones (TH)
Numerous lines of evidence indicate that thyroid dysfunction plays an essential role in AD pathology [167]. TH suppresses the expression of APP genes in the brain and its diminished action on the APP gene promotes AβPP overexpression and the number of processed AβPP products [168–170]. Hyperthyroidism has been associated with AD development [171], and research indicates that hypothyroidism increases vulnerability to the development of amyloid deposits [172]. Animal research has found that hypothyroidism promotes cerebral atrophy, brain proinflammatory cytokines, Aβ production, and tau hyperphosphorylation [173], and TH supplementation can reverse these conditions [174]. Lowered thyroid stimulating hormone (TSH) has been found to increase the vulnerability to develop AD [175], and evidence suggests that low serum free thyroxine (fT4), and high serum TSH levels are differentially associated with AD pathology [176, 177]. Furthermore, thyrotropin-releasing hormone (TRH) has been found to be depleted in AD brains [178]. Evidence suggests that TRH suppresses tau phosphorylation in hippocampal neurons and reduced TRH gene expression results in elevated tau and GSK-3β, a critical enzyme necessary for the phosphorylation of tau and tau protein [179]. TRH administration results in significant reduction in tau phosphorylation in hippocampal neurons [178, 179].
Also, TH affects cognitive function in AD [173, 180]. TH is recognized to regulate hippocampal neurogenesis [181, 182], and normal thyroid function is recognized to be essential in maintaining optimal cognition in aging [183], indicating that TH status may contribute to the clinical manifestations of AD. TSH levels have been found to be positively correlated with glucose metabolism in the brain, which is negatively correlated to CSF t-Tau levels, in the brain of AD patients [184]. Neurons expressing message levels for TH receptor mRNA expressed in AD hippocampus have been found to be significantly reduced [180]. Evidence suggests that there may be a negative correlation between serum level of TSH with the right and left hippocampal and brain volume ratio, reinforcing the fact that TH may play a role in cognitive impairment observed in AD [185].
Also, TH status influences many of the risk factors of AD. TH has been found to play a role in neuropsychiatric manifestations in AD patients [180], and evidence suggests that TH dysfunction increases vulnerability to the occurrence of depression [181, 186]. TH plays a role in the development and protection of the basal forebrain cholinergic neurons involved in AD and its supplementation increases cholinergic markers as well as reverses cognitive impairment [186–188]. Neuroinflammation has been implicated in AD and evidence suggest that hypothyroidism upregulates the levels of pro-inflammatory cytokines in the hippocampus [173, 189]. Glutamate excitotoxicity has been implicated in AD and evidence suggests that TH is involved in the regulation of extracellular glutamate levels [190, 191].
In summary, evidence suggests that TH plays a critical role in Aβ deposition, tau hyperphosphorylation, cognitive decline, glutamate excitotoxicity, neuropsychiatric disorders, cytokines regulation, and many factors that increase the risk of AD.
Sex hormones
Sex hormone neurosteroids (estrogen, progesterone, and testosterone) have been found to ameliorate Aβ-induced mitochondrial deficits and AD tauopathy [192]. Findings from research indicate that they possess significant α-synuclein anti-aggregation properties and fibril-destabilizing effects [193].
In women, the risk of AD increases with aging due to age-related decline in estrogen and progesterone as well as elevated levels of luteinizing hormone (LH). Findings from animal studies have demonstrated that estrogen may play a vital role in suppressing the expression of AβPP mRNA [194]. Estrogen has been found to elevate the levels of soluble AβPP alpha (sAβPPα), the secreted form of AβPP, and to decrease cerebral Aβ levels [195]. Estrogen and progesterone have been found to modulate the expression levels of several Aβ clearance factors [196]. Furthermore, findings from research suggest that there is increased estrogen receptor-α (ERα)-tau interaction in AD brain, indicating that the sequestration of ERα by tau abnormality is at the heart of the loss of estrogen neuroprotection in AD [197]. Low levels of endogenous progesterone have been associated with dementia and progesterone supplementation have been demonstrated to reduce total tau levels [198]. Also, multiple lines of evidence suggest that LH modulates the processing of AβPP and Aβ deposits in the brain and may contribute to the development of AD via an amyloid-dependent mechanism [199, 200].
Importantly, evidence suggests that estrogen plus progestin therapy may confer an increased risk for dementia in postmenopausal women [201], indicating that estrogen and progesterone may not always be neuroprotective against Aβ deposits and AD development. However, research indicates that estrogen therapy within 5 years of menopause significantly lowers the risk of AD in women [202], indicating there might be a perimenopausal window in which estrogen or estrogen plus progesterone therapy is useful against dementia and AD development. Also, estrogen therapy has been found to be beneficial against early-onset AD [203], reinforcing the window hypothesis and suggesting that the older a patient, the less effective estrogen therapy may become. Notably, elevated LH levels in postmenopausal women, which promotes the deposition of Aβ plaques [204] may contribute to estrogen treatment failure in postmenopausal women.
In men, the risk of AD increases with aging due to a decline in the levels of testosterone and androgens [205]. Evidence suggests that low testosterone level is associated with an elevated risk of AD development in elderly men [206], and in brain levels of AD men, testosterone has been found to be negatively correlated with soluble Aβ [205]. Research indicates that testosterone counteracts Aβ toxicity via estrogen-independent mechanisms [207, 208]. In men with AD neuropathology, testosterone supplementation has been found to stimulate the secretion of the nonamyloidogenic AβPP fragment, sAβPPα, and decrease neuronal secretion of Aβ peptides [209]. Moreover, androgen deprivation therapy has been found to elevate plasma levels of Aβ and the risk of developing AD [210]. Several animal studies have found that androgen modulates Aβ levels in the brain and provides neuroprotection against the advancement of AD-like pathology [210, 211].
Furthermore, sex hormones affect the glutamate system. Research indicates that blood glutamate levels are negatively correlated with levels of plasma estrogen and progesterone [212], and evidence suggests that blood glutamate levels are linked with the levels of the brains’ extracellular glutamate [213]. Taken together, it can be inferred that estrogen and progesterone regulate the brain’s glutamate concentrations and their ideal levels keep both brain glutamate and intracellular CA levels low [212], preventing glutamate excitotoxicity.
Also, sex hormones affect cognition in AD. Research indicates that ovariectomy may cause poor memory performance and estrogen and progesterone therapy have been shown to independently and synergistically reverse this effect [214]. Estrogen has been demonstrated to reduce microglial activation and the loss of neurons in the hippocampus [215]. Moreover, testosterone has been demonstrated to improve cognition in older men and men with AD [216, 217] as well as promote hippocampal neurogenesis. Research on hippocampal neurons has demonstrated that testosterone preserves neurites, the expression of presynaptic proteins, and ameliorates both Aβ-induced impairments of synaptic exocytosis and reduction of HSP70 [218]. LH has many receptors in the brain, particularly in the hippocampus, and evidence from animal study suggests that the ablation or elimination of LH may reverse cognitive decline in AD [199, 219].
Furthermore, sex hormones affect many risk factors of AD. Evidence suggests that low estrogen levels increase the risk of depression in female AD patients [220]. Findings from animal research indicate that progesterone treatment can reverse depressive behavior in AD neuropathology [221], indicating that low levels may play an important role in the risk of depression in AD patients. Testosterone is associated with neuropsychiatric symptoms including hallucinations, agitation, and delusions in elderly men with AD [222]. Moreover, evidence suggests that ovarian hormones play a role in maintaining cholinergic function [223], and long-term estrogen treatment in postmenopausal women can enhance cholinergic function [224]. Animal models indicate that estrogen and progesterone neuroprotective effects in AD may be connected to their amelioration of the cholinergic deficits [225]. Also, estrogen has anti-inflammatory properties and is involved in the inhibition of proinflammatory cytokines, as well as the regulation of cytokine and chemokine-mediated neuroinflammatory response in the CNS [226, 227].
In summary, evidence suggests that sex hormones play a critical role in Aβ deposition, tau hyperphosphorylation, α-synuclein anti-aggregation, cognitive decline, glutamate excitotoxicity, neuropsychiatric disorders, cytokines regulation, and many factors that increase the risk of AD development.
SYNERGISTIC INTERACTIONS BETWEEN FACTORS
These five factors work synergistically, and their synergisms are essential to understanding AD pathogenesis and pathophysiology.
VD interactions
Multiple lines of evidence indicate that VD deficiency is associated with thyroid disorders [228, 229]. VD receptors protect mitochondria optimal function and play a central role in protecting cells from OS [230]. Research indicates that VD deficiency/insufficiency is associated with increased OS [231] and OS plays a role in VD metabolism [232]. Furthermore, research indicates that VD is essential in the production of estrogen and progesterone [233]. Lower VD levels are associated with low levels of testosterone and hypogonadism [234]. Additionally, evidence suggests that VD levels have a strong inverse association with LH levels [235].
Mitochondria dysfunction and OS interactions
Evidence suggests that mitochondria are essential in steroidogenesis, and sex steroid hormones modulate mitochondrial biogenesis and function, indicating that impairment of mitochondrial function could affect optimal sex steroid hormones action and vice versa [236]. Research indicates that sex hormones modulate OS in AD [237]. Also, evidence suggests that OS can reduce testosterone and gonadotropin levels [238], and several lines of evidence indicate that testosterone can attenuate OS and cell damage [239, 240]. Numerous lines of evidence have described the antioxidant properties of estrogen indicating that it counteracts OS [241, 242]. However, it has been found to be capable of inducing OS [243]. Furthermore, research has found that progesterone administration attenuates OS [244] and increases levels of glutathione in the brain [244]. However, evidence suggests that progesterone can suppress cellular antioxidant action and promote OS [245]. The relationship between sex hormones and OS is complex and a better understanding will greatly enhance knowledge of AD pathophysiology and treatment.
Melatonin interactions
Evidence suggests that melatonin suppresses LH in postmenopausal women [246]. Findings from animal research indicate that melatonin modulates the number of estrogen and progesterone receptors [247]. Melatonin is well recognized to attenuate OS and preserve mitochondrial function [248, 249]. Also, melatonin increases thyroglobulin mRNA and protein expression and confers protection against thyroid disorder by attenuating OS involved in thyroid diseases [250, 251]. Furthermore, melatonin secretion has been found to be negatively correlated with variations in serum VD levels [252], suggesting that melatonin may mediate VD neuro-immunomodulatory and protective effects.
TH interactions
Evidence suggests that TH counteracts OS and can improve mitochondrial function [253]. Research indicates that OS plays an essential role result in different thyroid diseases [254]. Hyperthyroidism promotes a high metabolic rate triggering OS [255] and hypothyroidism is recognized to be a state of OS [256]. Research has found that hypothyroidism results in menstrual irregularities and depresses serum levels of estrogen and testosterone [257]. Further, evidence suggests that testosterone levels are significantly reduced in men with hypothyroidism [258]. Additionally, research indicates that both hypothyroidism and hyperthyroidism results in increased levels of LH [259].
The relationships between these factors indicate that they can act independently and synergistically in the etiopathogenesis and pathophysiology of AD.
GENETICS INTERACTIONS
Many genes have been implicated in AD but the ones that have been consistently found to be involved in AD are APP, PS1, PS2, and APOE [260, 261].
APOE
The ApoE ɛ4 allele has been shown to be associated with AD [260]. Research indicates that TH modulates APOE gene expression in the brain [262]. APOE ɛ4 polymorphism has been found to be associated with elevated levels of TSH and lower levels of free triiodothyronine and total triiodothyronine [263], indicating that TH status influences the expression of ApoE ɛ4 allele. Melatonin has been found to suppress the fibrillogenic action of ApoE ɛ4 on Aβ peptide [264], indicating that it modulates ApoE ɛ4 activity. Findings from research suggest that there is an interaction between testosterone and the ApoE ɛ4 allele on general cognition and executive functioning, working memory, and attention in older men [265], indicating that testosterone levels may modulate ApoE ɛ4 activity. Moreover, research indicates that APOE gene expression is differentially regulated by estrogen receptor subtypes [266]. Higher progesterone levels have been found in women with the ApoE4 allele, indicating that progesterone may modulate ApoE4 expression [267]. Research indicates that the ApoE4 allele is associated with higher VD levels, indicating that VD might affect its expression [268]. Moreover, evidence suggests that the ApoE4 genotype promotes OS [269, 270], indicating that OS/total antioxidant capacity status may influence the expression of ApoE4.
AβPP, PS1, PS2
Melatonin has been found to suppress beta-secretase enzyme (BACE1) and PS1 and it is involved in the activation of α-secretase (ADAM10) mRNA level and protein expression [271]. Evidence suggests that VD is involved in the transcriptional regulation of multiple genes involved in AβPP including PS1, PS2, Nicastrin, BACE1, ADAM10, and APP [42]. Mutations in the PS1 and PS2 genes are believed to be responsible for most cases of early-onset familial AD, and evidence suggest that OS conditions could alter these genes [272], indicating that OS may play a role in the epigenetic mutation of these genes. Furthermore, multiple lines of evidence suggest that TH regulates the APP gene as well as the processing and secretion of the AβPP peptides [168, 169]. Additionally, estrogen has been found to reverse the apoptotic actions of mutant PS1 [273], indicating that estrogen may play a role in modulating the action of this gene.
MAJOR CHALLENGES FOR THE HYPOTHESIS
The current review is a bold approach to integrate most of the recognized risk factors of AD. However, there are certain limitations in this theory that must be addressed. Firstly, the theory discussed the interaction between the factors and a few genes associated with AD. However, evidence suggests that many more genes play a role in AD risk [274]. The theory proposes that many AD risk factors including smoking, infections, nerve growth factor, metabolic diseases (obesity, diabetes), depression, etc. are connected to AD via OS. Although it true that these conditions are states of high OS and OS might be the likely pathogenic mechanism behind their association with AD, there is no direct and definitive proof that OS is the underlying factor that drives their pathogenic mechanism. Clearly, more research is needed to know their definitive pathogenic mechanism in AD.
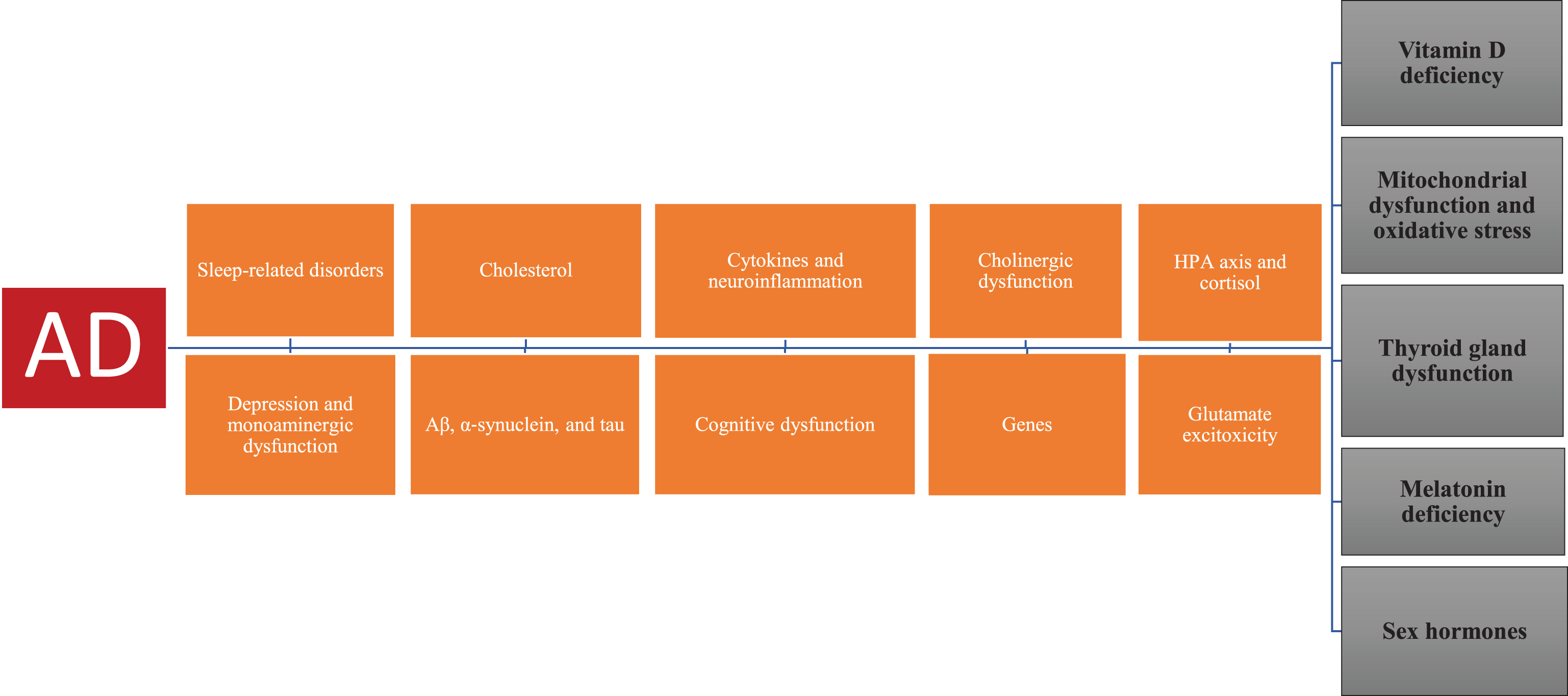
These five factors act independently and synergistically in AD pathogenesis and pathophysiology.
Moreover, although there is significant evidence that these factors in the quintuple framework play a vital role in AD pathogenesis and progression, evidence of their ability to reverse or prevent the disease are mixed. Research indicates that antioxidants therapy or supplementation can help prevent and reverse many of the symptoms of AD [275–277]. However, numerous lines of evidence have found that antioxidants supplementation fails to prevent AD [278, 279]. Furthermore, evidence from animal models suggests that VD supplementation may decrease amyloid burden and improve neurogenesis and cognition in AD [48, 280]. However, evidence from some research has found no association between VD status and long-term risk of dementia or cognitive dysfunction [281], as well as doubts about the role of VD in neurodegenerative diseases [282, 283]. Also, research has found no association between TSH and TH and risk of AD [284], and thyroid medication has been found to increase the risk of developing AD [285]. In addition, results from hormonal replacement therapy are mixed [286]. One reason for the failure of antioxidant supplementation may be because antioxidant supplements do not offer the same health benefits as antioxidants in food [287]. Also, the lack of precise knowledge of the specific antioxidants that may confer benefit for a particular condition/disease over the others [288] may play a role in the failure of antioxidant therapy. The optimal doses of antioxidants that are required to achieve a therapeutic effect are poorly understood [288], and this may contribute to their therapeutic failure. In addition, research indicates that antioxidant supplementation may have a “therapeutic window” during which supplementation may still have a preventive effect of reversing oxidative damage [288], and a lack of knowledge of such a window may contribute to their therapeutic failure. This window hypothesis may also apply to many other treatment failures observed in AD. Evidence suggests that estrogen therapy within 5 years of menopause significantly lowers the risk of AD in women [202], but may confer an increased risk for AD in postmenopausal women [201]. Another reason for treatment failure is that AD is a multifaceted disease with myriads of factors involved which creates a situation where tackling one factor may not produce significant benefit due to a similar role play by another factor. As an example, LH levels are elevated in postmenopausal women, and it promotes the deposition of Aβ plaques [204]. Consequently, estrogen therapy or VD supplementation may not result in a significant reduction in amyloid burden or AD symptom relief in postmenopausal women because of LH. The most important takeaway from these failures is that AD is a very complex disease that needs more research to fully understand and any solution must involve strategies that tackle multiple pathways involved in AD. In other words, there is no silver bullet that can cure AD and any successful strategy must include trying to reverse or treat amyloid burden, sleep disorders, depression, cholinergic dysfunction, genetic polymorphisms, OS, and other major factors involved in the disease concomitantly.
Another limitation is that most of the articles in the review are observational, and RCTs are lacking to definitively show the relationship between AD and some of the factors as well as the usefulness of using them to treat the disease, particularly thyroid dysfunction. So, large double-blind RCTs are clearly needed to strengthen the hypothesis.
LINKAGE TO OTHER MAJOR THEORIES
The quintuple framework goes further than previous theories on AD. As a matter of fact, OS, which is the most important factor in the quintuple framework, can be used to account for most theories of AD. Penke and colleagues discussed the role of protein dyshomeostasis in neurodegenerative diseases including AD [289], and multiple lines of evidence indicate that OS affects the preservation of organelle-homeostasis and proteostasis in the cells of mammals [290–292]. Bell and Zlokovic proposed the role of neurovascular dysfunction and the blood-brain barrier in the onset and progression of AD [293]. Research indicates that vascular cells are very sensitive to OS, and OS plays a key role in the neurovascular dysfunction observed in AD [294–297]. Also, OS can result in neurodegeneration and breakdown of the blood-brain barrier via disruption of tight junction proteins [298, 299]. Ashraf et al. discussed the role of different infectious agents (bacteria, fungi, and viral infections) in AD causation and progression [300]. Evidence suggests infectious agents that cause acute or chronic diseases including viral, bacterial, and parasitic infections induce OS which promotes neurodegeneration [301–304]. Moreover, several theories have discussed the role of metals (copper, zinc, aluminum) in neurotoxicity and neurodegeneration [303, 305–307], and evidence suggests that OS underlies their role in AD pathogenesis and pathophysiology [122, 308].
CONCLUSIONS AND CLINICAL RECOMMENDATIONS
Aging is considered the greatest risk factor of AD and the number of old people in the total population is increasing worldwide [6]. According to the United Nations (UN), by 2050, the global population of older persons (60 years or over) is projected to reach nearly 2.1 billion (more than double its size in 2015), and the number of people aged 80 years or over is expected to triple to almost 450 million from just 125 million in 2015 [309]. This projected increase in the number of old people has a monumental implication for the incidence and prevalence of AD and dementia. Furthermore, air pollution is a problem in many cities around the world because of decades of fossil fuel usage [310]. One of the most important effects of air pollutants is the promotion of OS [311], and evidence suggests that long-term exposure to air pollution may significantly increase the risk of AD [312, 313]. In addition, low social economic status has been implicated as a risk factor for AD [314], and income inequality is reported to be increasing steadily in the US since the 1970 s [315] and in many parts the world. Taken together, it can be predicted that the world is about to witness a monumental AD crisis unless adequate steps are taken to address the problem. To counteract the looming AD crisis, more must be done to highlight the effects of air pollution, on the risk of AD. Furthermore, more must be done to combat income inequality and poverty especially amongst the elderly population. Social support system should be improved for the elderly to reduce the risk of depression and AD. Additionally, the high prevalence of thyroid dysfunction, VD deficiency, and OS enhancing activities such as smoking, alcohol abuse, and obesity in many populations, particularly the elderly (people aged 60 and older) [316], calls for action to create better public awareness of their long-term effects on the risk of AD.
The quintuple framework described in this article could help improve the treatment of AD and slow the progress of the disease. Research indicates that early AD is characterized by cognitive deficits, usually in short-term memory, which is then followed by a slow decline in other cognitive functions [317]. Consequently, when such symptoms present themselves in the elderly (people aged 60 and older), clinicians should check and treat thyroid dysfunction, total antioxidant capacity, melatonin levels, VD deficiency, sleep-related disorders, and depression. Lifestyle changes to reduce OS such as exercising to maintain ideal body weight and abstinence from alcohol and smoking should be recommended for the elderly. Also, a diet with high glycemic index or high glycemic load should not be advised because evidence suggests that it increases OS [238, 318]. To improve sleep, a warm shower performed before bedtime should be recommended [319]. Further, to slow cognitive decline and risk of AD, cognitive and leisure activities should be encouraged for people 60 and older [125]. Additionally, adequate intake of essential minerals (magnesium, calcium, zinc, and iron) should be recommended to attain the full benefit of VD and reduce uptake of toxic elements (lead, arsenic, aluminum, cobalt, and strontium) that can increase the risk of developing AD [320].
DISCLOSURE STATEMENT
The author’s disclosure is available online (https://www.j-alz.com/manuscript-disclosures/18-1052r2).