
Abstract
Traumatic brain injury (TBI), a brain disorder that causes death and long-term disability in humans, is increasing in prevalence, though there is a lack of protective or therapeutic strategies for mitigating the damage after TBI and for preserving neurological functionality. Microglia cells play a key role in neuroinflammation following TBI, but their regulation and polarization by a member of the vascular endothelial growth factor (VEGF) family, VEGF-C, is unknown. Here, we show that VEGF-C induced M2 polarization in a murine microglia cell line, BV-2, in vitro, by a mechanism that required signaling from its unique receptor, VEGF receptor 3 (VEGFR3). Moreover, in a TBI model in rats, VEGF-C administration induced M2 polarization of microglia cells, significantly improved motor deficits after experimental TBI, and significantly improved neurological function following TBI, likely through a reduction in cell apoptosis. Together, our data reveal a previously unknown role of VEGF-C/VEGFR3 signaling in the regulation of post-TBI microglia cell polarization, which appears to be crucial for recovery from TBI.
INTRODUCTION
Traumatic brain injury (TBI) causes death and long-term disability in humans, and its prevalence increases each year [1]. Although the majority of TBI cases only lead to mild to moderate injury, about 10% of severe cases require long-term health care and support services for substantial and lifelong cognitive, physical, and behavioral impairments [2–4]. The elderly appears to be much more vulnerable to TBI, and suffer more severe outcomes in terms of disability and the development of neurodegenerative and neuropsychiatric issues [5]. Recent studies have shown that TBI can predispose patients to develop neural disorders like Parkinson’s disease (PD) and Alzheimer’s disease (AD) [6]. Since there is a significant lack of therapeutic options for TBI, it is imperative to seek any protective or therapeutic strategies for alleviating the damage after TBI and for preserving neurological functionality.
TBI-triggered pathological processes involve a broad spectrum of cellular and molecular pathways, and can be classified into two phases [7]. The first phase is a primary damage phase from the initial insult. The pathological changes in this phase include contusion, laceration, injury to axons, swelling of brain tissue, intracranial hemorrhage, and neural cell death [7]. The first phase is followed by the second phase, which lasts weeks. The pathological changes in the second phase include breakdown of the blood-brain barrier (BBB), increases in oxidative stress and glutamate excitotoxicity, progressive neurodegeneration, and delayed cell death [7]. Importantly, the inflammatory response during the first and second phase after TBI is often disorganized, which sometimes lead to further tissue injury and impairment of tissue repair [8].
Neuroinflammation is mediated by a number of cell types from both the CNS and the circulation. Astrocytes and microglia are cells from the CNS that play a pivotal role in the post-TBI inflammatory response [8]. Microglia, derived from erythromyeloid precursor cells, are specialized immune cells in the brain with phagocytic and antigen-presenting capabilities [7]. Extreme heterogeneity has been found in microglia and is largely a function of their activation state [9].
In the healthy brain, microglia are described as ‘ramified’ in a resting or quiescent state. Upon activation, microglia transform to a hypertrophic morphology for antigen-presentation, phagocytosis or the release of cytokines or hormones for tissue repair [9]. Previous studies have shown that microglia may have various roles after injury, through their complex responses in the context of brain injury [9]. The plasticity of microglia likely results from their phenotypic changes, much like macrophages outside the brain. However, the polarization of microglia has been minimally studied, especially during TBI [9].
Vascular endothelial growth factor (VEGF) receptor signaling plays a central role in vascularization and angiogenesis-related events [10]. The VEGF family is composed of 6 members: VEGF-A, VEGF-B, VEGF-C, VEGF-D, VEGF-E, and placental growth factor (PlGF) [11]. VEGFR3 mediates lymphangiogenesis in response to VEGF-C and VEGF-D [12]. Very recently, VEGF-C/VEGFR3 signaling has been shown to be involved in macrophage differentiation in non-brain tissue [13–17]. Moreover, VEGF-C/VEGFR3 signaling has been shown to regulate glial cell responses in mesenchymal stem cell transplantation in the rat brain [18]. Based on these previous reports, we hypothesized that VEGF-C/VEGFR3 signaling may play a role in the microglia cell response in TBI, and this hypothesis was examined in the current study.
MATERIALS AND METHODS
Protocol approval
The study protocol and experimental design were approved by the institutional Ethics Committee at Shanghai Jiao Tong University. All the animal experiments were approved by the Institutional Animal Care and Use Committee at Shanghai Jiao Tong University, and were performed strictly according to the guideline.
Cell culture and treatment with VEGFR3 inhibitor
BV-2 is a murine cell line generated by infecting primary microglia cells with a v-raf/v-myc oncogene-carrying retrovirus [19]. We obtained BV-2 cells from The Chinese Academy of Sciences (Beijing, China), and cultured them in Dulbecco’s Modified Eagle’s Medium supplied with 10% fetal bovine serum (FBS, Invitrogen, Carlsbad, CA, USA) in a 37°C incubator with 5% CO2. Recombinant VEGF-C was purchased from R&D Systems (West Grove, PA, USA), and was used at a dose of 100 ng/ml. For suppression of VEGFR3 signaling, SAR131675 (Selleckchem, Houston, TX, USA) was applied to BV-2 cells at a concentration of 50 nM.
Cell transfection
The BV-2 cells were transfected with either short hairpin small interfering RNA for VEGFR3 (shVEGFR3) or a scrambled sequence under the control of a cytomegalovirus (CMV) promoter. The plasmids were prepared from a backbone pcDNA3.1-CMV-GFP plasmid (Clontech, Mountain View, CA, USA). The sequence for shVEGFR3 is 5′-CCCAGTATTGTGTGGTACAAA-3′. The sequence for the scrambled is 5′- GTGCCAAGAC GGGTAGTCA -3′, as described [20]. Transfection was performed using the Lipofectamine-3000 assay (Invitrogen).
Flow cytometry
F4/80 + Iba-1 + microglia cells were isolated by flow cytometry, after the fixed cells were incubated with APC-conjugated anti-F4/80 and Pacific blue-conjugated anti-Iba-1 antibodies. CD206 + M2 cell analysis and sorting were performed after the living cells were labeled with PEcy7-conjugated anti-CD206 antibodies (Becton-Dickinson Biosciences, San Jose, CA, USA). For analysis of cell apoptosis, dissociated living cells were double-stained with FITC-Annexin V and propidium iodide (PI) with an FITC Annexin V Apoptosis Detection Kit I (Becton-Dickinson Biosciences). Annexin V + PI-cells were determined to be (early) apoptotic cells. Flow cytometry was performed using a FACSAria flow cytometer (Becton-Dickinson Biosciences). Negative controls were applied to remove background noise and to confirm positive cells. Data were analyzed and quantified using Flowjo software (Flowjo LLC, Ashland, OR, USA).
Arginase activity measurement
Arginase activity was assessed in cell lysates indirectly by measuring urea concentration generated by the arginase-dependent hydrolysis of l-arginine, using an arginase activity assay kit (Sigma-Aldrich). The samples and standards (100μL/well) were transferred in triplicate to a 96-well plate for checking the optical density at 540 nm with a 690 nm correction. Sample concentrations were determined from the standard curve and converted to Arginase units.
RT-qPCR
RNA was extracted using the RNeasy kit (Qiagen, Hilden, Germany), and then was used as a template for cDNA synthesis. Quantitative PCR (RT-qPCR) was performed in duplicates using the QuantiTect SYBR Green PCR Kit (Qiagen). Data were assessed using 2-ΔΔCt method. The primers used for quantitative PCR sequences were as follows: VEGFR3: GCCCGAGGACGAGGGTGACT, CCTGGCTGCGCCTATCCTGC; Arg-1: TGAGAGACCACGGGGACCTG, GCACCACACTGACTCTTCCATTC; Fizz1 (Retnla: resistin-lke-α): CCATAGAGAGATTATCGTGGA, TGGTCGAGTCAACGAGTAAG; Ym1 (Chi3l3: chitinase 3-like 3): TGGAATTGGTGCCCCTACAA, AACTTGCACTGTGTATATTG; β-actin: AAATCTGGCA CCACACCTTC, GGGGTGTTGAAGGTCTCAAA. The values of genes were determined by sequential normalization to β-actin and experimental controls.
Western blotting and immunocytochemistry
Cell or tissue extracts were lysed in radioimmunoprecipitation assay (RIPA) buffer (Invitrogen) supplied with the Complete Protease Inhibitor Cocktail (Roche, Indianapolis, IN, USA). Protein concentrations were determined with a BCA protein assay kit (Bio-rad, Bejing, China). Primary antibodies were rabbit anti-caspase 3, anti-brain-derived neurotrophic factor (BDNF), and anti-β-actin (Cell Signaling, San Jose, CA, USA). The secondary antibody was HRP-conjugated anti-rabbit (Jackson ImmunoResearch Labs, West Grove, PA, USA). β-actin was used as a protein loading control. Immunoreactivity was detected by the chemiluminescence method (Thermo Scientific, San Jose, CA, USA). Protein levels were obtained by sequential normalization to β-actin and experimental controls. The primary antibodies used in immunocytochemistry were rabbit anti-Iba-1, anti-VEGFR-C and anti-VEGFR3 (R&D Systems). DAPI (4′,6-diamidino-2-phenylindole) was used to stain nuclei.
Rat manipulations
Male Sprague–Dawley Rats (260–300 g) were obtained from the SLAC Laboratory Animal Co. Ltd (Shanghai, China). Rats were kept at 25°C, 50–60% humidity, and were fed on standard pellet chow and water. The TBI model was developed as described using a weight— drop device [14]. Briefly, a midline incision was made on the anaesthetized rats, after which the skull, right parietal craniotomy and dura were exposed with a high— speed microdrill. Then, a 40— G steel weight fell freely through a vertical tube from 2.5 m onto the motor cortex to induce TBI. Following injury, the bone flap was placed in situ and the scalp was sutured. Control rats (CTL) received a sham operation without TBI. Recombinant VEGF-C was injected intracranially at a dose of 5μg.
Rotarod task
A rotarod was used to assess motor function as previously described (15). Briefly, a 7 cm diameter cylinder was positioned 1.2 m above a foam pad while speed and acceleration were controlled by computer interface (San Diego Instruments, San Diego, CA, USA). For each trial, the rat was placed on the rotating barrel, the speed was accelerated from 4 to 40 rpm over a period of 5 min, and the latency to fall was recorded. Pre-training occurred once a day for the 3 days preceding injury. Following injury, rats were re-tested on the rotarod task on days 1, 3, 5, 7, and 14 post-TBI. Each rat underwent four test trials per day. The average latency of the total of the four trials was calculated.
Neurological scores
Neurological function was evaluated on the basis of Modified Garcia Scale [21].
Brain water content
Rat brains were rapidly removed and weighed immediately at sacrifice (wet weight). The brains were then dried in an oven at 105°C for 24 h and weighed again (dry weight). Brain water content was calculated as [(wet weight - dry weight)/wet weight]×100%.
Determination of BBB permeability
BBB permeability was quantitatively evaluated by Evans blue (EB; Sigma-Aldrich) extravasation analysis. Briefly, rats were injected intravenously with EB at a dose of 2% wt/vol in saline; 4 mL/kg body weight. EB was allowed to circulate in each animal for 40 min prior to determination of sample EB concentration using external standards ranging from 50 to 1000 ng/mL, prepared in the same solvent. The supernatant was quantified for absorbance of EB dye using a spectrophotometer (excitation 620 nm, emission 680 nm).
Statistical analysis
All of the statistical analyses were performed using the GraphPad Prism 6 (GraphPad Software, San Diego, CA, USA). Statistical analysis of group difference was carried out using a one-way analysis of variance (ANOVA) test followed by Turkey multiple comparison post-hoc analysis. All values represent the mean±standard deviation (SD). A value of p < 0.05 was considered significant after Bonferroni correction.
RESULTS
BV-2 microglia cells express both VEGF-C and its receptor VEGFR3
The BV-2 cell line is a broadly used murine microglia cell line. First, we analyzed the properties of the BV-2 cells. By flow cytometry, we found that BV-2 cells expressed both F4/80 and CD11b, two surface markers for macrophages (Fig. 1A). Moreover, by immunocytochemistry, we found that BV-2 cells expressed Iba-1, a specific marker for microglia in the brain (Fig. 1B). Since Iba-1 was not expressed by macrophages with a non-brain origin, our data suggest that BV-2 is a specific microglia cell line, which is distinguishable from normal macrophages.
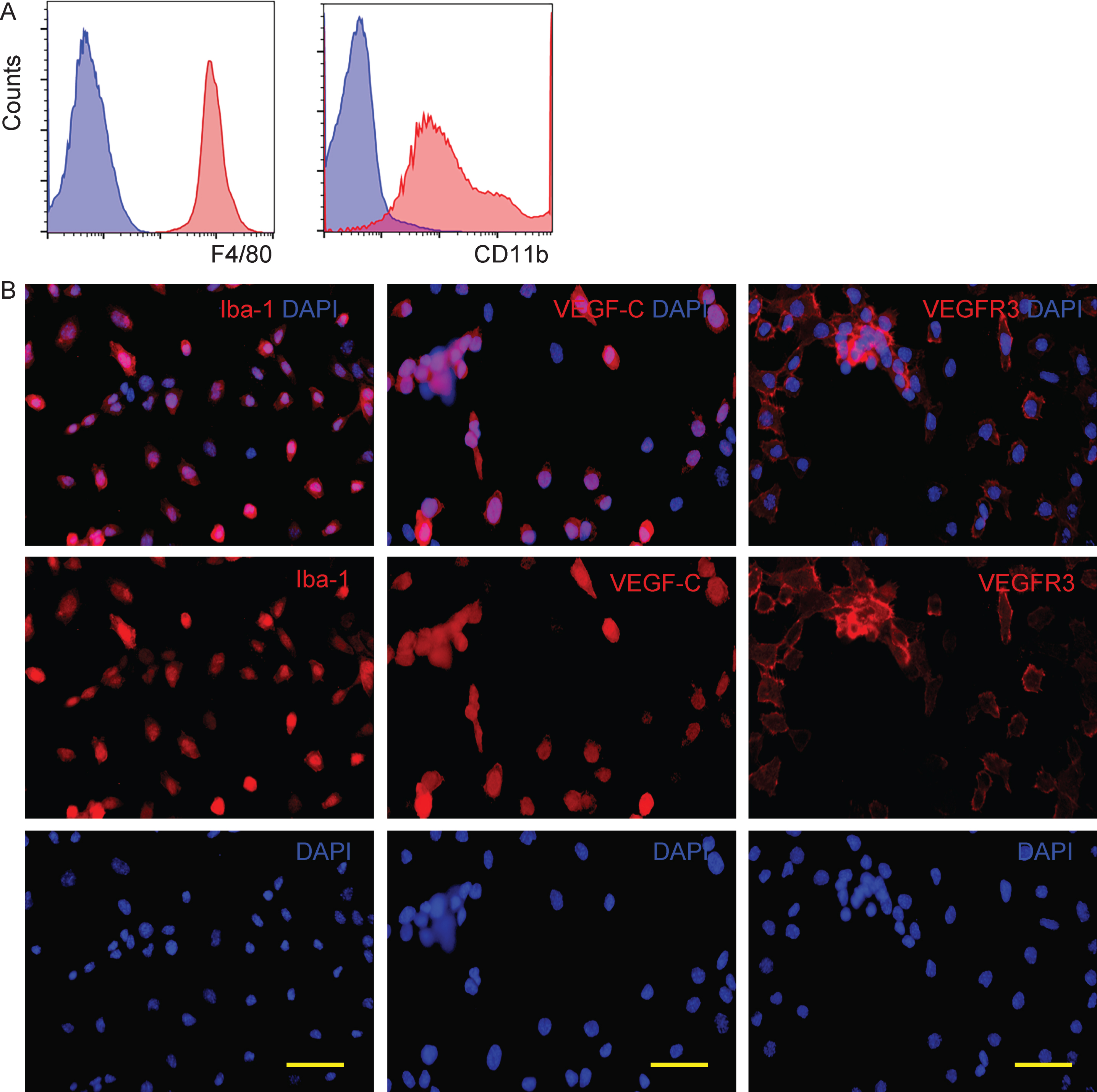
BV-2 microglia cells express both VEGF-C and its receptor VEGFR3. A) We analyzed the expression of F4/80 and CD11b on BV-2 cells by flow cytometry, shown as representative flow charts. Blue: isotype control. Red: with antibody. B) Immunocytochemistry for Iba-1, VEGF-C and its unique receptor VEGFR3 in BV-2 cells. Scale bars are 20μm.
Since a previous study has shown that VEGF-C and its unique receptor VEGFR3 are expressed by glial cells [18], we examined whether BV-2 cells also express VEGF-C and VEGFR3. We found that nearly all BV-2 cells are positive for VEGFR3, and the majority of BV-2 cells also expressed VEGF-C (Fig. 1B), consistent with previous reports that suggest an autocrine secretory pattern for VEGF-C/VEGFR3 in glial cells [18].
VEGF-C induces M2 polarization of BV-2 cells in a VEGFR3-dependent manner
Next, BV-2 cells were used as microglia cells to be tested for the possibility of VEGF-C-induced cell polarization. First, BV-2 cells were transfected with shVEGFR3, resulting in about 80% knockdown of VEGFR3 (Fig. 2A). Next, a co-culture system was applied. In the first condition, which was called BV (1), the BV-2 cells were cultured with media control. In the second condition, which was called BV (2), the BV-2 cells were cultured with recombinant VEGF-C. In the third condition, which was called BV (3), the BV-2 cells were cultured with recombinant VEGF-C in the presence of a specific VEGFR3 inhibitor, SAR131675. In the fourth condition, which was called BV (4), the BV-2-shVEGFR3 cells were cultured with recombinant VEGF-C (Fig. 2B). To assess for M2 macrophage polarization by VEGF-C, we checked the mRNA levels of Arginase 1 (Arg-1), Ym-1, and Fizz-1, three M2 macrophage markers. We found that VEGF-C induced an approximately 25 times’ increase in Arg-1 mRNA (Fig. 2C), approximately 250 times’ increase in Ym-1 mRNA (Fig. 2D), and approximately 13 times’ increase in Fizz-1 mRNA (Fig. 2E). A typical characteristic of M2 macrophages is their potential role in converting L-arginine to ornithine and urea through the action of Arg-1. Thus, arginase activity was determined using a urea-based assay, showing significant increases in VEGF-C-treated macrophages (Fig. 2F). CD206 is a surface marker exclusively expressed by M2 macrophages. We found that CD206 was induced in VEGF-C-treated-macrophages by flow cytometry (Fig. 2G). All these M2-like changes in BV-2 cells were significantly attenuated by either the presence of SAR131675, or using VEGFR3-depleted BV-2 cells (Fig. 2C–G), suggesting that VEGF-C induces M2 polarization of BV-2 cells in a VEGFR3-dependent manner.

VEGF-C induces M2 polarization of BV-2 cells in a VEGFR3-dependent manner. A) RT-qPCR for VEGFR3 on BV-2 cells transfected with plasmids carrying scrambled sequence (BV-2) and BV-2 cells transfected with shVEGFR3 (BV-2-shVEGFR3). B) Schematic of a co-culture system. In the first condition, which was called BV (1), the BV-2 cells were cultured with media control. In the second condition, which was called BV (2), the BV-2 cells were cultured with recombinant VEGF-C. In the third condition, which was called BV (3), the BV-2 cells were cultured with recombinant VEGF-C in the presence of a specific VEGFR3 inhibitor, SAR131675. In the fourth condition, which was called BV (4), the BV-2-shVEGFR3 cells were cultured with recombinant VEGF-C. C-E) RT-qPCR for Arginase 1 (Arg-1; C), Ym-1 (D) and Fizz-1 (E) on BV (1), BV (2), BV (3), and BV (4). F) Measurement of arginase activity. G) CD206 levels on BV (1), BV (2), BV (3), and BV (4) by flow cytometry. *p < 0.05. NS, non-significant. N = 5.
VEGF-C administration improves motor deficits after experimental TBI
TBI was induced in mice. VEGF-C or control saline were intracranially injected into the rat brain. The rats were separated into 3 groups. Group 1 comprised a sham operation with saline injection (CTL). Group 2 comprised of TBI with saline injection (TBI). Group 3 comprised of TBI with VEGF-C injection (TBI + VEGF–C). Brain function was assessed by a timed rotarod task method, and neurologic score was evaluated at 14 days post-TBI (Fig. 3A). We found that the latency to fall was significantly shortened in rats after TBI, a finding which was significantly attenuated in TBO-rats that had received VEGF-C (Fig. 3B). Thus, VEGF-C administration improves motor deficits after experimental TBI.
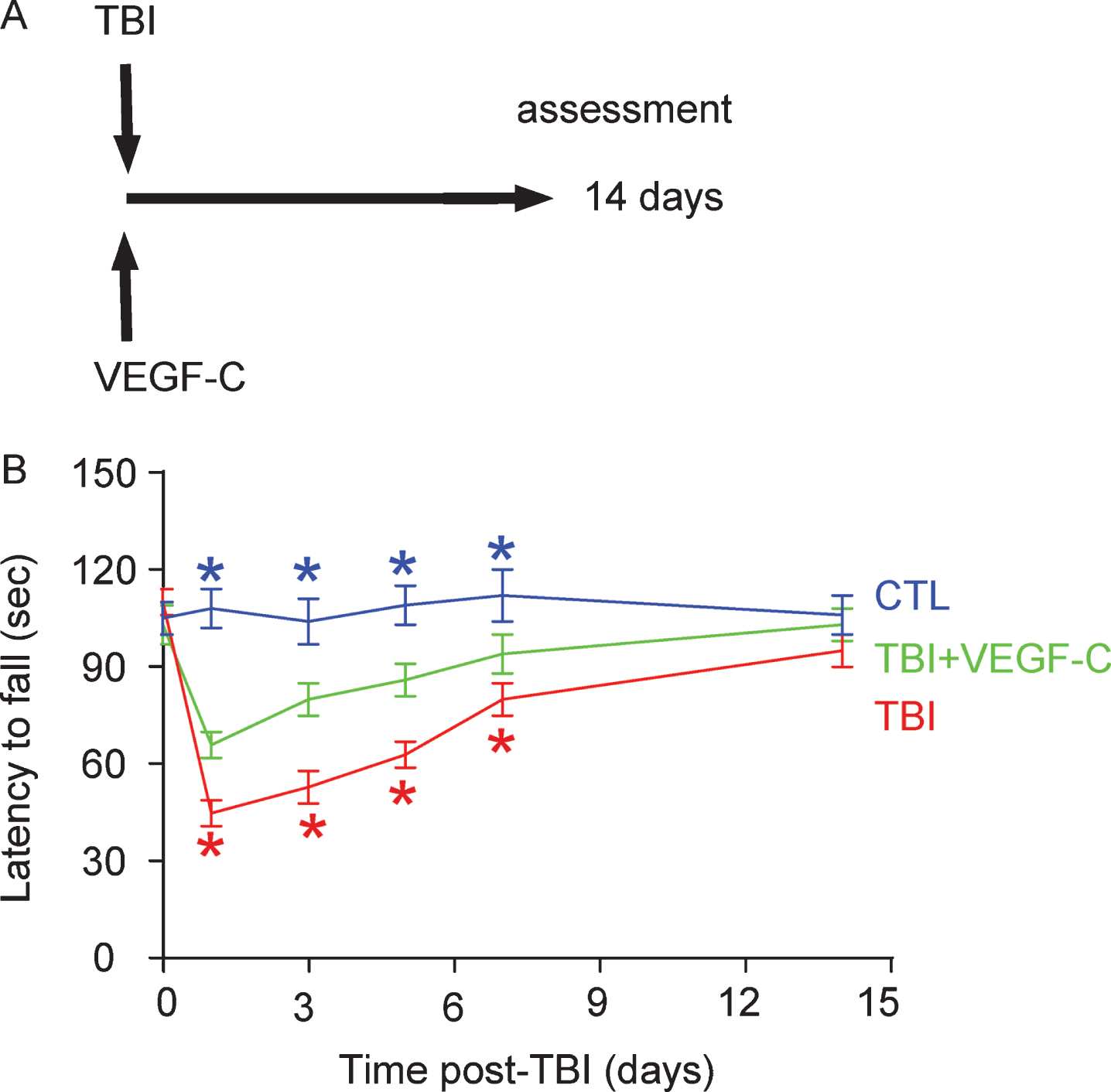
VEGF-C administration improves motor deficits after experimental TBI. A) Schematic of the experiment. TBI was induced in mice. VEGF-C or control saline was intracranially injected into rat brain. The rats were separated into 3 groups. Group 1 comprised a sham operation with saline injection (CTL). Group 2 comprised TBI with saline injection (TBI). Group 3 comprised TBI with VEGF-C injection (TBI+VEGF-C). Brain function was assessed by a timed rotarod task method, and the neurologic score was evaluated at 14 days post-TBI. B) Measurement of the latency to fall in the rotarod task. Red *p < 0.05, TBI versus TBI + VEGF-C; Blue*p < 0.05, CTL versus TBI + VEGF-C; N = 10.
VEGF-C administration improves brain function after experimental TBI
At 14 days after TBI, the neurological score in TBI rats was significantly reduced, a finding which was significantly attenuated in TBO-rats that had received VEGF-C (Fig. 4A). We then assessed the brain water content. We found that TBI significantly increased the brain water content, a finding which was significantly attenuated by VEGF-C (Fig. 4B). BBB permeability was also evaluated using an EB extravasation assay. We found that TBI significantly increased EB extravasation, a finding which was significantly attenuated by VEGF-C (Fig. 4C). BDNF is a marker for neural regeneration and high BDNF levels are often associated with preservation of neural function. We found that BDNF levels in the TBI-rat brain were significantly upregulated by VEGF-C (Fig. 4D). Together, these data suggest that VEGF-C administration improves brain function after experimental TBI.
VEGF-C reduces cell apoptosis in the TBI-brain
The effects of VEGF-C on cell apoptosis were then assessed by Annexin V assay on dissociated brain cells. We found that TBI significantly increased apoptotic cells in the brain, which was significantly attenuated by VEGF-C administration, shown by representative flow charts (Fig. 5A), and by quantification (Fig. 5B). Moreover, a key protein for apoptosis, cleavage caspase 3, was significantly upregulated in the TBI-brain, which was found to be significantly attenuated by VEGF-C (Fig. 5C), while a key anti-apoptotic protein, Bcl-2, was significantly downregulated in the TBI-brain, which was significantly attenuated by VEGF-C (Fig. 5D). These data suggest that VEGF-C administration may improve the function of the TBI-brain through reduction in cell apoptosis.

VEGF-C administration improves brain function after experimental TBI. A) The neurological score in TBI rats was assessed at 14 days after TBI. B) Brain water content at 14 days after TBI. C) EB extravasation assay. D) Western blotting for BDNF levels in the rat brain. *p < 0.05. N = 10.
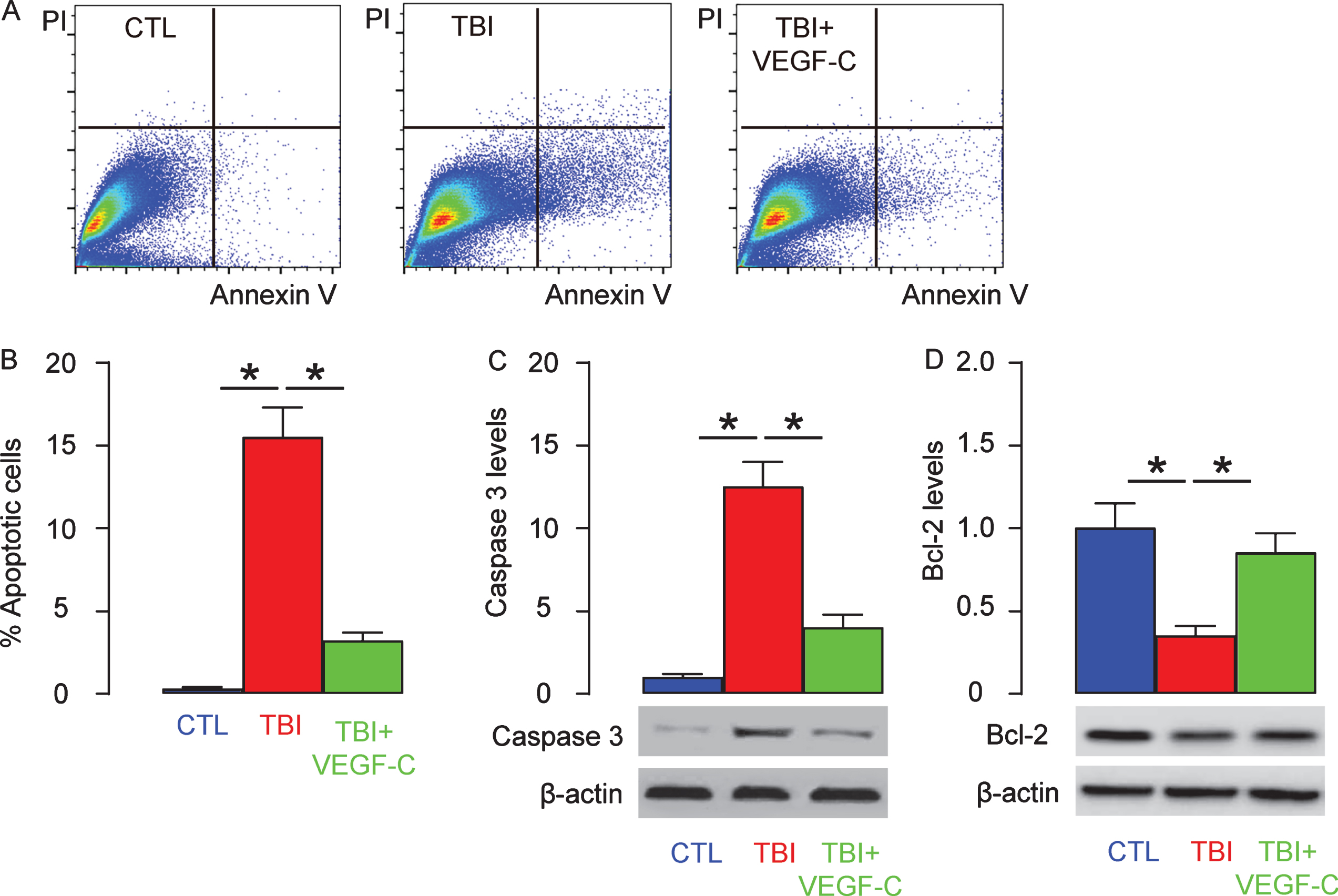
VEGF-C reduces cell apoptosis in the TBI-brain. The effects of VEGF-C on cell apoptosis were then assessed by Annexin V assay on dissociated brain cells, shown by representative flow charts (A), and by quantification (B). Western blotting for cleavage caspase 3 (C) and Bcl-2 (D) in dissociated brain cells. *p < 0.05. N = 10.

VEGF-C reduces microglia polarization to M2 in vivo. A) The microglia cells were purified from the dissociated rat brain post-TBI by double positivity for F4/80 and Iba-1, shown by a representative flow chart. B) Flow cytometry for CD206 on these microglia cells in three conditions (CTL; TBI; TBI+VEGF-C). C-E) RT-qPCR for Arginase 1 (Arg-1; C), Ym-1 (D) and Fizz-1 (E) on F4/80 + Iba-1 + microglia cells. *p < 0.05. NS, non-significant. N = 10.
VEGF-C reduces microglia polarization to M2 in vivo
Finally, we examined the effects of VEGF-C on microglia polarization in vivo. The microglia cells were purified from the dissociated rat brain post-TBI by double positivity for F4/80 and Iba-1 (Fig. 6A). First, we examined expression of CD206 on those microglia cells in three conditions (CTL, TBI, TBI + VEGF-C). We found that CD206 was induced in the microglia cells from VEGF-C-treated-TBI-rats by flow cytometry (Fig. 6B). Moreover, significant increases in Arg-1 mRNA (Fig. 6C), in Ym-1 mRNA (Fig. 6D), and in Fizz-1 mRNA (Fig. 6E) were also induced in the microglia cells from VEGF-C-treated-TBI-rats. Together, these data suggest that VEGF-C reduces microglia polarization to M2 in vivo.
DISCUSSION
The increasing prevalence of TBI correlates with increasing societal burden, though there is currently a lack of protective or therapeutic strategies for alleviating the damage after TBI and for preserving neurological functionality [5, 6]. Microglia cells play a pivotal role in the post-TBI inflammatory response [8]. Microglia cells express a large diversity of cell surface markers, which allow them to be distinguished from peripheral macrophages. Unlike peripheral macrophages, microglia cells express low CD45 [22], high CX3CR1 [23], and specially ionized calcium binding adapter molecule 1 (Iba-1) [24]. The latter was used in the current study to isolate microglia cells from the rat brain.
Microglia cells often display a prolonged and persistent pattern of activation after TBI, and may have both beneficial and detrimental effects on the recovery of brain tissue [9]. Of note, these multiple effects of microglia cells largely stem from their differentiation potential [25]. Indeed, as a specific tissue-resident macrophage in the brain, microglia cells share a lot of common properties with peripheral macrophages, including M1/M2 polarization. The microglia response in TBI highly depends on the timing and nature of the injury, in which microglia acquire heterogeneous phenotypes in response to insults with certain environmental cues. As in macrophages, M1 microglia exhibit some phagocytosing capability, while M2 microglia are considered neuroprotective [26]. However, in previous studies, VEGF-C has not been shown to be a trigger for M2 polarization of microglia.
Here, consistent with a previous study which showed both VEGF-C and VEGFR3 expression on glial cells [18], we showed that VEGF-C and VEGFR3 were both expressed in a microglia cell line. Since VEGFR3 seemed to be expressed by all the cells, while VEGF-C was only produced by some cells, we hypothesized that VEGF-C expression may represent a more active status of microglia cells, and that this VEGF-C/VEGFR3 signaling may function in an autocrine manner to amplify the response to VEGF-C. Most interestingly, triggering of this pathway by an external source of VEGF-C induced M2-like polarization of microglia cells in vitro, supporting our hypothesis.
To confirm our hypothesis, we used a TBI model in rats to assess the effects of VEGF-C administration. In order to analyze a clean microglia cell population, we had to use F4/80 and Iba-1 as markers for purification, since F4/80 is able to isolate peripheral macrophages and microglia cells from other cell types in the brain, while Iba-1 is only expressed on microglia cells, but not macrophages [24]. Iba-1 is not a surface marker, so we had to fix and perform penetration of the cells before analysis. Therefore, we were not able to perform an arginase activity assay on these fixed cells. However, the RT-qPCR data and results for expression of the cell surface marker CD206 supported an M2 polarization of microglia cells by VEGF-C. Of note, the beneficial effects of VEGF-C on protection and recovery of brain function, as well as on reduction in cell apoptosis, may result from both M2 polarization of microglia cells and augmentation of lymphangiogenesis [27], a classical function of VEGF-C, although so far there is no evidence that lymphangiogenesis plays a role in the pathological events post TBI [28].
To the best of our knowledge, the current study is the first to demonstrate a beneficial role of VEGF-C in treating TBI, which also suggests a possible mechanism of M2 polarization of microglia cells. Future studies may be designed to further characterize this model, in order to assess the possible use of VEGF-C for therapy in humans.
DISCLOSURE STATEMENT
Authors’ disclosures available online (https://www.j-alz.com/manuscript-disclosures/19-0063r1.