
Abstract
Metformin is used for the treatment of insulin resistant diabetes. Diabetics are at an increased risk of developing dementia. Recent epidemiological studies suggest that metformin treatment prevents cognitive decline in diabetics. A pilot clinical study found cognitive improvement with metformin in patients with mild cognitive impairment (MCI). Preclinical studies suggest metformin reduces Alzheimer-like pathology in mouse models of Alzheimer’s disease (AD). In the current study, we used 11-month-old SAMP8 mice. Mice were given daily injections of metformin at 20 mg/kg/sc or 200 mg/kg/sc for eight weeks. After four weeks, mice were tested in T-maze footshock avoidance, object recognition, and Barnes maze. At the end of the study, brain tissue was collected for analysis of PKC (PKCζ, PKCι, PKCα, PKCγ, PKCɛ), GSK-3β, pGSK-3βser9, pGSK-3βtyr216, pTau404, and APP. Metformin improved both acquisition and retention in SAMP8 mice in T-maze footshock avoidance, retention in novel object recognition, and acquisition in the Barnes maze. Biochemical analysis indicated that metformin increased both atypical and conventional forms of PKC; PKCζ, and PKCα at 20 mg/kg. Metformin significantly increased pGSK-3βser9 at 200 mg/kg, and decreased Aβ at 20 mg/kg and pTau404 and APPc99 at both 20 mg/kg and 200 mg/kg. There were no differences in blood glucose levels between the aged vehicle and metformin treated mice. Metformin improved learning and memory in the SAMP8 mouse model of spontaneous onset AD. Biochemical analysis indicates that metformin improved memory by decreasing APPc99 and pTau. The current study lends support to the therapeutic potential of metformin for AD.
INTRODUCTION
The incidence of Alzheimer’s disease (AD) is on the rise. To date there are no effective treatments to stop or reverse the progression of the disease. The primary focus is now on early detection and prevention [1]. Obesity and diabetes mellitus have been identified as risk factors for dementia [2]. Studies have focused on repurposing of drugs used to treat diabetes as possible treatments for AD [3]. Metformin is a biguanide class drug that has been a mainstay in the treatment of diabetes. Human studies suggest that type II diabetics on metformin are less likely to develop dementia than patients on other forms of treatment [4–6]. Preliminary data suggests that metformin may be useful in the treatment of dementia [5, 7].
Studies of metformin on dementia in rodents have been promising [6]. In diabetic mice, metformin was found to restore abnormal blood-brain barrier transport of amyloid-β (Aβ) along with improving memory [8]. Metformin was found to activate the protein kinase C-CREB binding protein (PKC-CBP) pathway to promote neurogenesis and increase atypical PKC in non-diabetic mice [9]. PKC plays a critical role in learning and memory and has been suggested to play a critical role in dementia [10]. Activation of the PKC pathway in AD leads to a decrease in GSK3β activation as well as an increase in ERK1/2 leading to increase α secretase and ECE-1 [11]. These functions lead to decreases in Aβ. In addition, metformin decreased BACE-1, the rate limiting enzyme responsible for the conversion of amyloid-β protein precursor (AβPP) to Aβ, in cells and non-diabetic mice [12].
In humans, there are reports of positive effects on the prevention of dementia with metformin in older patients with diabetes [4, 14], there have been some negative reports. In a study of a database in Taiwan, long-term metformin use was associated with an increase in neurocognitive disorders [15]. A separate study found the negative effects on cognition with metformin use was linked to decreased vitamin B12 after chronic metformin usage [16]. In rodents, metformin in drinking water at a dose resulting in 300 mg/kg/day did not improve memory in transgenic tau mice [17], whereas in D-galactose-induced age-related dementia metformin was able to prevent dementia [18]. Based upon the contradictory results in the literature, a carefully controlled study is warranted to fully elucidate the beneficial effects of metformin in age-related dementia.
The SAMP8 mouse is a model of sporadic AD that develops deficits in learning and memory starting at approximately eight months of age [19, 20]. SAMP8 mice exhibit an age-related increase in Aβ, tau phosphorylation, and oxidative stress [21–24]. The cognitive deficits can be reversed by lowering Aβ with antisense directed at AβPP or an antisense directed against GSK-3β, the primary kinase that phosphorylates tau [21, 26].
In the current study, we examined if chronic metformin treatment can reverse age-related dementia in the SAMP8 mouse model of sporadic AD. Mice were treated with metformin for eight weeks and behavioral tests were performed starting after four weeks of treatment. Biochemical analyses of the PKC pathway were performed to help elucidate the mechanism of action of metformin in the CNS.
MATERIALS AND METHODS
Animal subjects
The SAMP8 male mice were 12-month-old males at the time of behavioral testing originating from our breeding colony. 12-month-old mice were used as we have years of data from our colony demonstrating the age related deficit in learning and memory compared to the young SAMP8 mice, and the stability of our phenotype [27]. We had two sets of 12-month-old SAMP8 mice. The first set received metformin (20 mg/kg/d, 200 mg/kg/d) or vehicle for 8 weeks. Behavioral testing began after 4 weeks of treatment. The first set were tested in T-maze, novel object recognition (N = 10∼11 mice per group). The second set of mice was given the same 8-week treatment regimen. Behavioral testing began after 4 weeks of treatment in the Barnes maze (N = 11 mice per group). Mice were individually housed for four weeks during behavioral testing to ensure each mouse was equally aroused and equally exposed to external stimuli (T-maze buzzer) during testing. All studies were conducted with the approval of the Animal Care and Use Committee at the VA Medical Center, St. Louis, MO. Sentinels from the facility were tested regularly to ensure our facility is virus-and pathogen-free. Food (Richland 5001) and water were available on an ad libitum basis and the rooms had a 12 h light-dark cycle with lights on at 0600 h. Behavioral experiments were conducted between 0730 and 1100 h.
Metformin
Metformin hydrochloride (1,1-dimethylbiguanide hydrochloride) was purchased from Sigma-Aldrich (St. Louis, MO, USA). Metformin was dissolved in saline and administered subcutaneously (sc) daily at 8 am for 8 weeks. The doses of metformin used were 20 and 200 mg/kg based on previous studies using rodents examining effects of metformin on dementia [28, 29].
Chemicals and materials
All chemicals were of the highest purity and purchased from Sigma-Aldrich (St. Louis, MO, USA) unless stated otherwise. Nitrocellulose membranes, polyacrylamide gels, XT MES electrophoresis running buffer, Precision Plus Proteintrademark, and all Blue Standards were purchased from Bio-Rad (Hercules, CA).
Behavioral testing
Behavioral testing started at the end of 4th week of treatment (see Table 1).
T-Maze training and testing procedures
Acquisition was tested in an aversive T-maze. The T-maze is both a learning task based on working-memory and a reference-memory task. The T-maze consisted of a black plastic alley with a start box at one end and two goal boxes at the other. The start box was separated from the alley by a plastic guillotine door that prevented movement down the alley until raised at the onset of training. An electrifiable floor of stainless steel rods run throughout the maze to deliver a mild scrambled foot-shock.Mice were not permitted to explore the maze prior to training. A block of training trials began when a mouse was placed into the start box. The guillotine door was raised and a cue buzzer sounded simultaneously; 5 s later, foot-shock was applied. The arm of the maze entered on the first trial was designated “incorrect” and the mild foot-shock was continued until the mouse entered the other goal box, which in all subsequent trials was designated as “correct” for the particular mouse. At the end of each trial, the mouse was returned to its home cage until the next trial.Mice were trained until they made one avoidance. Training used an inter-trial interval of 60 s, the buzzer was of the door-bell type sounded at 55 dB, and shock was set at 0.35 mA (Coulbourn Instruments scrambled grid floor shocker model E13-08). Retention was tested one week later by continuing training until mice reached the criterion of five avoidances in six consecutive trials. The results were reported as the number of trials to first avoidance for acquisition and the number of trials to criterion for the retention test.
Novel Object Recognition
Novel object recognition (NOR) was tested the five days following T-maze retention testing. NOR is a declarative memory task that involves the hippocampus when, as performed here, the retention interval is 24 h after initial exposure to the objects [30]. Mice were habituated to an empty apparatus for 5 min a day for 3 days prior to entry of the objects. During the training session, the mouse was exposed to two identical objects which it was allowed to examine for 5 min. The apparatus and the objects were cleaned between each mouse. 24 h later, the mouse was exposed to one of the original objects and a new novel object in a new location and the percent of time spent examining the new object was recorded. The novel object was made out of the same material as the original object and of the same size, but a different shape. This eliminated the possibility of smell associated with a particular object being a factor. The underlying concept of the task is based on the tendency of mice to spend more time exploring new, novel objects than familiar objects. Thus, the greater the retention/memory at 24 h, the greater the discrimination index (DI). The time with new object (tn) and time spent with the old object (to) was used to calculate the DI [DI = (tn–to)/(tn + to)] [31].
Barnes maze
The Barnes maze is a spatial learning and memory test. The Barnes maze (Otto Environmental, LLC, Milwaukee, WI) consists of an open circular platform with a small escape recessed chamber located under the platform called the “target box”. This technique has previously been used by us [32]. The mouse was first given one habituation session in the maze. After the habituation trial, the mouse was given four training trials. The training trial consisted of placing the mouse in the start chamber. After 10 s, the chamber was removed and the mouse was allowed to explore the maze freely for a maximum of 3 min. A trial ended when the mouse entered the target box or 3 min had elapsed, whichever happened first. The number of errors, the latency to reach the target hole, and distance travelled to reach the target hole were recorded with an ANY-maze video tracking system (San Diego Instruments, CA).Each mouse received four trials per day for four days. Probe trials: 24 h after the last training session and seven days after the first probe, trial mice were tested in a probe trial to determine if they remembered where the target box had been located. The target hole was closed and the mouse was allowed to explore the maze for 90 s. The number of errors (nose pokes in holes other than the target box), as well as the latency and distance travelled to reach the target hole, were recorded.
Sample preparation
Brains were cut in half and flash frozen. The assays were performed on
Aβ assay
Aβ was measured using Kit #294-62501 (FUJIFILM Wako Chemical Europe, Meuss, Germany).
G3DPH assay
Glycerol-3-phosphare dehydrogenase (G3PDH) activity was measured using assay kit #ab174095 (Abcam Biotechnology Company, Cambridge, MA). The assay was performed by Confluence Discovery Technologies (St. Louis, MO).
Western blots
Antibodies used in western blot analyses: pGSK3β ser9, PKCα, PKCɛ, PKCγ, PKCι, and PKCζ (Cell Signaling Technologies, Danvers, MA), pGSK3β tyr216 and total GSK3β (BD Biosciences, San Jose, CA), pTau404 (Abcam Biotechnology Company, Cambridge, MA), and β actin (Sigma, St. Louis, MO). The secondary antibodies were anti-rabbit IgG HRP (Cell Signaling), anti-mouse IgG HRP (BD Biosciences), and anti-mouse IgG HRP (BD Biosciences).After protein content was determined, the lysate was combined with NuPage reducing agent, Nupage sample loading buffer, and deionized water. The total volume was 150μL for each sample where the lysate was added such that the final concentration of total protein would be 50μg/20μL. All the prepared samples were then frozen at – 80°C. To perform electrophoresis, samples were thawed and then boiled at 100°C for 5 min. Samples were allowed to cool and then 20μL was loaded into the wells of a 4–12% bis-tris SDS-PAGE gel. The samples were run for 35 min at 200 V constant. Following separation, the proteins were transferred from the gel onto a 0.2μm pore nitrocellulose membrane via Invitrogen Turboblot transfer module. The gels were transferred in duplicate with 30 V applied constant for 90 min. After transfer, the membranes were dried then blocked in Li-Li-Cor Blocking Buffer (Li-Cor, Lincoln, NE) for 1 h at room temperature. The membranes were then probed for PKCα (1:1000), PKCɛ (1:1000), PKCγ (1:1000), PKCι (1:1000), PKCζ (1:1000), pGSKβ ser9 (1:1000), pGSK3βtyr216 (1:1000), total GSK3β (1:1000) or pTau404 (1:1000), and β-actin (1:1000) at 4°C for 24 h overnight at 4°C in Li-Cor Blocking buffer 0.2% TWEEN–20. The following day the membranes were washed with TBS–0.1% tween–20 4× for 5 min each. After washes the membranes were probed with anti-mouse (1:2000) and anti-rabbit (1:2000) secondary antibodies diluted in Li-Cor Blocking Buffer 0.2% tween-20. The incubation was for 1 h at room temperature with gentle shaking. After secondary detection, the membranes were washed again in TBST and imaged on the Li-Cor Odyssey (Li-Cor, Lincoln, NE) scanner. All samples were run in duplicates. All samples were processed and analyzed by persons that were blinded to the group assignment. Westerns were performed by Confluence Discovery Technologies (St. Louis, MO).
Blood glucose levels
Blood glucose levels were measured prior to the start of treatment and at the time of sacrifice using an ACCU-Chek (Roche, Indianapolis, IN). Eight weeks of metformin treatment at 20 mg/kg and 200 mg/kg in 12-month-old SAMP8 mice improved acquisition (A) and retention at one week (B) in T-maze and 24 h retention in NOR (C) (N = 10–11/group). *p < 0.05, **p < 0.01, ***p < 0.001.
Statistical analysis
Results from the T-maze were analyzed by a T-test. Statistical significance for comparing changes in PKC (PKCζ, PKCι, PKCα, PKCγ, PKCɛ), GSK-3β, pGSK-3βser9, pGSK-3βtyr216, pTau404, and AβPP expression and G3PDH activity between treated and control SAMP8 mice were analyzed using independent samples t-test. Statistical significance for comparing changes in GSK-3β expression in transgenic control, transgenic treated, and wild-type control mice were analyzed using one-way ANOVA. Bonferroni post-hoc tests were used if the one-way ANOVA was significant. Expression statistical analyses were conducted using R Studio. Significant differences were set at p < 0.05.
RESULTS
Metformin on learning and memory in SAMP8 mice
T-maze
12-month-old SAMP8 mice administered metformin at 20 or 200 mg/kg or vehicle were tested in T-maze footshock avoidance acquisition. One week later, the mice were tested for retention. The ANOVA for trials to first avoidance during acquisition in the T-maze showed a significant effect F(2,27) = 15.50, p < 0.0001. Bonferroni’s post hoc test indicated that the mice that received metformin at 20 and 200 mg/kg took significantly fewer trials to reach their first avoidance than the mice that received vehicle. There was no difference between the mice that received 20 mg/kg and the mice that received 200 mg/kg (Fig. 1A) The ANOVA for trials to criterion on the retention test showed a significant effect F(2,27) = 56.92, p < 0.0001. Bonferroni’s post hoc test indicated that the mice that received 20 and 200 mg/kg took significantly fewer trials on the retention test than the mice that received vehicle. There was no difference between the mice that received 20 mg/kg and the mice that received 200 mg/kg (Fig. 1B).
NOR
The ANOVA for time spent with the object on day one was not significant F(2,30) = 1.96, p NS. The ANOVA for discrimination index for exploration of the two objects was significant F(2,30) = 8.802 p < 0.001. Bonferroni’s post hoc test indicated that the mice that received 20 and 200 mg/kg spent significantly more time with the novel object that the old object compared to the vehicle control group. There was no difference between the group that received 20 mg/kg and the group that received 200 mg/kg (Fig. 1C).
Barnes maze
The two-way ANOVA for treatment×day was significant for treatment F(2, 252) = 3.472, p < 0.01. There was no effect of day or an interaction of treatment×day. Tukey’s post hoc test indicated that on day 4 the latency to reach the target in the mice that received 20 mg/kg was significantly shorter than the mice that received vehicle (Fig. 2A). The ANOVA for time spent in the target quadrant on the 24 h probe test was not significant F(2, 27) = 0.05, p NS (Fig. 2B) The ANOVA for the time spent in the target quadrant on the one week probe test probe test was not significant F(2, 27) = 1.523, p NS (Fig. 2C).
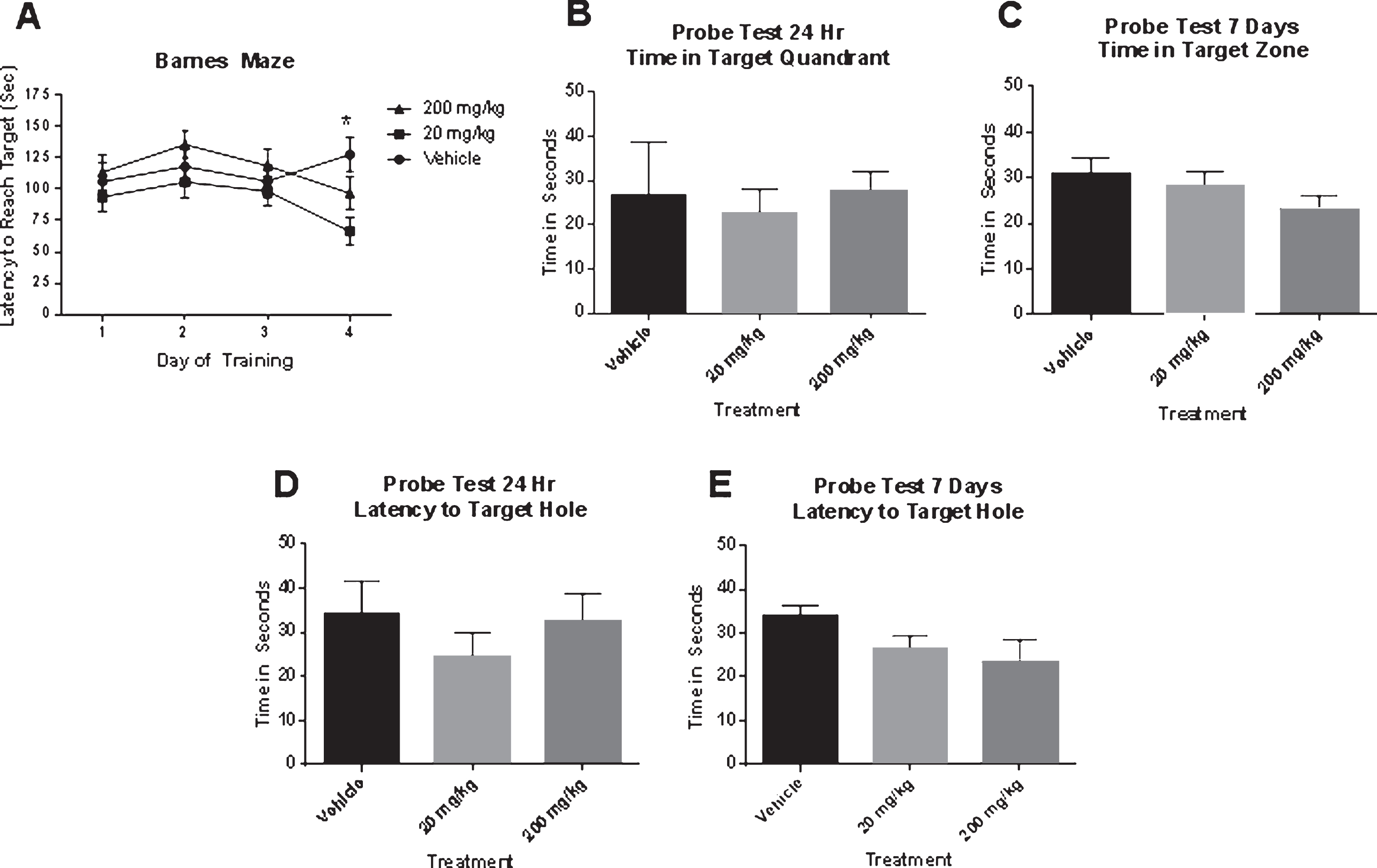
Eight weeks of metformin treatment in 12-month-old SAMP8 mice at 20 mg/kg improved performance on day 4 of training in the Barnes maze (A); however, there was no difference on time spent in the target quadrant on the 24 h or the 7-day probe test (B–E) (N = 11/group). *p < 0.05.
Effects of metformin on APPc99, AβPP, Aβ, G3DPH, and pTau404 in SAMP8 mice
Western blot analysis was used to determine the effect of metformin treatment on expression of APPc99 and pTau. The ANOVA for APPc99 expression was significant F(2,28) = 14.63, p < 0.0001. Bonferroni’s post hoc test indicated that the mice which received 20 mg/kg and 200 mg/kg had significantly lower APPc99 than the mice which received vehicle (Fig. 3A) The ANOVA for Aβ protein levels was significant (F,2,28) = 4.19, p < 0.03. Bonferroni’s post hoc test indicated that the mice that received 20 mg/kg had significantly lower Aβ than the mice that received vehicle. There was no difference between the mice which received 200 mg/kg and the vehicle treated mice or the mice which received 20 mg/kg. The ANOVA for pTau404 expression was significant F(2,28) = 4.73, p < 0.02. Bonferroni’s post hoc test indicated that the mice that received 20 and 200 mg/kg had significantly lower levels of pTau404 than the mice that received vehicle. There was no difference between the mice that received 20 mg/kg and the mice that received 200 mg/kg (Fig. 3B) The ANOVA for AβPP expression was not significant F(2, 27) = 1.42, p NS (Fig. 3C) The ANOVA for G3PDH activity was not significant F(2,26) = 1.92, p NS (Fig. 3D).
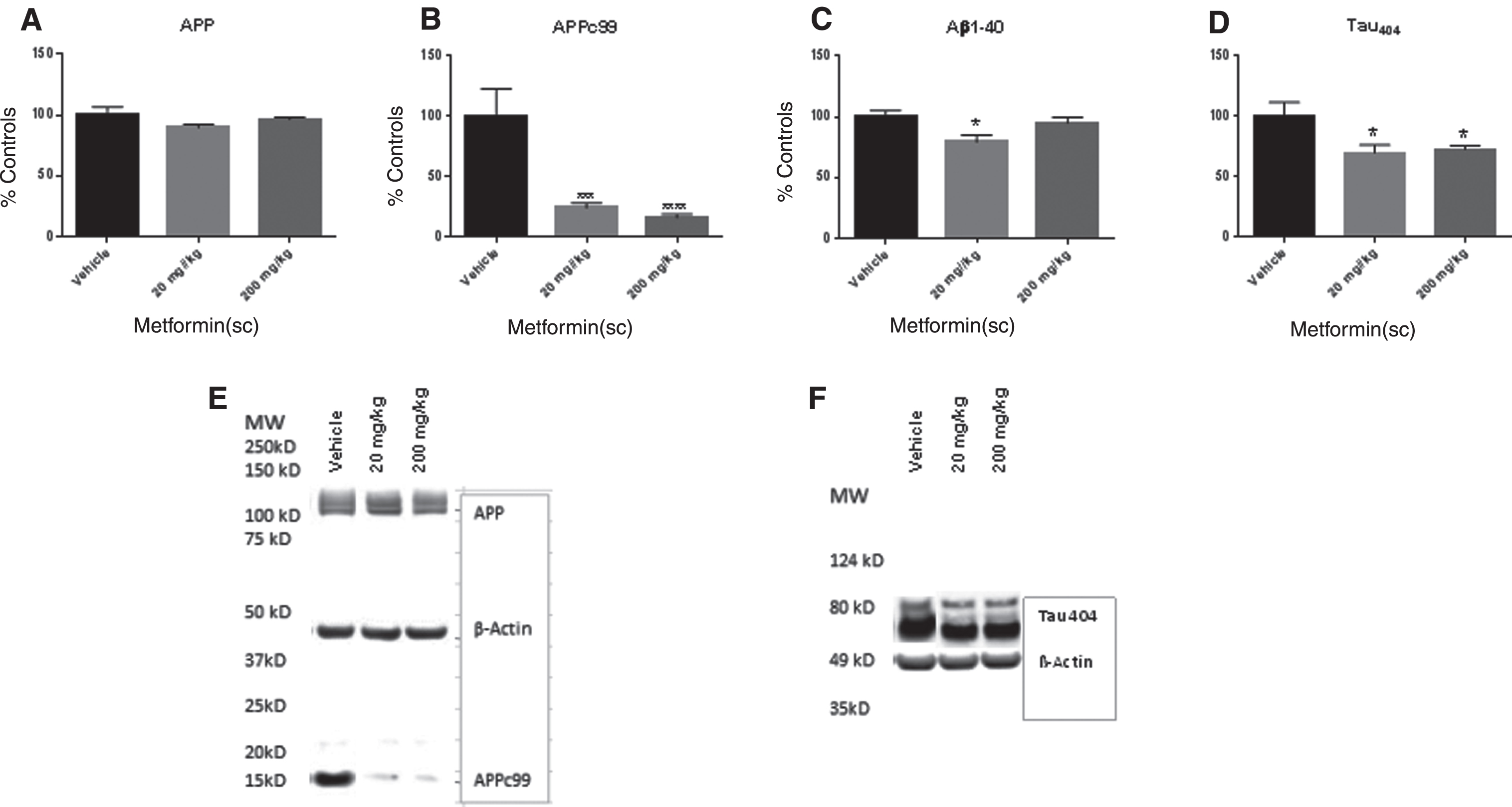
Metformin treatment at both 20 mg/kg and 200 mg/kg significantly decreased APPc99 expression, and pTau404 expression in 12-month-old SAMP8 mice (B and D). Metformin decreased Aβ levels at 20 mg/kg (C). Metformin did not affect AβPP expression or G3PDH activity (A and D). Panel E is a representative blot of AβPP and APPc99 expression and panel F is a representative blot of pTau404 expression. *p < 0.05, ***p < 0.001, ****p < 0.0001 (N = 9–11/group).
Effects of metformin on levels of total GSK-3β, pGSK-3βser9, and pGSK-3βtyr216 in SAMP8 mice
Phosphorylated GSK-3β leads to the phosphorylation of 88% of tau. A reduction in GSK-3β could be a mechanism by which metformin decreases pTau expression. The ANOVA for pGSK-3β ser9/total was significant F(2,26) = 5.059, p < 0.01. Bonferroni’s post hoc testing indicated that the mice that received 200 mg/kg of metformin had significant higher levels of pGSK-3β ser9/total than the mice which received saline and the mice which received 20 mg/kg. There was no difference between the mice that received 20 mg/kg and the mice which received saline (Fig. 4A) The ANOVA for pGSK-3βtyr216/total expression was F(2, 26) = 4.11, p < 0.02. Bonferroni’s post hoc test indicated that the mice which received 20 mg/kg had significantly higher pGSK-3β tyr216/total than the mice which received saline. There was no difference between the mice which received 20 mg/kg than the mice which received 200 mg/kg of metformin (Fig. 4B). The ANOVA for total GSK-3β was significant F(2,26) = 4.132, p < 0.03. Bonferroni’s post hoc test indicated that the mice that received 200 mg/kg had significantly lower total GSK-3β expression than the mice which received vehicle. There was no difference between the mice that received 20 mg/kg and the mice that received 200 mg/kg (Fig. 4C).

Metformin treatment significantly increased the percent of pGSK-3β ser9 expression at 200 mg/kg (A) and pGSK-3β tyr216 (B) and total GSK-3β (4C) expression at 20 mg/kg in 12-month-old SAMP8 mice compared to the vehicle treated SAMP8 mice. Panels D–F are representative blots of the isoforms of GSK-3β expression. *p < 0.05 (N = 9–11/group).
Effect of metformin on PKC
PKC is a precursor to many kinases in the brain including GSK-3β. PKC is a family of enzymes that have come in several forms. The ANOVA for PKCζ expression was significant F(2,26) = 3.78, p < 0.03. Bonferroni’s post hoc test indicated that the mice which received metformin 20 mg/kg had significantly higher levels of PKCζ than the mice which received vehicle. The mice which received 200 mg/kg were not significantly different from the mice which received 20 mg/kg (Fig. 5A) The ANOVA for PKCα expression was significant F(2,29) = 7.844, p < 0.002. Bonferroni’s post hoc test indicated that the mice which received metformin at 20 mg/kg had significant higher PKC levels than the mice which received vehicle. The mice which received metformin 200 mg/kg were not significantly different from the mice that received 20 mg/kg or the mice that received vehicle (Fig. 5B) The ANOVA for PKCɛ was significant F(2,30) = 4.28, p < 0.02. Bonferroni’s post hoc test did not find a significant effect (Fig. 5C) The ANOVA for PKCγ was not significant F(2,30) = 0.45, p NS (Fig. 5D) The ANOVA for PKCι was not significant F(2,30) = 1.14, p NS (Fig. 5E).
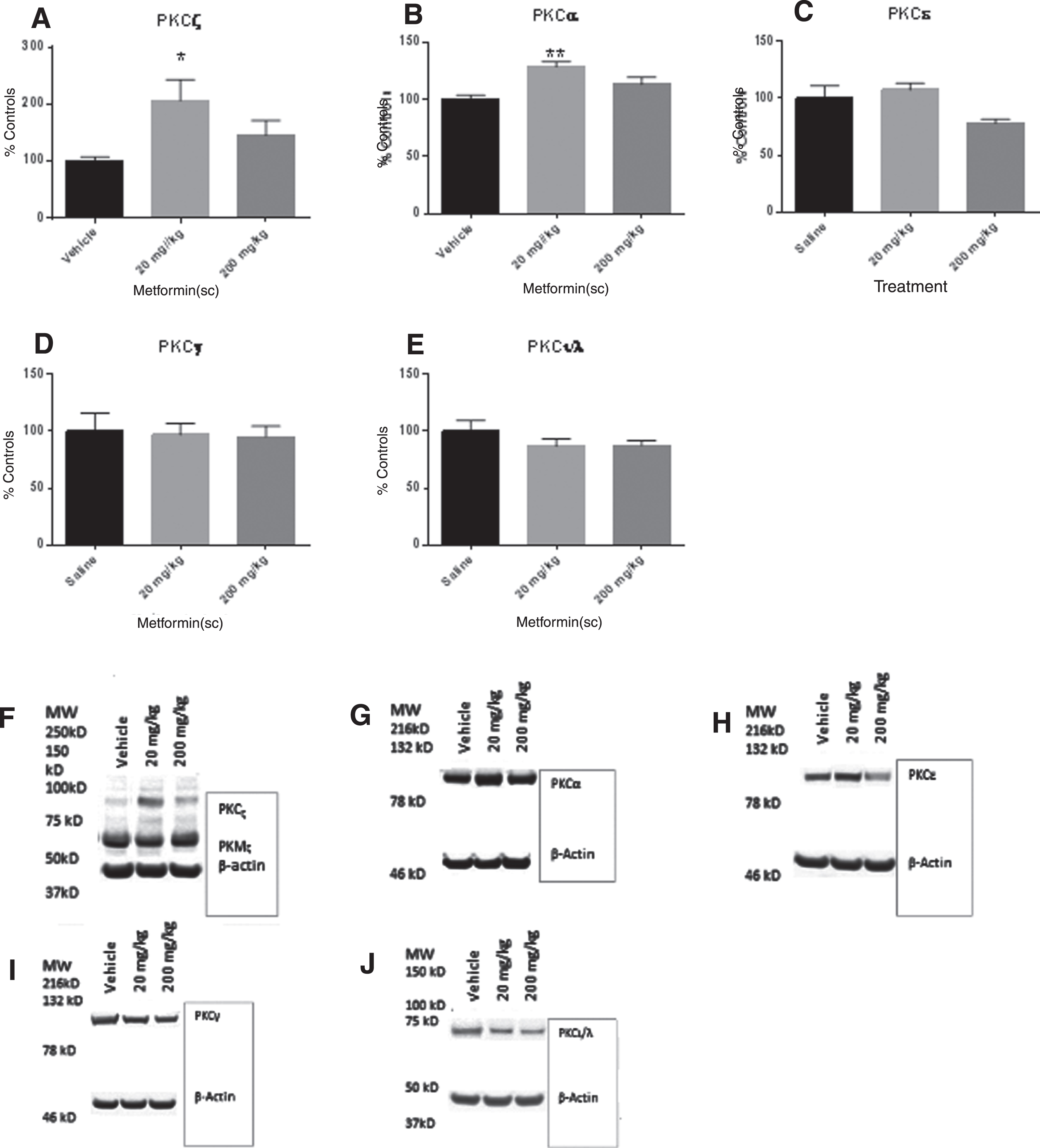
Eight weeks of metformin in 12-month-old SAMP8 mice significantly increased PKCζ and PKCα expression at 20 mg/kg (A and B). Metformin treatment had no effect on PKCɛ, PKCγ, or PKCι/λ expression (C–E). Panels F–J are representative blots of PKC expression for each isoform of PKC in panels A–E. *p < 0.05, **p < 0.01 (N = 9–10/group).
The effect of metformin on blood glucose levels
The ANOVA for blood glucose levels prior to treatment was not significant F(2, 30) = 0.11, p NS. (Vehicle = 133.5 mg/dL, 20 mg/kg = 127.6 mg/dL, 200 mg/kg = 129.6 mg/dL). The ANOVA for blood glucose levels at the end of treatment were not significant F(2,30) = 0.16, p NS. (Vehicle = 145.5 mg/dL, 200 mg/kg = 142.0 mg/dL, 200 mg/kg = 150.3 mg/dL).
DISCUSSION
Metformin is a first line type II diabetes treatment. Recent studies have found that in diabetics, metformin can prevent dementia [33] which supports it as a drug of focus in the current interest in repurposing pharmaceuticals [34]. Metformin has become a candidate for possible use in neurological disorders [3]. Here we examined metformin in the SAMP8 mouse model of sporadic AD. We found that metformin at 20 and 200 mg/kg improved memory in 12-month-old SAMP8 mice in both T-maze and NOR. We chose 12-month-old SAMP8 mice as we have shown that they have a stable age-related impairment in learning memory as well as elevated AβPP, Aβ, and hyperphosphorylated tau [35–38]. Metformin decreased APPc99 and pTau404 in the 12-month-old SAMP8. The effects on PKC, Aβ, and the forms of GSK-3β were less clear, indicating their role is variable.
Here we chose to use 20 and 200 mg/kg. These doses are commonly used in rodent studies examining metformin and dementia [28, 39]. Based on a PubMed search, there has only been one pilot study examining metformin in patients with dementia [14]. The authors in that study chose to use 2,000 mg/70 kg which is the typical dose for type II diabetes [14]. This dose is comparable to our 20 mg/kg dose. The authors did find beneficial effects in this pilot study. Our results suggest that the dose similar to doses given to patients with diabetes is likely sufficient to improve memory in AD with no effect on blood glucose levels which is what we found in our mice.
In the current study, we found that metformin had no effect on AβPP and only decreased Aβ at the 20 mg/kg. While this was somewhat surprising, we do know that very small changes in Aβ can affect memory. We have shown that giving very low doses to young mice actually improves memory while very high doses impairs memory indicating there is an optimal dose-range for Aβ for memory enhancement [40]. In addition, there were changes in APPc99 and pTau at both does suggesting that their effects may play a greater role in the effect of metformin in memory than Aβ at these doses. Our previous studies have shown that targeting AβPP with an antisense can reverse learning and memory deficits [41, 42]. APPc99 is formed from the cleavage of AβPP in the mitochondrial-associated endoplasmic reticulum (ER) by β-secretase to form the C-terminal fragment (c99) [43]. APPc99 is then cleaved by γ-secretase to generate Aβ [44]. In AD, APPc99 accumulates in the ER, resulting in mitochondrial dysfunction [43]. APPc99 has been found to be a key factor in interneuronal pathology found in the 3×TgAD mouse model of AD and has been identified as the first AβPP to accumulate in the hippocampus of these mice [45]. Metformin was able to decrease APPc99 at both doses in our study without affecting AβPP suggesting improvement in memory may be related to changes in mitochondrial function.
In the current study, we found that metformin decreased pTau. Hyperphosphorylated tau is known to be a major factor in AD. Hyperphosphorylated tau leads to neurofibrillary tangles and mitochondrial dysfunction [46, 47]. The SAMP8 have hyperphosphorylated tau [48]. Studies indicate that treatments which decrease phosphorylated tau improve memory in mouse models of AD [41]. Antisense which targets the primary kinase GSK-3β that phosphorylates 88% of tau improves memory in 12-month-old SAMP8 mice [26, 49]. Here we found a slight increase in pGSK-3β inactive ser9 form at the low levels of metformin and an increase in active tyr216 form at the 200 mg/kg, yet we found improvement at both 20 and 200 mg/kg suggesting the effects on pGSK-3β are not the primary mechanism of action behind the improvement. This is consistent with previous data with metformin in neurons which found a decrease in ser9, the inactive form of pGSK-3β, still resulted in a decrease in tau [50]. That study found that metformin decreased phosphorylation of tau via mTOR/protein phosphatase 2A signaling in primary neurons from human tau mice [50]. The PI3K/Aklt/mTor pathway alterations occur early in the progression of AD [51]. We have found the genes in this pathway to be altered in the hippocampus of SAMP8 mice with age [52]. Our results would suggest that this pathway should be investigated further in vivo.
PKC isozymes are known to be decreased in humans with AD and in mouse models of AD [53, 54]. PKC plays a crucial role in memory formation and insulin function in the brain [55]. Potentially, its involvement in memory formation may be due to PKC’s role in promoting synaptogenesis [55]. PKC has been found to decrease with age as Aβ and tau increase [56]. Metformin has been shown to increase PKC [9, 57]. Here, we found an increase in PKC alpha and zeta, suggesting that increases in these two forms of PKC were probably not the primary factors associated with the improvement in memory.
In the current study, we examined the effects of metformin on memory in 12-month-old SAMP8 mice. Metformin treatment for 8 weeks improved memory in the SAMP8 mouse model of sporadic AD. The improvement was associated with decreases in APPc99 and pTau404. Decreases in these proteins have been linked to improvement of mitochondrial function [43, 47]. The biochemical results suggest that future studies of metformin in dementia should focus on mitochondria. The findings from the current study suggest that metformin is a possible treatment for dementia.