
Abstract
Background:
Presenilin1 (PSEN1) is the most common gene related to familial Alzheimer’s disease (AD). Only several mutation types from Chinese have been reported, with less biological function research conducted.
Objectives:
We explore the pathological function of PSEN1 M139L, a mutation located at α-helix of PSEN1 transmembrane 2, using predictive programs and in vitro study and compare its effects on Aβ production to those of an artificial PSEN1 S141G located at non α-helix mutation face.
Methods:
APP, PSEN1, and PSEN2 genes were screened for mutations using Sanger sequencing in the DNA samples of the proband and additional available family members. Disease-mutation cosegregation analysis and three software programs were performed to predict the mutation’s pathogenicity. In vitro, we investigated the impact of these mutations on Aβ production in HEK293-APPswe cells using lentiviral vectors harboring PSEN1 WT, PSEN1 M139L, the positive control (PSEN1 M139V) and the non α-helical mutation (PSEN1 S141G). In addition, we co-transfected PSEN1 and tau into cells to determine the mutations’ impact on tau phosphorylation.
Results:
PSEN1 M139L mutation was discovered in the index patient and four affected siblings. Cosegregation analysis and silicon prediction suggested the mutation was probably disease causing. In vitro studies demonstrated that both PSEN1 M139L and PSEN1 S141G caused elevated ratios of Aβ42/Aβ40, but changes of tau phosphorylation were not detected.
Conclusion:
The novel PSEN1 M139L mutation found in familial AD increases the Aβ42/Aβ40 ratio significantly. Mutations at non α-helical mutation face of PSEN1 TM2 can affect Aβ production and the region may play a key role in PSEN1 function.
INTRODUCTION
Mutations in the PSEN1 gene are the most common cause of autosomal dominant Alzheimer’s disease (AD), with more than 200 pathogenic mutations reported worldwide. Although most variants of PSEN1 are found throughout the whole protein as missense mutations with only one amino acid substitution, there is wide heterogeneity of clinical and image phenotypes even at the same codon [1–3]. Discovery and characterization of more novel variants would widen the mutation spectrum and help demonstrate a clearer relationship between PSEN1 mutations and AD.
PSEN1 is the catalytic center of γ-secretase, an enzyme complex which includes other three subunits, Nicastrin, Pen-2, and Aph-1 [4–7]. γ-secretase serves as an intramembranous aspartic protease, which cleaves a number of type I membrane proteins including amyloid-β protein precursor (AβPP) and Notch [8–10]. Previous studies have found that PSEN1 mutations could increase the longer and more toxic Aβ forms through γ-secretase processing of AβPP [11]. In addition, some studies have suggested that PSEN1 mutations could decrease the activity of γ-secretase and lead a reduced total Aβ production [12]. Hence, it is important to investigate the effect of PSEN novel mutation on AβPPprocessing.
The transmembrane domain of PSEN1, where a number of mutations related to AD were discovered, has been considered functionally fundamental. With regards to PSEN1 TM2, it is interesting that all discovered mutations line up along the α-helix [13], suggesting that these residues were more conserved and vulnerable than the non α-helical region. Electron microscopy density readings reveal a high flexibility of PSEN1 TM2, and structural analysis suggests that TM2 may play a role in the regulation of substrate entry and cleavage [14]. Biological investigation of mutations at the α-helical face and non-α-helical face may provide evidence for further analysis of relationship between structure and function.
In this study, we present a novel missense PSEN1 M139L mutation in a Chinese family with early-onset AD. Soft programs and disease-mutation cosegregation analysis were performed to demonstrate the pathogenicity of the mutation. In vitro studies, we investigate the effects of PSEN1 M139L on Aβ production. An artificial mutation PSEN1 S141G was introduced to investigate the biological role of non α-helical face of PSEN1 TM2.
MATERIALS AND METHODS
Family members
Families were enrolled through the Chinese Familial Alzheimer’s disease Network (CFAN, http://cfan.wydztech.com/FrontPage/Index.aspx). Diagnosis of AD was established according to the revised criteria of the NIA-AA (National Institute on Aging and Alzheimer’s Association) in 2011. Early-onset AD were defined as age of onset < 65 years. A battery of cognitive rating scale and brain magnetic resonance image (MRI) scan were performed on all available family members. This study was approved by the Ethics Committee of Xuan Wu Hospital. Written informed consent was obtained from each participant or his/her legal guardian.
Genetic sequencing, variant analysis, and cosegregation analysis
DNA was collected from blood samples from all available familial members. Polymerase chain reaction (PCR) was performed to amplify exons 3–12 of PSEN1 and PSEN2 and exons 16–17 of APP, followed by Sanger sequencing. The APOE genotype was determined using the Sanger sequencing method.
Population databases were searched for the PSEN1 variant, and included the Exome Aggregation Consortium (ExAC, http://exac.broadinstitute.org/), Exome Variant Server (http://evs.gs.washington.edu/EVS), 1000 Genomes Project (http://browser.1000genomes.org), and the disease database (ClinVar, http://www.ncbi.nlm.nih.gov/clinvar). Th-ree computation predictive programs, PANTHER (http://www.pantherdb.org/tools/csnpScoreForm.jsp), Mutation Taster (http://mutationassessor.org), and PolyPhen-2(http://genetics.bwh.harvard.edu/pph2) were used to aid in the interpretation of sequence variant. The algorithm used for cosegregation analysis was based on a published paper, which provided the quantitative criteria for pathogenic classification of the genomic variant [15].
Construction of expression plasmids
The vector pLVX-IRES-ZsGreen1 (Code No.632187, Clontech) (kind gift of Professor Hongzhi Li, Wenzhou Medical University) and human WT PSEN1 were ligated using pEASY-Uni Seamless Cloning and Assembly. Positive clones were identified by colony PCR and double enzyme digestion, followed by Sanger sequencing for further identification. PSEN1 M139L, S141G, and M139V mutations were introduced into the pLVX-IRES-ZsGreen1 expression vector encoding the WT PSEN1 by the site-directed mutation method. M139L denotes the novel clinical variant observed in this Chinese pedigree, M139V is a known clinical variant, and S141G was artificially constructed to study its effect on the non α-helical face of PSEN1. The human WT microtubule-associated protein tau (MAPT) complementary DNA (cDNA) was amplified using total RNA from human neuroblastoma SH-SY5Y cells. Both WT and mutant cDNA sequences were verified by direct sequencing.
Lentiviral vector particles production
pLVX-IRES-ZsGreen1 vectors encoding WT or mutant PSEN1, with packaging plasmids pMD2G (#50487, addgene) and psPAX2 (#12260, addgene), were transiently co-transfected into HEK293T cells. The media was changed 6 h post-transfection. Supernatants were collected at 30 and 54 h after transfection, then centrifuged at 3000 rpm for 5 min to remove cell debris, and subsequently passed through 0.45-μm filters, and frozen at –80°C until needed.
Cell culture and transfection
HEK293 cells stably transfected with APP KM670/671NL (APP/swe) were cultured in Dulbecco’s modified Eagle medium supplemented with 10% fetal bovine serum (Gibco, Grand Island, NY, USA) at 37°C in a humidified incubator with 5% CO2. For lentiviral transduction, cells were grown to 40% –50% confluence and were infected with 30μ lentivirus solution per 2 cm2 cultivation area. After 24 h, the media containing the virus was removed and replaced with fresh medium. After 3 days post infection, the media was changed. After an additional 48 h, the media was collected and centrifuged at 3000rpm for 5 min, and the supernatant was frozen at –80°C for further analysis. To determine tau phosphorylation, cells overexpressing WT or MT PSEN1 were transfected with MAPT plasmid using Lipofectamine 3000 (L3000015, Invitrogen). Cells were harvested 48 h post-transfection.
pLVX-IRES-ZsGreen1 vectors express green fluorescent protein ZsGreen1, and transfection efficiency was demonstrated by the average fluorescence intensity per cell. Briefly, three fields of view were randomly selected from each WT or mutant transfection. Fluorescence intensity measurement and cell count were performed using Image J (64-bit Java 1.8.0_112) to calculate the average fluorescence intensity per cell.
ELISA assay
Human Aβ40 and Aβ42 ELISA kits (Aβ40/Aβ42 ELISA kits, IBL, Hamburg, Germany) were used to determine Aβ levels in the cell media according to the manufacturer’s instructions. Briefly, cell media was added into the wells of a 96-well plate for incubation at 4°C overnight. Plate wells were then sequentially incubated with the secondary antibody for 2 h at room temperature. Reaction substrate was then added into plate wells, followed by stop solution. Within 10 min, color intensity was measured at 450 nm. Levels of Aβ were normalized by the concentration of total protein which was detected using the BCA assay (Applygen, China).
Western blotting analysis
Cells were lysed in RIPA buffer with 1×protease inhibitors cocktail (Applygen, China) and 1×phosphatase inhibitors cocktail (Applygen, China) on ice for 30 min. The lysate was centrifuged at 12,000 rpm for 30 min at 4°C and then the supernatant was transferred to a fresh tube and stored at –80°C. Protein concentrations were determined using the BCA assay (Applygen, China). Protein lysates were separated in 8%–12% SDS-PAGE and transferred onto PVDF membrane. After blocking nonspecific sites with 5% skim milk, the membranes were incubated with primary and secondary antibodies sequentially. Immunodetection was performed using enhance chemiluminescent (ECL) substrates for HRP following the manufacturer’s instructions (Millopore, German). Antibodies used in this study are listed in Supplementary Table 1.
Statistical analysis
Aβ levels and quantitative data of western blots were presented as mean±standard error. Statistical significance was tested by using SPSS23 (IBM, Armonk, NY, US) or GraphPad Prism 7.0 software (Graphpad Software Inc., La Jolla, CA, US). Multiple comparisons were tested with ANOVA followed by Turkey’s post hoc test. Two groups of data were compared by Student’s t-test. p < 0.05 was considered to be statistically significant.
RESULTS
Index patient and family members
The index patient (III-11, Fig. 1A) consulted the Memory Clinic of Xuan Wu Hospital at age 49, with a complaint of memory impairment. She began to have memory problems at 45 years old and developed a decline in daily life ability about 2 years later. Additional symptoms included visuospatial impairment and emotional irritability. She was almost bedridden when she turned 50 years old. She had a strong family history of dementia with 11 affected family members spanning three generations (Fig. 1A). Family members had early-onset AD with age of onset ranging from 51 to 56 years old. In addition, the family members had similar clinical features to the index patient (Table 1).
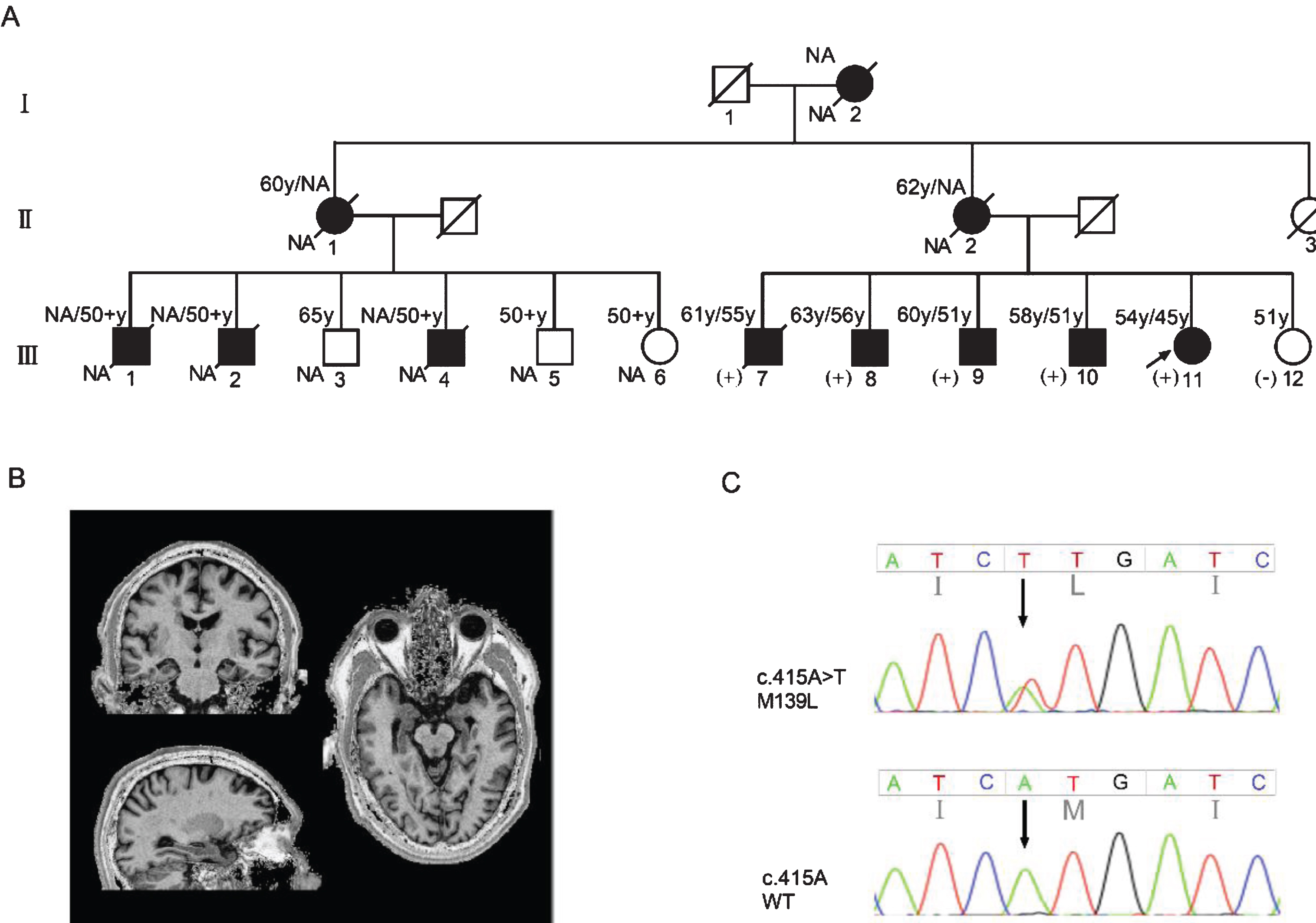
A) Pedigree with p.M139L (c.415 A > T) mutation. Roman numbers represent generations. Proband is marked by arrow, black symbols denote affected members, white symbols denote unaffected members, square denotes man, circle denotes women, and slashes indicate deceased members. Age1/age2 at the top left corner of symbol indicates current age or age of death/age at onset. (+) denotes mutation carrier, (-) denotes mutation non carrier. NA, not available. B) Coronal, axial, and horizontal MRI images of family member III-8. Bilateral hippocampus and mild cortex atrophy. C) The sequencing chromatogram indicates a heterozygous PSEN1 c.415A>T mutation (nomenclature according to National Center for Biotechnology Information. Reference Sequence: NM_000021) in the patients.
Genetic and clinical features of family members with PSEN1 M139L mutation
AD, Alzheimer’s disease; AAO, age at onset; CN, cognitively normal; “#” indicates hippocampus atrophy is present on MRI, while for others brain MRI was not obtained; N/A, not available.
For the eldest brother (III-7) of the proband, short-term memory decline was noticed around 55 years old, accompanied by visuospatial dysfunction, behavioral abnormalities, uncontrolled diet and aggression. He developed prosopagnosia later and could only recognize his wife before passing away. He died of the disease at the age of 62. The second eldest brother (III-8) of the index patient began to have episodic memory impairment at age of 56, with a gradual decline in executive function. His visuospatial function began to decline at 57 years old, accompanied by repeating simple and meaningless actions. At the age of 59, his personality became irritable and subsequently, developed generalized cognitive impairment, accompanied by motor symptoms such as stagger and myoclonus. At present, his self-care ability is significantly declined and he needs to be taken care of at all times. Magnetic resonance imaging performed on III-8 showed atrophy of the bilateral hippocampus and cerebral cortex (Fig. 1B). The third eldest brother (III-9) of the proband has lost self-care ability and is incontinent. No further details were available and clinical characteristics of the other family members were not obtained.
Genetic sequencing and cosegregation analysis
We screened mutations in APP, PSEN1, and PSEN2 genes from the DNA sample of the index patient. A heterozygous PSEN1 c.415A>T mutation (reference sequence: NM_000021) was discovered, which resulted in a substitution of Methionine to Leucine at condon 139 (M139L) (Fig. 1C). The same mutation was identified in DNA samples from family members III-7, 8, 9, 10, all of whom were affected by AD, except for III-12 who was the only unaffected sibling of the proband. Under the dominant inheritance pattern, PSEN1 M139L was cosegregated with disease in this family. Based on the public algorithm, the probability was N = (1/2)6= 1/64, which yielded an evidence level of strong pathogenesis.
The family had no mutations in PSEN2 or APP. In addition, PSEN1 M139L mutation was not discovered in either 100 cognitively normal controls or 100 late-onset AD patients. The APOE genotypes of the family members were listed in Table 1. Except for III-7 whose APOE genotype is APOE ɛ3/ɛ3, his siblings all carried an APOE ɛ4 allele (Table 1).
Sequence variant interpretation
To verify the frequency of PSEN1 M139L in large populations, three population databases (ExAC, Exome Variant Server, 1000 Genome) and one disease database (ClinVar) were searched. The variant was not found in all the databases. To predict disease pathogenesis of PSEN1 M139L, three computational programs were utilized. Using PANTHER, based on evolutionary analysis, preservation time scored 842, suggested “probably damaging” for PSEN1 M139L. Using Mutation taster, PSEN1 M139L was suggested to affect protein features and lead to disease. Using PolyPhen-2, the mutation was predicted to be probably damaging with a score of 0.963. All of the above information suggests that PSEN1 M139L was associated with the onset of AD.
PSEN1 mutation (M139L) affects Aβ production but not tau phosphorylation
We overexpressed PSEN1 in HEK293-APPswe cells to investigate the effect of M139L mutation on Aβ production. The transfection efficiency was not different among the constructs, as determined by PSEN1 expression using western blot (Fig. 2A–2C, p > 0.05). Hence, the cell lines transfected with equivalent expression of PSEN1 were used for further comparison. Compared to PSEN1 WT, the Aβ40 levels of PSEN1 M139L were decreased (p < 0.05) while Aβ42 levels had an increasing trend. The Aβ42/Aβ40 ratios of M139L were increased by 1.6 times compared to WT PSEN1 (Fig. 3). PSEN1 M139V, which has been investigated using H4-APPswe and HEK293-APP695 cells lines in previous studies, was used as the positive control in our study. The Aβ40 levels decreased while Aβ42 levels remained unchanged, resulting in Aβ42/Aβ40 ratio increasing by around 1.7 times, comparing M139V with WT. Meanwhile, beta-secretase1 (BACE1) and soluble amyloid-β protein precursor β (sAβPPβ), the main enzyme and production of AβPP through β-site cleavage, showed no difference between groups (Fig. 4). In order to determine the effect of the mutations on tau phosphorylation, we co-transfected PSEN1 and MAPT into HEK293-APPswe cells. Phosphorylated tau specified by T181 and AT8 (s202/205) did not differ between cells transfected with WT or the mutants (data not shown). Furthermore, most common phosphorylation kinases of tau, GSK3β (and phosphorylated form, GSK3β phospho S9), and CDK5, showed no difference of expression between WT and mutation groups (data not shown). Based on these results, PSEN1 M139L increases the ratio of Aβ42/Aβ40 but we did not find changes of tau phosphorylation.

Transfection efficiency indicated by fluorescence intensity per cell and PSEN1 expression. A) Fluorescence intensity per cell, normalized to WT group. B) Western blotting of full length of PSEN1 (PSEN1-FL) and the carboxy-terminal fragment of PSEN1 (PSEN1-CTF). C) Quantitative analysis of PSEN1-CTF. Bars indicate mean±standard error based on three independent measurements. ns, not significant.

Quantitative analysis of Aβ40, Aβ42, and Aβ42/Aβ40 levels. A) Concentrations of secreted Aβ40 in media of HEK293/APPswe cells. B) Concentrations of secreted Aβ42 in media of HEK293/APPswes cells. C) Aβ42/Aβ40 ratios in media of HEK293/APPswe cells. Bars indicate mean±standard error based on three independent experiments. **p<0.001, ***p<0.0001 relative to WT PSEN1 according to ANOVA and Turkey’s post hoc test.
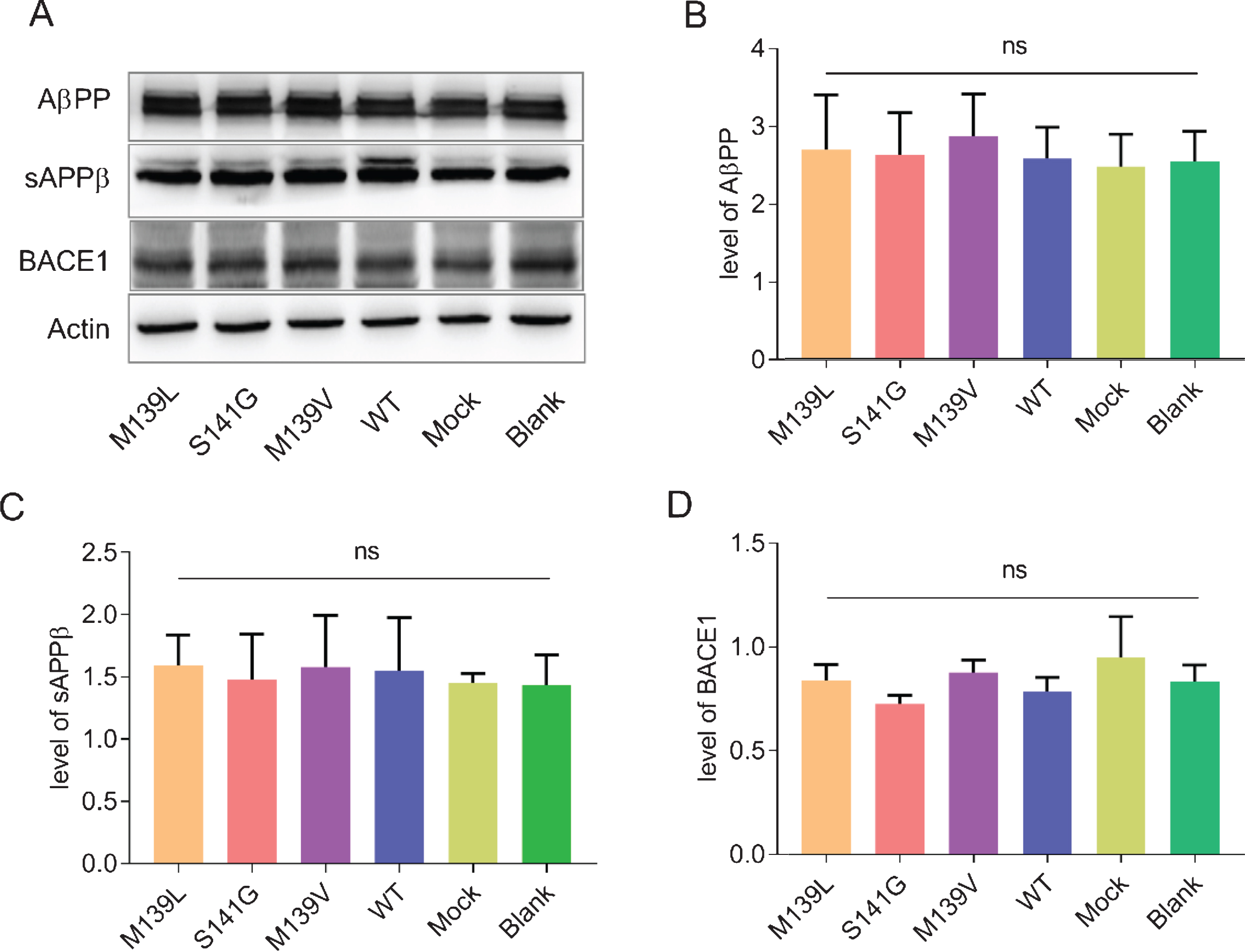
Western blotting analysis of proteins, sAβPPβ and BACE1, involved in AβPP processing. A) Representative western blot of AβPP, sAβPPβ, and BACE1. B-D) Quantitative analysis of the AβPP, sAβPPβ, and BACE1. Bars indicate mean±standard error based on three independent experiments. ns: not significant.
PSEN1 mutation (S141G) at the non-α-helical face of transmembrane II affects Aβ production but not tau phosphorylation
To investigate the biological effect of the mutation at the non α-helix region of PSEN1 TM2, the mutation PSEN1 S141G was introduced into HEK293-APPswe cells. PSEN1 S141G increased both the levels of Aβ40 and Aβ42 by 2.1 and 2.7 times, respectively, compared to PSEN1 WT. The ratio of Aβ42/Aβ40 in cells transfected with S141G increased by 1.4 times (Fig. 3). Furthermore, compared to PSEN1 M139L and M139V, the Aβ40 and Aβ42 levels of PSEN1 S141G were increased. Similarly, PSEN1 S141G did not affect tau phosphorylation at residues 181 and 202/205 (data not shown). Thus, PSEN1 S141G, which was a mutation at the non α-helix of TM2, increased both the total Aβ and Aβ42/Aβ40 ratio but had no effect on tau phosphorylation in this in vitro model.
DISCUSSION
In this study, we presented a Chinese early-onset familial AD with a heterozygous PSEN1 c.415A>T mutation, which led to a substitution of Methionine to Leucine of PSEN1 at condon 139. This mutation was not found in either 100 normal controls or 100 patients with late-onset AD, and it has not been reported in AD&FTD database (http://www.molgen.ua.ac.be/ADmutations/), ALZFORUM (https://www.alzforum.org/) or PubMed. PSEN1 M139L was also not discovered in various population and disease databases. These results indicate that PSEN1 M139L (c.415A>T) is a novel mutation, which adds a new member to the PSEN1 mutations pool.
Condon 139 of PSEN1 is a mutation hot spot, and at least 18 families carrying other four mutation types (M139V, M139K, M139T, M139I) have been reported [16–20]. We reviewed and summarized the clinical and imaging characteristics of patients with PSEN1 mutations at condon 139 (Table 2). In the studies, age of onset (AAO) of patients ranged from 30s to 60s, which covered the 45-56-year-old age group in our familial AD. Clinical presentations in our pedigree showed prominent memory problem, accompanied by visual spatial dysfunction and personality changes as disease progressing, which had some similarity to a reported family [21]. There are other families showing atypical AD such as early personality changes, psychosis and abnormal behavioral symptoms [22, 23]. The wide heterogeneity may be due to the different amino acid substitutions. Moreover, with regards to the families from at least eight different countries, different genetic background and (or) environmental factors may account for this issue [24]. MRI scan of the brain in our study showed atrophy of bilateral hippocampus, a well-established imaging biomarker of AD. In addition, mild white matter lesions could also be discovered in brain dissectional imaging of patients carrying mutations at 139 condon. [20, 22].
Clinical and imaging features of patients carrying mutations at condon 139 of PSEN1
AAO, age at onset; DD, disease duration; CT, computed tomography; MRI, magnetic resonance imaging; NA, not available.
In this family, disease information and sequencing results of six family members were accessible, allowing us to perform cosegregation analysis. Based on the guidelines for the interpretation of sequence variants [25] and the simplified algorithm [15], the probability N = (1/2)6 = 1/64, suggested a strong evidence for pathogenesis of M139L. Furthermore, three silicon programs based on the evolutionary conservation of the nucleotide or amino acid, the biochemical characteristics of the amino acid substitution, and the location or context within the protein, predicted M139L to be probably damaging. Based on these results, we propose that PSEN1 M139L is a very likely pathogenic mutation.
Amyloid is one of the hallmarks in AD. We investigated the effect of M139L mutation on Aβ production in vitro by using HEK293/APPswe cells, a cell model widely used in AD mutation research [21, 26–28]. PSEN1 M139L increased the Aβ42/Aβ40 ratio by about 1.6 times, while levels of Aβ42 increased but not significantly. As reported, mutations at condon 139 could decrease Aβ40 by 1.6 to 1.8 times and increase Aβ42/Aβ40 by 1.9 to 2.9 times. Our results are in consistence with previous reports. Similar results of PSEN1 M139V were observed in our study. To investigate the biological role of non-α-helix of PSEN1 TM2, we constructed a PSEN1 S141G mutation at non-α-helical face. Unexpectedly, this mutation increased both Aβ42 and Aβ40, even higher than M139L and M139V groups, resulting in a significant increase of Aβ42/Aβ40 ratios. In the cell lysate, sAβPPβ and BACE1 showed no difference among M139L, S141G, or WT groups. This indicated that the PSEN1 M139L and PSEN1 S141G did not affect β-secretase processing of AβPP. As well-accepted, the mutations may be involved in the processing of AβPP through γ-secretase. A recent study proposed that AD mutations could increase Aβ42/Aβ40 ratios by destabilizing γ-secretase-Aβn interactions [29]. Overall, in vitro studies demonstrated that PSEN1 M139Lincreased the Aβ42/Aβ40 ratio significantly. Mutation at the non-α-helical face also had an effect on Aβ production and increased the Aβ42/Aβ40 ratio. The increased Aβ42/Aβ40 ratio is the hallmark of many PSEN1 mutations [30] and is a promising biomarker for AD [31]. It is reported that Aβ42 is more easily aggregated and the oligomers of Aβ42 are more toxic [32]. However, there is also some researches indicating that PSEN1 mutations fail to increase the Aβ42/Aβ40 ratios and the mechanism between familial AD mutants and AD is independent of Aβ [33, 34]. The exact mechanisms between familial AD mutants, Aβ and AD remain to be deciphered.
Neurofibrillary tangles consisting of phosphorylated tau is another hallmark of AD. However, in our study, changes of tau phosphorylation and phosphorylation kinases were not detected for the mutations at PSEN1 TM2 using HEK293/APPswe cells, which is in accordance with previous researches using CHO, COS, SH-SY5Y, and N2A cells [35, 36]. It suggests that PSEN1 mutations may not change phosphorylation of tau through direct interaction. Furthermore, in vitro cell lines may not be ideal models to investigate pathological changes of phosphorylated tau. The above-mentioned cell lines cannot accurately mimic the pathological changes of neurons in AD patients’ brain, which may be explained by the different expression of agents and the lack of functional membrane receptors or ion channels [37–39]. In recent studies, it has been illustrated that neurons derived from induced pluripotent stem cell (iPSC) lines of patients with AD are more suitable for investigating tau phosphorylation and other pathogenic changes related to AD [40, 41]. Future studies using iPSC or in vivo models are needed.
APOE ɛ4 is the strongest risk factor for both early-onset AD and late-onset AD [42]. It not only lowers the age of AD onset [43], but also accelerates disease progression [44].In our familial AD, except for family member III-7 whose APOE genotype was ɛ3/ɛ3, his affected siblings all carried a single allele of ɛ4. In this pedigree, AAO of III-7 was larger than the mean AAO, but he had the shortest disease duration. It suggests that APOE ɛ4 maybe a modifier for AAO but not for disease progression. In addition, other factors such as nursing care conditions and comorbidities may influence the disease course. There is also research suggesting that early-onset familial AD is not modifiable by APOE genotype [45]. We plan to perform further follow up studies with the family and obtain genotypes from additional family members to investigate the role of APOE genotype on disease progression.
In summary, we present a Chinese early-onset familial AD characterized by typical AD, with prominent symptoms of personality changes and visual spatial dysfunction during disease progression. Interpretation analysis and in vitro studies demonstrate that the novel PSEN1 M139L mutation is probably pathogenic. The artificial mutation PSEN1 S141G at the non-α-helical face affected Aβ production significantly, suggesting non-α-helix of TM2 may play a key role in PSEN1 function. Further investigation is needed to decipher the exact pathogenic mechanism.
Footnotes
ACKNOWLEDGMENTS
This study was supported by the Key Project of the National Natural Science Foundation of China (81530036); the National Key Scientific Instrument and Equipment Development Project (31627803); Mission Program of Beijing Municipal Administration of Hospitals (SML20150801); Beijing Scholars Program; Beijing Brain Initiative from Beijing Municipal Science & Technology Commission (Z161100000216137); Innovation Base Training and Development Special Program (Z171100002217007); CHINA-CANADA Joint Initiative on Alzheimer’s Disease and Related Disorders (81261120571), Beijing Municipal Commission of Health and Family Planning(PXM2018_026283_000002), and Project for outstanding doctor with combined ability of Western and Chinese medicine.