
Abstract
Chronically elevated basal glutamate levels are hypothesized to attenuate detection of physiological signals thereby inhibiting memory formation and retrieval, while inducing excitotoxicity-mediated neurodegeneration observed in Alzheimer’s disease (AD). However, current medication targeting the glutamatergic system, such as memantine, shows limited efficacy and is unable to decelerate disease progression, possibly because it modulates postsynaptic N-methyl-D-aspartate receptors rather than glutamate release or clearance. To determine if decreasing presynaptic glutamate release leads to long-term procognitive effects, we treated AβPP/PS1 mice with LY379268 (3.0 mg/kg; i.p.), a metabotropic glutamate receptor (mGluR)2/3 agonist from 2–6 months of age when elevated glutamate levels are first observed but cognition is unaffected. C57BL/6J genetic background control mice and another cohort of AβPP/PS1 mice received normal saline (i.p.) as vehicle controls. After 6 months off treatment, mice receiving LY379268 did not show long-term improvement as assessed by the Morris water maze (MWM) spatial learning and memory paradigm. Following MWM, mice were isoflurane anesthetized and a glutamate selective microelectrode was used to measure in vivo basal and stimulus-evoked glutamate release and clearance independently from the dentate, CA3, and CA1 hippocampal subregions. Immunohistochemistry was used to measure hippocampal astrogliosis and plaque pathology. Similar to previous studies, we observed elevated basal glutamate, stimulus evoked glutamate release, and astrogliosis in AβPP/PS1 vehicle mice versus C57BL/6J mice. Treatment with LY379268 did not attenuate these responses nor diminish plaque pathology. The current study builds upon previous research demonstrating hyperglutamatergic hippocampal signaling in AβPP/PS1 mice; however, long-term therapeutic efficacy of LY379268 in AβPP/PS1 was not observed.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) is an age-related neurodegenerative disorder resulting in gradual accumulation of extracellular amyloid-β (Aβ) plaques and intracellular neurofibril tangles composed of hyperphosphorylated tau protein [1]. Over time, the accumulation of these proteins either coincides with, or causes, alterations in neurotransmitter dynamics, synapse loss, and cerebral atrophy that culminates in the eventual cognitive and functional decline associated with AD [2]. Current pharmacotherapy options target cholinesterase inhibitors, to increase acetylcholine levels, and N-methyl-D-aspartate (NMDA) receptor antagonism, to prevent glutamate mediated excitotoxicity [3, 4]. However, these therapies have limited efficacy, are symptomatic, and do not decelerate disease progression, possibly because they are administered at advanced AD stages when synapse loss is too pronounced. To slow or stop cognitive decline, future therapeutics should target neurological components that are altered during the prodromal phase of AD. Increasing evidence supports the glutamatergic system as a possible early target that meets these criteria [5 –10].
Glutamate, the predominant excitatory neurotransmitter in the mammalian central nervous system, has a strong prevalence in neocortical and hippocampal pyramidal neurons, playing a critical role in learning and memory. However, altered glutamate release, clearance, or both may lead to the cognitive and functional decline observed in AD. For example, postmortem analysis has revealed that vesicular glutamate transporter 1 boutons were elevated in pre-clinical AD cases [11] while glutamate transporters were decreased in AD patients [12]. As such, a prevailing hypothesis in AD research supports persistent, excessive activation of NMDA receptors impedes detection of physiological signals initiating cognitive impairment [13, 14]. While the mechanistic link behind the elevated glutamate is not fully elucidated, data supports that accumulation of soluble Aβ isoforms are the bioactive component initiating synaptic dysfunction and the eventual neurodegeneration [15, 16]. Our laboratory and others have demonstrated that soluble Aβ42 elicits glutamate release through the α7 nicotinic acetylcholine receptor (α7nAChR) [17 –19]. We have also demonstrated that double transgenic mice expressing the amyloid precursor protein (Mo/HuAPP695swe) and Presenilin 1 (PS1-dE9) genes (AβPP/PS1) have elevated hippocampal glutamate as early as 2–4 months of age; prior to the onset of cognitive decline [10] that continues through at least 12 months of age [20] when cognitive deficits are more pronounced. These data support the hypothesis that elevated synaptic glutamate levels may, over time, result in excitotoxicity and the eventual atrophy observed in AD. To attenuate the glutamatergic tone, targeting the metabotropic glutamate receptor (mGluR), may provide promising therapeutic interventions for early AD treatments.
The G-protein-coupled mGluRs modulate pre-and postsynaptic glutamate release and consist of various subtypes including group I (mGluR1/5), group II (mGluR2/3), and group III (mGluR4/6/7/8). Group I is positively coupled to phospholipase C and potentiates glutamate release whereas Groups II and III typically inhibit adenylate cyclase activity thereby suppressing glutamate release [21]. The mGluRs are expressed throughout the brain particularly in regions associated with neurodegenerative disorders including the hippocampus [22]; however, their cellular distribution varies. For example, mGluR2 is predominantly expressed on preterminal extrasynaptic sites whereas mGluR3 is more widely distributed on pre-and postsynaptic neurons as well as glia. These receptors are well positioned to monitor extrasynaptic spillover of glutamate, and therefore, act as a negative feedback loop that maintains physiological levels of glutamatergic neurotransmission to prevent excitotoxicity [23]. In support of this, mGluR2/3 agonists, specifically 1R,4R,5S,6R)-4-amino-2-oxabicyclo[3.1.0]hexane-4,6-dicarboxylic acid (LY379268), has been shown to attenuate basal as well as stimulus-evoked glutamate release [24, 25] while providing long-lasting neuroprotective properties against apoptotic and excitotoxic stimuli [26, 27].
The purpose of the present study was to determine if starting LY379268 treatment in 2-month-old AβPP/PS1 mice could provide long-term procognitive effects that were mediated through reduction of the hippocampal glutamatergic tone. To do this, intraperitoneal (i.p.) injections of 3.0 mg/kg body weight (b.w.) of LY379268 was given to AβPP/PS1 mice from 2–6 months of age, prior to the onset of cognitive deficits [8, 28], but when elevated hippocampal glutamate has been observed [10]. C57BL/6J and AβPP/PS1 mice receiving vehicle (normal saline; i.p.) were used as controls. Starting at 12 months of age, when AβPP/PS1 mice typically present with elevated Aβ plaque burden that correlates with cognitive deficits [29], mice were tested for cognitive performance using the Morris water maze (MWM) paradigm. In vivo hippocampal glutamate signaling was assessed using an enzyme-based microelectrode array (MEA) coupled with constant potential amperometry. Immunohistochemistry (IHC) was used to examine hippocampal astrogliosis and Aβ plaque burden. The results presented here support that LY379268 does not have long-term procognitive efficacy nor reduce hippocampal glutamatergic tone, astrogliosis, and plaque pathology in AβPP/PS1 mice.
METHODS
Animals
Male C57BL/6J (RRID:IMSR_JAX:000664) and AβPP/PS1 (RRID:MMRRC_034832-JAX) mice were obtained from Jackson Laboratory (Bar Harbor, ME). Protocols for animal use were approved by the Laboratory Animal Care and Use Committee at Southern Illinois University School of Medicine. Mice were group housed on a 12:12 h light: dark cycle, and food and water were available ad libitum. A timeline of the experimental design is presented in Fig. 1. From 2–6 months of age, a randomly assigned cohort of AβPP/PS1 mice were given twice weekly i.p. injections of LY379268 (3.0 mg/kg b.w.) in normal saline (n = 7). C57BL/6J (n = 8) and another cohort of AβPP/PS1 (n = 8) mice received twice weekly i.p. injections of normal saline as vehicle controls. All mice received a total of 32 injections of LY379268 or normal saline from 2–6 months of age. The dosing strategy was based on previously published in vivo studies [26 , 31]. All mice underwent cognitive assessment, in vivo glutamate recordings, and IHC analysis except for one C57BL/6J mouse where the MEA failed during glutamate recordings. Immediately following anesthetized glutamate recordings, mice were euthanized with an overdose of isoflurane followed by decapitation. Mouse genotypes were confirmed by collecting a 5 mm tail snip that was analyzed by TransnetYX®, Inc (Cordova, TN).

Experimental design. A graphical outline of the experimental design. LY379268 treatment was conducted when mice were 2–6 months of age. At 6 months of age, treatment was discontinued for the remainder of the study. MWM, Morris water maze.
Based on our previous studies, a power calculation supports n = 10 mice per group to detect differences with 95% confidence (α= 0.05) and 0.8 power [20]. We included 15 mice per treatment group to account for potential loss of AβPP/PS1 mice that is typically observed with age. Mice were then divided into 3 cohorts (n = 5 mice per treatment group) to reduce the time between MWM and electrochemical measurements. After the second cohort was completed, the MWM data was pooled and analyzed to determine if the study warranted inclusion of the third cohort to enable achieving the null hypothesis addressing procognitive effects with systemic LY379268 administration. No procognitive effects were observed during data analysis of the first two cohorts thereby inhibiting achievement of our null hypothesis, and we subsequently discontinued the study prior to beginning the third cohort.
Chemicals
All chemicals were prepared and stored according to manufacturer recommendations unless otherwise noted. L-glutamate oxidase (EC 1.4.3.11) was obtained from Cosmo Bio Co. (Carlsbad, CA) and diluted in distilled, deionized water to make a 1 U/μl stock solution for storage at 4°C. Sodium phosphate monobasic monohydrate, sodium phosphate dibasic anhydrous, 1,3 phenylenediamine dihydrochloride (mPD), sodium chloride, calcium chloride dehydrate, and H2O2 (30% in water) were obtained from Thermo Fisher Scientific (Waltham, MA). L-glutamic acid sodium salt, bovine serum albumin (BSA), glutaraldehyde, KCl, dopamine hydrochloride (DA), L-ascorbic acid (AA), were obtained from Sigma-Aldrich Co. (St. Louis, MO). LY379268 was obtained from Tocris Bioscience (Bristol, United Kingdom), while Amylo-Glo® RTD™ was obtained from Biosensis (Temecula, CA).
Morris Water Maze (MWM) training and probe challenge
At approximately 12 months of age, mice underwent cognitive assessment using the MWM spatial learning and memory recall paradigm. For this test, mice were trained to utilize visual cues placed around the room to repeatedly swim to a static, hidden escape platform (submerged 1 cm below the opaque water surface) regardless of starting quadrant [10, 32]. The MWM paradigm consisted of 5 consecutive training days with three, 90 s trials/day and a minimum inter-trial-interval of 20 min. Starting quadrant was randomized for each trial. After two days without testing, the escape platform was removed and all mice entered the pool of water from the same starting position for a single, 60 s probe challenge to test long-term memory recall. The ANY-maze video tracking system (Stoelting Co., Wood Dale, IL; RRID:SCR_014289) was used to record mouse navigation during the training and probe challenge. The three trials for each training day were averaged for each mouse for analysis.
Enzyme-based microelectrode arrays
Enzyme-based MEAs with platinum (Pt) recording surfaces were fabricated, assembled, coated, and calibrated for in vivo mouse glutamate measurements as previously described [33 –35]. One of the R2 MEA Pt sites was coated with BSA, glutaraldehyde, L-glutamate oxidase solution that aides in enzyme adhesion to enzymatically degrade glutamate to α-ketoglutarate and H2O2, the electroactive reporter molecule. The second Pt recording site (self-referencing or sentinel site) was coated with a BSA and glutaraldehyde solution, which is unable to enzymatically generate H2O2 from L-glutamate. A potential of +0.7V versus an Ag/AgCl reference electrode was applied to the Pt recording surfaces resulting in oxidation of the H2O2 reporter molecule. The subsequent current generated from the two electron transfer was amplified and digitized by the Fast Analytical Sensing Technology (FAST) 16mkIII (Quanteon, LLC; Nicholasville, KY) electrochemistry instrument.
mPD electropolymerization
After enzyme coating, all Pt recording surfaces were electroplated with 5 mM mPD in 0.05 M phosphate buffered saline (PBS). FAST electroplating software applied a triangular wave potential with an offset of – 0.5V, peak-to-peak amplitude of 0.25V, at a frequency of 0.05 Hz, for 20 min. This created a size exclusion layer that restricts the passage of AA, DA, uric acid and 3,4-dihydroxyphenylacetic acid to the Pt recording surface [32].
Calibration
Each MEA underwent an in vitro calibration prior to implantation to create a standard curve for the conversion of current to glutamate concentration. The Pt recording sites and a glass Ag/AgCl reference electrode (Bioanalytical Systems, Inc., West Lafayette, IN) were placed in a continuously stirred solution of 40.0 mL of 0.05 M PBS maintained at 37°C with a recirculating water bath (Stryker Corp., Kalamazoo, MI). Final beaker concentrations of 250μM AA, 10, 20, 30, and 40μM L-glutamate, 2μM DA, and 8.8μM H2O2 were used to assess MEA performance. A total of 24 MEAs (8 unique) were used in the present study. The average±standard error of the mean (SEM) for glutamate sensitivity was 5.9±0.1 pA/μM (R2 = 0.998±0.001), selectivity ratio of 400±99 to 1, and limit of detection of 0.18±0.03μM based on a signal-to-noise ratio of 3.
Microelectrode array/micropipette assembly
A glass micropipette (1.0 mm outer diameter, 0.58 mm internal diameter; World Precision Instruments, Inc., Sarasota, FL) was used to locally apply solutions to the mouse hippocampal subfields. Glass micropipettes were pulled using a vertical micropipette puller (Sutter Instrument Co., Novato, CA) and the tip was “bumped” to create an internal diameter of 12–15μm. The tip of the micropipette was positioned between the pair of recording sites and mounted 100μm above the MEA surface. The micropipettes were filled with sterile filtered (0.20μm) 70 mM KCl (70 mM KCl, 79 mM NaCl, and 2.5 mM CaCl2, pH 7.4). Fluid was pressure-ejected from the glass micropipette using a Picospritzer III (Parker-Hannafin, Cleveland, OH), with pressure (5–15 psi) adjusted to consistently deliver volumes between 100–200 nl over 1-2 s intervals. Ejection volumes were monitored with a stereomicroscope (Luxo Corp., Elmsford, NY) fitted with a calibrated reticule [36].
Reference electrode
An Ag/AgCl reference electrode was prepared by stripping ∼5 mm of the Teflon off the silver wire (200μm bare, 275 μm coated; A-M Systems, Carlsberg, WA) from both ends. One stripped end was soldered to a gold-plated test connector (Newark element14, Chicago, IL) and the other end was coated with AgCl by placing the tip of the stripped sliver wire (cathode) into a 1 M HCl plating bath saturated with NaCl containing a stainless steel wire (anode) and applying +9 V DC using a power supply to the cathode versus the anode for 15 min.
In vivo anesthetized recordings
Beginning one week after MWM, mice were anesthetized using 1.5–2.0% isoflurane (Abbott Lab, North Chicago, IL) in a calibrated vaporizer (Vaporizer Sales & Service, Inc., Rockmart, GA) and prepared for in vivo electrochemical recordings [32]. The mouse was placed in a stereotaxic frame fitted with a mouse anesthesia mask (David Kopf Instruments, Tujunga, CA) and body temperature was maintained at 37°C with a water pad (Braintree Scientific Inc., Braintree, MA) connected to a recirculating water bath. A craniotomy was performed to access the dentate (DG; AP: – 2.0, ML: ±1.0, DV: – 2.2 mm), CA3 (AP: – 2.0, ML: ±2.0, DV: – 2.2 mm), and CA1 (AP: – 2.0, ML: ±1.0, DV: – 1.7 mm) from Bregma [37]. A Ag/AgCl reference wire was positioned beneath the skull and rostral to the right hemisphere craniotomy. Constant voltage amperometry (4 Hz) was performed by using a potential of +0.7 V versus the Ag/AgCl reference electrode applied by the FAST16mkIII electrochemical instrument. MEAs were allowed to reach a stable baseline for 60 min before a 10-s basal glutamate determination and pressure ejection studies commenced. The FAST software saves amperometric data, time, and pressure ejection events for all recording sites. Calibration data, in conjunction with a MATLAB (MathWorks, Natick, MA; RRID:SCR_001622) graphic user interface program was used to calculate basal glutamate, stimulus-evoked glutamate release, and glutamate uptake rate. Five reproducible signals were evoked in each hippocampal subfield and averaged into a representative signal for treatment comparisons.
Immunohistochemical staining and semi-quantification
Following in vivo electrochemistry, the brains were removed and post-fixed in 4% paraformaldehyde for 48 h and then transferred into 30% sucrose in 0.1 M PB for at least 24 h prior to sectioning. A cryostat (Model HM525 NX, ThermoFisher Scientific) was used to obtain 20μm sections of the hippocampus. Serial sections (every 6th) underwent IHC using chicken polyclonal glial fibrillary acidic protein (GFAP) antibody (Biosensis; 1:1000; RRID:AB_2492333) and Amylo-Glo® RTD™ amyloid plaque stain reagent (Biosensis; 1:100; TR-300-AG) or glutamate transporter 1 (Glt-1) antibody (ThermoFisher Scientific; 1:100; RRID: AB_10980198). Slides were treated with 10% H2O2 in 20% methanol for 10 min and then transferred to a 70% ethanol solution for 5 min followed by a 2 min wash in PBS. Sections were incubated for 10 min in Amylo-Glo® RTD™ and rinsed in 0.9% saline for 5 min without shaking followed by rinsing (3 2 min) in PBS. Sections were permeabilized in phosphate buffered saline with 0.25% TritonX-100 (PBST) followed by washes (3×10 min) in sodium borohydride in PBS (1 mg/ml) for antigen retrieval. To control for nonspecific binding, sections were washed (3×10 min) with PBST and incubated in 10% normal goat serum for 1 h followed by overnight incubation (4°C) with primary antibody. The next day, sections were washed (3×10 min) in PBST and incubated at room temperature for 1 h with AlexaFlour 594 goat anti-chicken (ThermoFisher Scientific; 1:1000; RRID:AB_2534099) or AlexaFlour 594 goat anti-rabbit (ThermoFisher Scientific; 1:1000; RRID: AB_2534079). Afterwards, sections were washed (3×10 min) in PBST and coverslipped using Fluoromount-G (SouthernBiotech; Birmingham, AL). To control for staining intensity, all sections were allowed to develop overnight and imaged the following day. Staining intensity of hippocampal plaque formation was determined using National Institutes of Health Image J Software (v. 1.48; RRID:SCR_003070) to measure a gray scale value within the range of 0–256, where 0 represents white and 256 represents black. Individual templates for the DG, CA3, and CA1 were created and used on all brains similarly. Images were captured with an Olympus 1×71 microscope equipped with an Olympus-DP73 video camera system, and a Dell Optiplex 7020 computer. Measurements were performed blinded, and approximately five sections were averaged to obtain one value per subject. Staining density was obtained when background staining was subtracted from mean staining intensities on every sixth section through the hippocampus.
Data analysis
Prism (GraphPad Software, Inc., La Jolla, CA; RRID:SCR_002798) software was used for statistical analyses. For glutamate measurements and IHC, hippocampal subregions were examined independently because of different cell types and afferent inputs. Mouse weights and MWM training were analyzed using a two-way analysis of variance (ANOVA), while a one-way ANOVA was used for the probe challenge, glutamate measures, and GFAP IHC. When the ANOVA indicated a statistically significant main effect, a Fisher’s LSD post-hoc test was used to determine treatment differences. A two-tailed unpaired t-test was used for amyloid plaque analysis. Outliers were identified with a single Grubbs’ test (alpha = 0.05) per group. Data are represented as mean±SEM and statistical significance was defined as p < 0.05.
RESULTS
LY379268 mouse weights
For each mouse, weight was determined twice weekly during the four-month treatment with LY379268 or control and then again during the MWM (12 months of age). The average weight during the first week of treatment served as the initial weight for the percent change calculations shown in Fig. 2. A main effect of treatment groups was observed (F2,20 = 6.19; p = 0.01). A post-hoc analysis revealed that AβPP/PS1 vehicle mice had a significantly higher percentage of weight gain compared to C57BL/6J vehicle mice throughout the duration of the treatment. By 12 months of age, AβPP/PS1 control mice gained significantly more weight compared to both C57BL/6J vehicle and the AβPP/PS1 LY379268 experimental group. No differences were observed between C57BL/6J vehicle and AβPP/PS1 LY379268 treated mice.
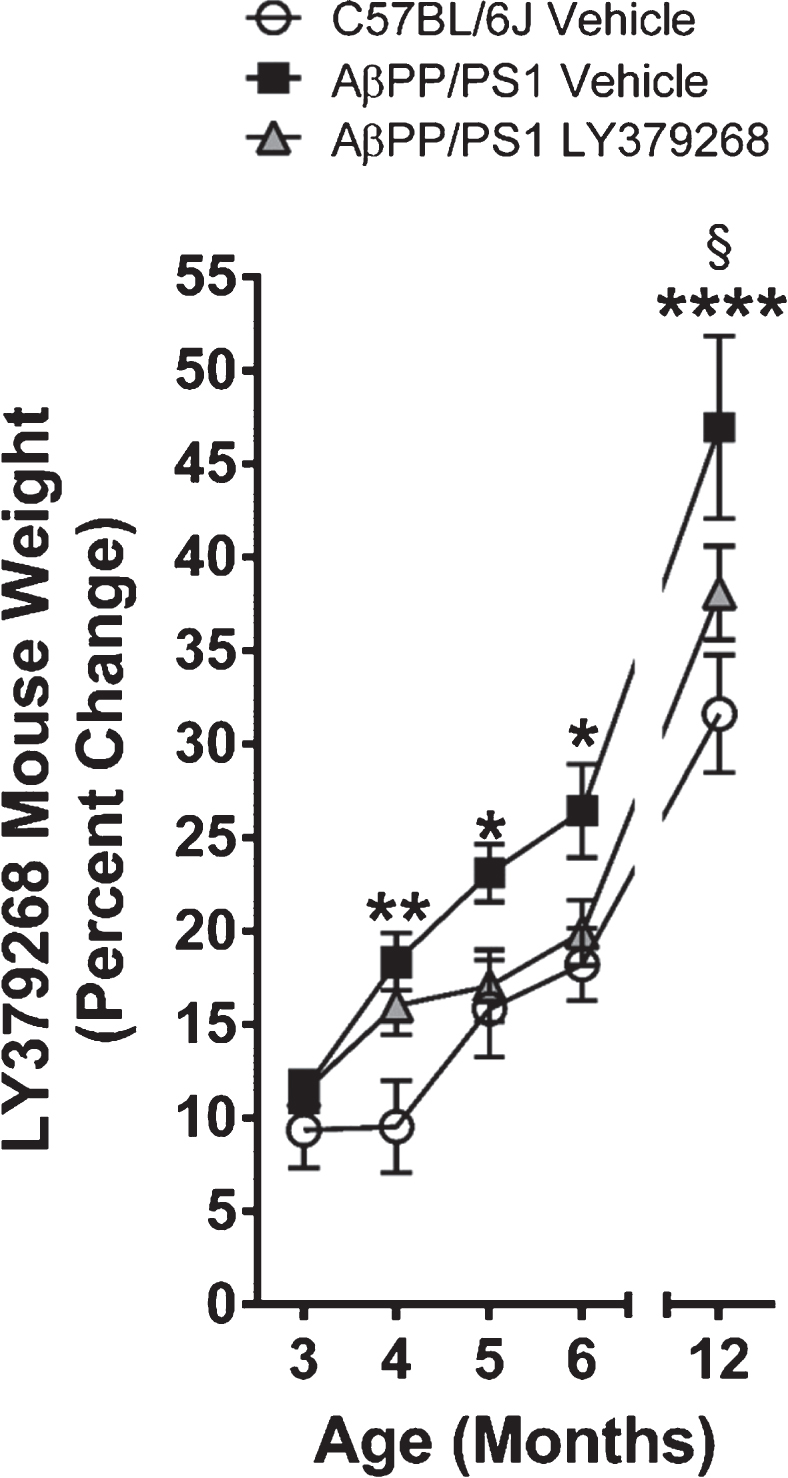
Mouse weight gain. Analysis of the percentage of mouse weight gain from the first week of treatment through 12 months of age. * p < 0.05, ** p < 0.01, **** p < 0.0001 AβPP/PS1 vehicle (n = 8) versus C57BL/6J vehicle (n = 8); § p < 0.05 AβPP/PS1 vehicle versus AβPP/PS1 LY379268 (n = 7).
LY379268 cognitive assessment
Six months post treatment, cognitive performance was assessed on all mice using the MWM learning and memory recall behavioral paradigm. Over the five-day training session, a significant main effect of the corrected integrated path length (CIPL; Fig. 3A; F4,80 = 73.12, p < 0.0001) and cumulative distance from the platform (Fig. 3B; F4,80 = 72.14, p < 0.0001) was observed indicating all treatment groups had learned the location of the hidden escape platform. Additionally, a main effect of treatment groups was also observed for the CIPL (F2,20 = 4.03, p = 0.03) and cumulative distance from the platform (F2,20 = 4.08, p = 0.03). A post-hoc analysis revealed on the first training day, AβPP/PS1 vehicle mice found the location of the escape platform slower compared to C57BL/6J vehicle and AβPP/PS1 LY379268 mice. No differences were observed over subsequent training days. After the 5 training days and 2 rest days, the hidden escape platform was removed and mice were subjected to a 60 s probe challenge. Representative track plots are shown in Fig. 3C. During the probe challenge, an increased trend of platform crossings was observed in C57BL/6J vehicle mice compared to both AβPP/PS1 mouse groups. When examining the cumulative distance from the platform, the main effect of treatment groups approached significance (F2,20 = 3.115; p = 0.06) with C57BL/6J vehicle searching in closer proximity to the former platform location compared to both AβPP/PS1 mouse groups. Furthermore, C57BL/6J mice spent more time moving towards the former platform location (F2,20 = 3.933; p = 0.03) compared with AβPP/PS1 vehicle (p = 0.02) and AβPP/PS1 LY379268 (p = 0.02) treated mice. These data support that four months of LY379268 treatment had minimal improvement in learning, but did not improve long-term memory recall in AβPP/PS1 mice.

MWM training and probe challenge. During the 5-day MWM training, each day consisted of 3 trials that were averaged into a single data point for the CIPL (A) and cumulative distance from the platform (B) for each treatment group. C) Representative track plots are shown for each treatment group. D) The percentage of time each treatment group spent in the individual quadrants. E) The number of annulus 40 entries for each treatment group. F) The total time each treatment group spent moving towards the platform. A, B) *** p < 0.001 AβPP/PS1 vehicle (n = 8) versus C57BL/6J vehicle (n = 8) mice, § p < 0.05 AβPP/PS1 vehicle versus AβPP/PS1 LY379268 (n = 7) mice. D) * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001 versus target quadrant.
LY379268 glutamate measures
We used an enzyme-based MEA to measure basal glutamate and glutamate dynamics independently from the DG, CA3, and CA1. Representative traces from each hippocampal subregion for all treatment groups are presented in Fig. 4. A main effect of treatment group on basal glutamate (Fig. 5A) was observed in the DG (F2,19 = 3.637, p = 0.04) and CA1 (F2,17 = 3.919, p = 0.04), but not the CA3 (F2,19 = 2.607, p = 0.10). A post hoc analysis indicated that basal glutamate was significantly elevated in AβPP/PS1 LY379268 mice compared to C57BL/6J control mice in the DG and CA1. To stimulate glutamate release, consistent volumes of 70 mM KCl (100–200 nl) were pressure ejected across all treatment groups in the DG (F2,19 = 0.3314, p = 0.72), CA3 (F2,19 = 0.7110, p = 0.50), and CA1 (F2,17 = 0.5392, p = 0.59). A main effect of treatment group on stimulus-evoked glutamate release (Fig. 5B) was observed in the DG (F2,19 = 4.277, p = 0.03) and CA1 (F2,17 = 3.496, p = 0.05), but not the CA3 (F2,19 = 0.2301, p = 0.80). A post hoc analysis indicated elicited glutamate release was increased in the DG of AβPP/PS1 vehicle and LY379268 treatment groups compared to C57BL/6J vehicle mice. In the CA1, stimulus-evoked glutamate release was increased in AβPP/PS1 vehicle, but not LY379268 treated mice, compared to C57BL/6J vehicle mice. Clearance of stimulus-evoked glutamate release was evaluated by examining the linear portion of the signal decay that was observed between the T20 and T60 time points [38] as shown in Fig. 5C. In this regard, a main effect of treatment group was only observed in the DG (F2,19 = 3.561, p = 0.04). A post hoc analysis indicated glutamate clearance was significantly elevated in the AβPP/PS1 vehicle compared to C57BL/6J vehicle mice. To examine the extent of change in the amount of glutamate released with respect to time we analyzed the area under the curve (AUC). While a trend towards prolonged glutamate release was observed in the DG and CA1 of both AβPP/PS1 Saline and LY379268 treated mice, no main effects of treatment groups were observed (Fig. 5D). These data support that four months of early intervention with LY379268 was insufficient to reduce the hippocampal glutamatergic tone, and caused elevated basal glutamate compared to C57BL/6J and AβPP/PS1 vehicle mice. However, caution should be taken when interpreting these results as counterintuitive to the known pharmacology of LY379268 since the basal glutamate levels in both AβPP/PS1 treatment groups are in agreement with previously published results using similarly aged AβPP/PS1 mice [20].

Representative glutamate traces. Representative traces of glutamate release from 70 mM KCl stimulation. Columns indicate treatment groups while rows indicate hippocampal subfields. The inset trace at the top of each panel depicts the reproducibility of the glutamate signals. The single response shown beneath is a magnified view of the first inset signal (dashed box) designed to give a clearer presentation of glutamate dynamics. Concentration and time axes are consistent in all panels for comparative interpretation.
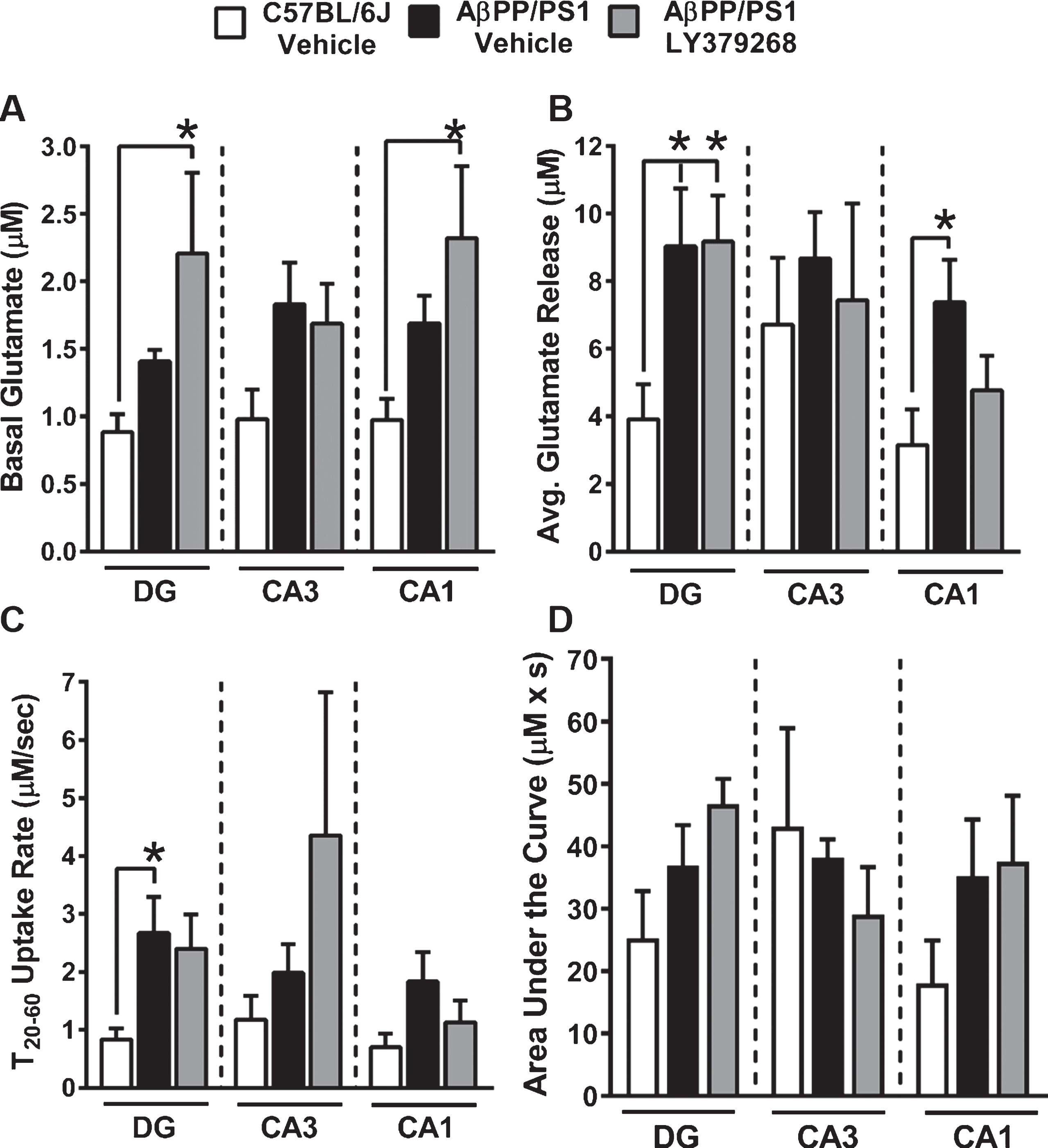
Stimulus-evoked hippocampal glutamate measures. A) Basal glutamate was determined prior to local application of stimulus in each hippocampal subfield. B) The average glutamate release from local application of 70 mM KCl was determined by subtracting the peak amplitude from the basal measure prior to ejection of stimulus. C) Glutamate uptake rate was calculated by determining the change in amplitude (μM) between 20–60% maximal amplitude divided by the corresponding length of time (s) for this signal decay. D) The change in extracellular glutamate concentration over time was determined by calculating the AUC for each evoked glutamate signal. * p < 0.05 C57BL/6J vehicle (n = 6-7) versus AβPP/PS1 LY379268 treated (n = 6-7) mice.
GLT-1 IHC
Glutamate clearance is predominantly mediated through glial high affinity amino acid transporters, of which GLT-1 accounts for ∼90% uptake [39]. To determine if differences in stimulus-evoked glutamate release were a result of transporter density, we used IHC to measure changes in GLT-1 expression. Representative 40x magnification images from the DG of C57BL/6J vehicle, AβPP/PS1 vehicle, and AβPP/PS1 LY379268 treated mice are shown. No differences in the average mean density of GLT-1 for each hippocampal subregion were observed (Fig. 6D), suggesting differences in stimulus-evoked glutamate release are not attributed to GLT-1 density.
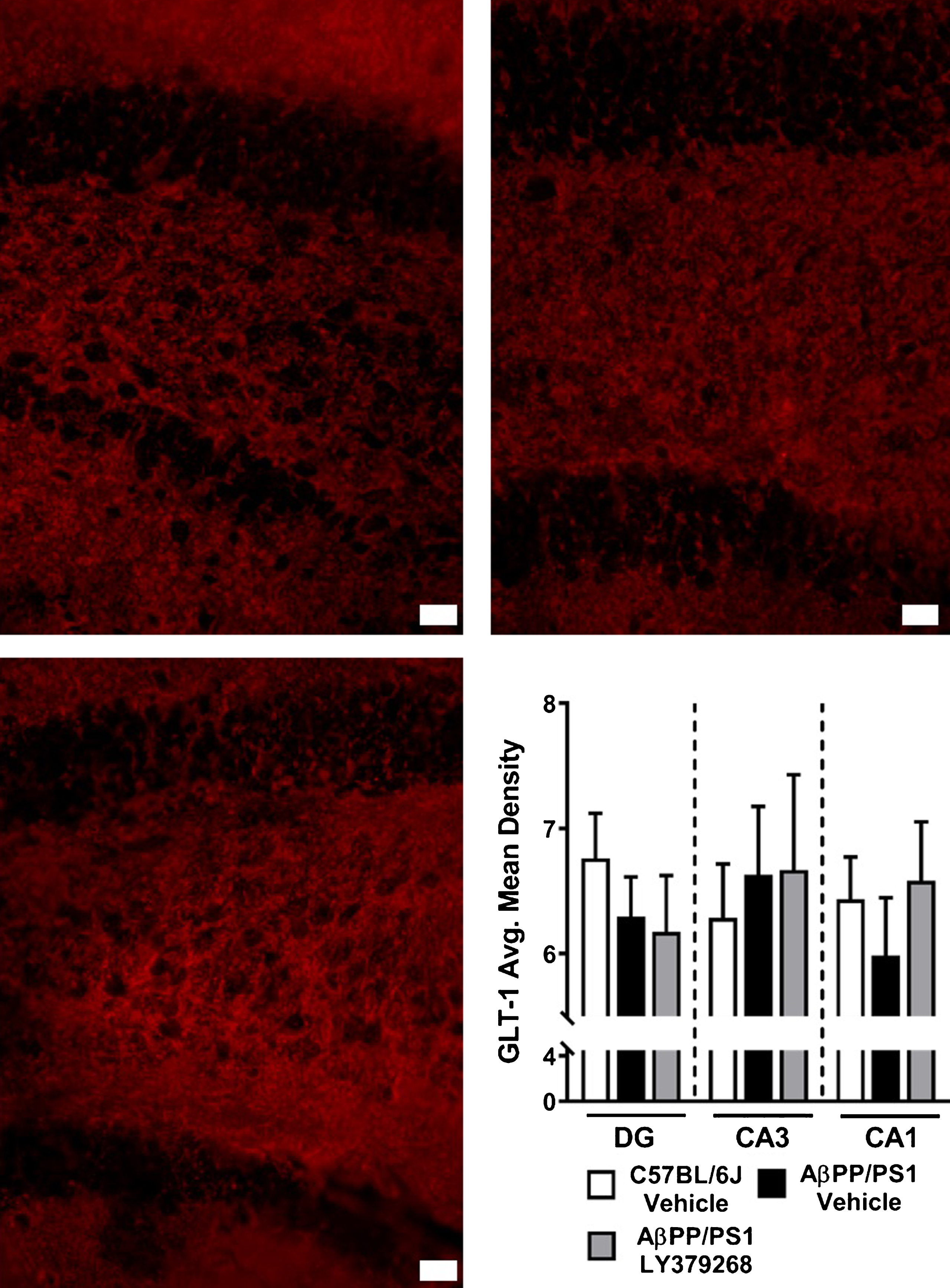
Hippocampal GLT-1 Immunohistochemistry. DG representative images at 40x magnification of GLT-1 in C57BL/6J vehicle (A), AβPP/PS1 Vehicle (B), and AβPP/PS1 LY379268 (C) treated mice. Scale bar represents 20μm. Average mean density of GLT-1 staining (D) in the DG, CA3, and CA1 of C57BL/6J vehicle (n = 8), AβPP/PS1 Vehicle (n = 8), and AβPP/PS1 LY379268 (n = 6) treated mice.
GFAP and amyloid plaque IHC
IHC was used to measure changes in GFAP expression and amyloid plaque pathology throughout the hippocampus. Representative 10x magnification images from the DG of C57BL/6J vehicle (Fig. 7A-C), AβPP/PS1 vehicle (Fig. 7D-F), and AβPP/PS1 LY379268 (Fig. 7G-I) treated mice are shown. Merged images highlight astrogliotic responses surrounding amyloid plaques in both cohorts of AβPP/PS1 mice. GFAP average mean density for each hippocampal subfield are presented in Fig. 7J. AβPP/PS1 vehicle mice showed a trend toward elevated astrogliosis compared to C57BL/6J vehicle mice throughout the hippocampus, particularly in the DG (F2,20 = 2.957; p = 0.075). LY379268 treatment in AβPP/PS1 mice did not significantly attenuate the astrogliotic response in any hippocampal subregion. Amyloid plaque hippocampal average mean density and plaque counts per slice for both cohorts of AβPP/PS1 mice are shown in Fig. 7K-L. Treatment with LY379268 had no effect on hippocampal amyloid plaque formation.

Hippocampal GFAP and amyloid plaque immunohistochemistry. DG representative images at 10x magnification of GFAP (left panels, red), amyloid plaques (middle panels, blue), and merged images (right panels) from C57BL/6J vehicle (A-C), AβPP/PS1 vehicle (D-E), and AβPP/PS1 LY379268 (H-I) treated mice. Scale bar represents 100μm and arrow heads (F, I) highlight plaques. Average mean density of GFAP staining (J) and amyloid plaque burden (K) in the DG, CA3, and CA1 of C57BL/6J vehicle (n = 8), AβPP/PS1 vehicle (n = 8) and AβPP/PS1 LY379268 (n = 7) treated mice. Average hippocampal plaque counts per slice in AβPP/PS1 vehicle (n = 8) and AβPP/PS1 LY379268 (n = 7) treated mice.
DISCUSSION
Group II (mGluR2/3) are expressed in the cortex and hippocampus [22] and act as extrasynaptic autoinhibitory modulators that suppress glutamate release from presynaptic neurons [40]. As such, agonists targeting mGluR2/3 receptors are ideal for therapeutic interventions and have good success at alleviating anxiety and stress in preclinical models [21, 41]. However, their role in neurodegenerative disorders and especially AD pathogenesis is less understood. Postmortem tissue from AD patients shows increased hippocampal expression of mGluR2 that is associated with degenerating neurons [42]. But the reason for this upregulation is unknown. Since we have previously demonstrated that Aβ42 elicits glutamate release [36] upregulation of mGluR2 could be a compensatory mechanism to prevent Aβ42-mediated excitotoxicity, as observed in cell culture [43]. However, other studies support that stimulation of mGluR2/3 triggers production and release of Aβ42, and therefore results in the plaque deposition and pathogenesis of AD [44]. These limited and conflicting reports indicated additional research was required to determine the potential therapeutic benefits from an mGluR2/3 agonist in mouse models of AD.
The mGluR2/3 agonist, LY379268, and dosing strategy was based upon previously published reports regarding the efficacy of the compound [26, 45]. LY379268 binds to the orthosteric site on mGluR2/3 receptors with high potency compared to Groups I and III [40] with minimal adverse events when administered systemically [31, 46]. Pharmacokinetic studies have shown that LY379268 crosses the blood-brain barrier with a peak brain concentration 30 min after i.p. injection and maintains cerebral receptor active concentrations for a minimum of 24-h post injection [26]. This long duration of receptor activation results in neuroprotective effects lasting up to 28 days post ischemia [26, 27]. Local application of LY379268 decreases basal and stimulus-evoked glutamate release [24, 25]. And, systemic administration of LY379268 for 5 consecutive days blocks ketamine-induced hippocampal glutamate efflux as measured by enzyme-based amperometric biosensors [45].
LY379268 prevents Aβ-induced toxicity [43], but the long-term effects of LY379268 on cognition and glutamatergic signaling in models of AD have not been addressed. For the current study, treatment with LY379268 began prior to the onset of typically reported AD-related pathology in AβPP/PS1 mice [28], but during a time point when hippocampal glutamate levels were elevated [10]. This therapeutic window was chosen to address our hypothesis that systemic administration of LY379268 could provide long-term procognitive effects that were mediated by reduction of the hippocampal glutamatergic tone in AβPP/PS1 mice. Since we did not observe long-term procognitive effects with LY379268 administration, we decided against allocating additional mice to elucidate potential mechanistic changes during acute treatment for ethical considerations. Although acute effects of LY379268 systemic administration in AD mouse models has not been addressed, previous studies support that 24 h pretreatment attenuates hippocampal stimulated glutamate release in male C57BL/6J mice [45]. The inclusion of C57BL/6J saline mice was used to determine if prodromal intervention with systemic administration of LY379268 resulted in procognitive benefits similar to age-matched genetic background control mice.
Weight gain was monitored throughout the study to determine tolerability of LY379268 since previous studies have shown mGluR2/3 agonists may be nauseous to rodents [41]. AβPP/PS1 vehicle mice gained more weight compared to C57BL/6J vehicle and AβPP/PS1 LY379268 treated mice throughout the study. Interestingly, by two months on LY379268 treatment, weight gain in AβPP/PS1 mice followed that of C57BL/6J vehicle mice for the remainder of the treatment, and was decreased at 12 months of age compared to AβPP/PS1 vehicle mice. While peripheral glucose was not monitored in this study, insulin resistance precedes cognitive decline in AβPP/PS1 mice [20 , 48] and may explain the increased b.w. But, the lower b.w. of AβPP/PS1 LY379268 treated mice are not necessarily attributed to a side-effect of the medication since averages were similar to C57BL/6J vehicle mice. Rather, LY379268 treatment may have altered the metabolic profile of AβPP/PS1 mice, because stimulation of mGluR2/3 on pancreatic β-cells causes insulin release [49].
Hippocampal basal glutamate and stimulus evoked glutamate release was elevated in AβPP/PS1 mice, similar to previous observations from our laboratory at this age. This elevated extracellular glutamate in AβPP/PS1 mice may result from the progressive accumulation of soluble Aβ42 [50] that can elicit glutamate release [18 , 36]. However, treatment with LY379268 did not attenuate hippocampal basal nor stimulus-evoked glutamate release, which has been previously demonstrated with local application studies [24, 25]. In fact, DG and CA1 basal glutamate was unexpectedly potentiated with LY379268 treatment, counterintuitive to its reported pharmacology. These elevated glutamate levels were not a result of changes in GLT-1 receptor density which accounts for 90% of glutamate clearance [39]. Additionally, we cannot rule out long term mGluR2/3 downregulation after repeated systemic administration leading to the elevated basal glutamate values. However, as noted in the results, these AβPP/PS1 basal glutamate values are similar to those reported elsewhere at 12–15 months of age [20]. Considering the basal glutamate levels are statistically similar between AβPP/PS1 treatment groups, the effects of cohort variance, rather than long-term LY379268 administration, may also be a factor.
The elevated basal glutamate levels in AβPP/PS1 LY379268 treated mice may also be attributed to the cellular distribution of hippocampal mGluR2/3 receptors and their mechanisms of action. Agonism of presynaptic mGluR2/3 immediately attenuates glutamate release by hyperpolarization of the neuron. But, agonism of postsynaptic mGluR2/3 is coupled to downstream regulators that increases surface expression of α-amino-3-hydroxy-5-methyl-4-isoxazole propionate (AMPA) and NMDA receptors and subsequent longer term modulation of glutamatergic neurotransmission [51, 52]. This suggests dual neuroprotective mechanisms to modulate hyperglutamatergic neurotransmission [53]. These receptors were not assayed because our main hypothesis was based on presynaptic modulation of glutamatergic signaling. Also, LY379268 agonism of glial mGluR3 has a separate function that induces production of neurotrophic factors [54] lasting 6-72-h post treatment [55, 56]. Although systemic administration of LY379268 is present in the brain at levels sufficient to activate mGluR2/3 for at least 24-h post treatment [26], the 6-month off-treatment duration may have allowed the AD pathophysiology to resume in AβPP/PS1 mice a few days after LY379268 treatment was discontinued despite long-term modulation through postsynaptic and glial mechanisms.
Besides differences in cellular distributions, mGluR2/3 are distinctly spread across the central nervous system, particularly within regions of the hippocampus. This regional localization, in combination the cellular distribution, helps to explain the basal glutamate and stimulus-evoked glutamate release observed in the present study. Hippocampal mGluR2 is localized to the presynaptic axons of the medial perforant pathway originating from the entorhinal cortex and terminating in the DG, CA3, and CA1 [57, 58]. The preterminal localization indicates mGluR2 directly contributes to hyperpolarizing axons thereby immediately attenuating glutamate release. Therefore, decreased hippocampal basal glutamate six months post LY379268 treatment would not be expected and is consistent with our present findings. However, mGluR3 is localized on glia in the CA1 and dorsolateral entorhinal cortex [57, 58] that would lead to production of neurotrophic factors and potentially cause long-term alterations of membrane excitability [59]. This would support the attenuated stimulus-evoked glutamate release observed in the CA1 six months post LY379268 treatment in AβPP/PS1 mice.
Since mGluR2/3 plays a role in hippocampal spatial working memory tasks [60], drugs stimulating this class of receptors prevents cognitive deficits associated with traumatic brain injury [31], alcohol exposure [61], and phencyclidine models of schizophrenia [62]. To test the procognitive effects of mGluR2/3 activation, LY379268 intervention began prior to the onset of cognitive deficits that have been reported as early as 6–8 months [63, 64] and continue throughout the lifespan of AβPP/PS1 mice [28]. LY379268 intervention in AβPP/PS1 mice only improved platform location on the first training day with subsequent training days similar between AβPP/PS1 cohorts. This first day MWM performance improvement may be due to the anxiolytic properties associated with agonism of mGluR2/3 by LY379268 [65], allowing the AβPP/PS1 mice to learn the location of the escape platform faster. Surprisingly, memory recall deficits during the probe challenge were slightly exacerbated by LY379268 treatment as indicated by fewer platform crossings and cumulative distance from the platform. The elevated extracellular hippocampal glutamate would cause over activation of the NMDA receptor, which is important during spatial navigation tasks [66]. This over activation disrupts the signal-to-noise ratio thereby impairing detection of phasic signaling and blocking formation of new memories [13].
Similar to our previous research, AβPP/PS1 vehicle mice had pronounced Aβ plaque deposition throughout the hippocampus by 12–15 months [20]. In AβPP/PS1 mice, Aβ plaque accumulation is typically observed starting at 6 months and increases with age [67, 68]. Yet, despite initiating LY379268 treatment at 2 months, no effect on hippocampal Aβ plaque formation was observed. Studies in isolated nerve terminals support activation of mGluR2/3 releases Aβ42 suggesting a possible trigger for Aβ plaque formation [44]. While our data does not support increased Aβ plaque deposition, an increased release of soluble Aβ42 may be responsible for the elevated hippocampal basal glutamate mediated through the α7nAChR as previously discussed.
Elevated GFAP expression, a marker of reactive astrocytes, was observed in AβPP/PS1 vehicle treated mice, which is in concordance with our previous research. Prodromal LY379268 treatment had a negligible effect on GFAP expression levels in AβPP/PS1 mice. In both cohorts of AβPP/PS1 mice, GFAP expression was prominent around amyloid plaques in agreement with previously published reports [67]. The astroglial juxtaposition is hypothesized to control Aβ plaque deposition [69] through a variety of clearance mechanisms [70].
Conclusion
The data presented here demonstrate that early intervention with LY379268 does not induce long-term procognitive effects nor reduce hippocampal glutamatergic tone in AβPP/PS1 mice. When it became apparent that the primary objectives of LY379268 treatment in AβPP/PS1 mice was not achievable, the study was discontinued for ethical considerations. The current study does, however, build upon previous research demonstrating hyperglutamatergic hippocampal signaling that begins at 2–4 months of age in AβPP/PS1 mice that may be mediated by progressive accumulation of soluble Aβ42. Attenuation of the glutamatergic tone may serve as a viable therapeutic strategy that ameliorates cognitive deficits while preventing the excitoxicity mediated neurodegeneration reported in AD. Unfortunately, the lack of therapeutic efficacy observed may be indicative of mGluR2/3 regional and cellular distribution and the subsequent mechanisms to attenuate hyperglutamatergic activity. As such, future studies will address whether other glutamatergic modulating compounds are effective at alleviating AD pathophysiology in AβPP/PS1 mice.
Footnotes
ACKNOWLEDGMENTS
Research reported in this publication was supported by the National Institute On Aging of the National Institutes of Health under Award Number R01AG057767 and R01AG061937, the Illinois Department of Public Health under Award Number 63282003D, the Center for Alzheimer’s Disease and Related Disorders at Southern Illinois University School of Medicine, the Kenneth Stark Endowment, the Fraternal Order of Eagles (KNH, CAF, ERH), and the Southern Illinois University Foundation Award (JB, ST), and does not necessarily represent the official views of the funding agencies.