
Abstract
Emerging evidence suggests that epigenetic dysregulation of gene expression is one of the key molecular mechanisms of neurodegeneration and Alzheimer’s disease (AD). However, little is known about the role of epigenetic dysregulation on synaptic dysfunction in humans, because of the difficulties of obtaining live human neurons. Here we generated mature human cortical neurons differentiated from human embryonic stem cells, and exposed them to amyloid-β (Aβ). We found that the histone methyltransferase, EHMT1, which catalyzes histone lysine 9 dimethylation (H3K9me2, a mark for gene repression), was significantly elevated in Aβ-treated human stem cell-derived neurons. Aβ treatment led to a significant reduction of AMPAR-mediated whole-cell current and excitatory postsynaptic current. Application of BIX01294, a selective inhibitor of EHMT1/2, restored AMPAR currents and glutamatergic synaptic transmission in Aβ-treated human cortical neurons. These results suggest that inhibition of the aberrant histone methylation is a novel approach to reverse Aβ-induced synaptic deficits in human neurons.
Keywords
INTRODUCTION
Alzheimer’s disease (AD) is the most prevalent neurodegenerative disorder in the world. One of its hallmarks is the presence of amyloid plaques in brain regions like cerebral cortex and hippocampus [1, 2]. A large body of literatures has shown that amyloid-β (Aβ) oligomers may play a central role in synapse damage underlying the cognitive deficits of AD [3–5]. Mutations in amyloid precursor protein (APP) that result in the excessive accumulation of Aβ have been linked to a subset of familial AD [6, 7]. Despite the intensive studies in rodent animal models, little research has been done to examine the impact of Aβ on synaptic function in humans, because of the difficulties of obtaining live human neurons.
Rapid development of the field of human pluripotent stem cells (hPSCs), especially in vitro neural differentiation, offers an unprecedented approach to generate unlimited amounts of human neurons. Robust differentiation protocol from hPSCs to cortical neuron has been well developed during the past two decades [8–11]. In a pioneer study, cortical neurons derived from familiar AD patient-specific induced pluripotent stem cells (iPSCs), which carry mutations in presenilin 1 and presenilin 2, exhibit increased Aβ secretion [12]. Neurotoxicity and synaptotoxicity of Aβ were also observed in human iPSC-derived neurons [13, 14], which enforces the long-holding Aβ toxicity hypothesis in the etiology of AD [5, 16]. The success of hPSC-derived cortical neurons in recapitulating major AD pathologies suggests that they can serve as a more biologically and clinically relevant system in AD research, comparing to the conventional overexpression-based animal models [17, 18].
Despite a few identified AD-related genetic risk factors, the lack of a genetic basis for the majority of this disease intrigues academic to find alternative etiology for AD. Emerging evidence suggests that epigenetic dysregulation of gene expression may play a key role in aging and neurodegeneration [19–22]. Histone methylation is one of the epigenetic mechanisms to control gene expression. Through adding or removing methyl groups on Lysine (K) residues of histones by histone methyltransferases or histone demethylases, the structure of chromatin is dynamically remodeled to allow or stop access of transcription regulatory proteins to genomic DNA, thus leading to gene activation or repression [23, 24]. Here we generated human embryonic stem (hES) cells-derived cortical neurons, and examined the potential of targeting histone methylation enzymes in rescuing glutamate receptor function in these human neurons exposed to Aβ.
MATERIALS AND METHODS
Differentiation of human ES cells to forebrain neurons
Human ES cells (H9, WiCell, Wisconsin) were routinely maintained on feeder cells in hES cell medium (DMEM/F12 supplemented with 20% knockout serum replacement, 0.1 mM nonessential amino acids (NEAA), 1x Penicillin/Streptomycin, 0.1 mM 2-Mercaptoethanol, 0.2 mM glutamine, 4 ng/ml bFGF) and passaged with 1 mg/ml dispase (Stemcell Technologies) every 7 days. Neural differentiation was performed using previously published protocols [8, 25] with modification. Briefly, hES cells around passage 30 were dissociated with 1 mg/ml dispase into small clumps which were gently transferred to non-treated EasYflasks (Nunc, cat# 169900) to form embryoid bodies (EB) in EB medium (DMEM/F12 supplemented with 20% knockout serum replacement, 0.1 mM nonessential amino acids (NEAA), 1x Penicillin/Streptomycin, 10μM SB431542 and 5μM Dorsomorphin) for 6 days. Then EBs were plated on Matrigel-coated 6-well plates in N2 Medium (DMEM/F12 with 1×N2 supplements, 0.1 mM NEAA, 10 ng/ml bFGF) for two weeks. Well-formed rosettes were isolated manually, dissociated with TrypLE and expanded in N2 medium on matrigel-coated plate. Upon reaching confluence, cells were dissociated with TrypLE into single cells and plated on polyornithine/matrigel-coated cover slips at 1×105-2×105/cm2 in neural differentiation medium containing: Neurobasal, 1×B27 supplements, GDNF (20 ng/ml), BDNF (20 ng/ml), NGF (20 ng/ml), ascorbic acid (200μM), and dcAMP (0.5 mM). DAPT (2.5μM) was added 7 days later. Medium was half changed every other day.
Rat primary neuronal culture
Rat frontal cortical cultures were prepared as described previously [26]. All experiments were carried out with the approval of State University of New York at Buffalo Animal Care Committee. Briefly, pregnant Sprague-Dawley rats were anaesthetized with isoflurane vapor and immediately sacrificed. Frontal cortex of rat embryos (E18) was dissected and cells were dissociated using trypsin and trituration through a Pasteur pipette. Neurons were plated on coverslips coated with poly-L-lysine in DMEM with 10% fetal calf serum at a density of 1×105 cells/cm2. When neurons attached to the coverslip within 24 h, the medium was changed to neurobasal media (Invitrogen) with B27 supplement. Cytosine arabinoside (Arac, 1.25μM) was added to culture media at DIV3 to stop glial proliferation. Neurons were maintained for 2 weeks before being used.
Aβ and BIX01294 treatment
Pre-aggregated Aβ1 - 42 peptides (Anaspec, AS72216) and BIX01294 (Sigma, B9311) was directly dissolved with PBS. Rat or human cortical cultures were subjected to Aβ (1μM) treatment for 3 days with or without overnight BIX01294 treatment (0.1μM for rat cortical cultures, 0.25μM or 0.5μM for human cortical cultures, added after Aβ treatment). BIX01294 was used as a specific EHMT1/2 inhibitor, because it specifically inhibits EHMT2 (G9a) and EHMT1 (GLP) with no significant activity at other histone methyltransferases [27].
Immunocytochemistry
Immunocytochemical staining of human neurons used similar procures as we described previously [28]. Human ES cell-derived cortical cultures (DIV120) were fixed in 4% paraformaldehyde in phosphate buffered saline (PBS) for 20 min at room temperature and washed 3 times with PBS. After permeabilization with 0.1% Triton X-100 in PBS for 15 min, neurons were incubated with 5% bovine serum albumin for 1 h to block nonspecific staining. Next, neurons were incubated with primary antibodies at 4°C overnight, including anti-Pax6 (1:200, Developmental Studies Hybridoma Bank, AB528427), anti-Nestin (1:5000, Millipore, abd69), anti-Tuj1 (1:1000; Abcam, ab78078), anti-MAP2 (1:500; Santa Cruz, SC74421), and anti-vGluT1 (1:1000; Synaptic System, 135–303). After washing, neurons were incubated with Alexa Fluor 594- or Fluor 488-conjugated secondary antibody (1:2000, Thermo Fisher) for 2 h at room temperature. After washing in PBS for three times, cell nuclei were visualized with DAPI (Sigma Aldrich, 1:10,000) and coverslips were mounted on slides with VECTASHIELD mounting medium (Vector Laboratories, Burlingame, CA). Fluorescence images were obtained using a 20×or 40×lens and were captured digitally using SPOT basic image capture software.
Real-time RT-PCR
Cultured neurons were lysed in cold Trizol reagent (Invitrogen) to extract total RNA. Then, the cDNA was obtained using iScript cDNA Synthesis Kit (Bio-Rad) and real-time PCR was performed using iQSYBR Green Supermix (Bio-Rad CFX96 Touch system) according to the manufacturer’s instruction. Fold changes in the target gene expression was determined by normalized CT value (2- Δ ( Δ CT), ΔCT = CTtarget - CTGAPDH, and Δ(ΔCT) =ΔCTAβ - ΔCTCon, GAPDH was used as housekeeping gene; CT means threshold cycle, which is defined as the cycle number getting detectable fluorescence signal. A PCR mixture of 20μl per well (96-well thin-wall PCR plate, Bio-Rad) was amplified according to the following cycling condition: 95°C for 5 min, followed by 40 cycles of 95°C for 30 s, 55°C for 30 s, and 72°C for 60 s. Quantitative real-time PCR was performed in double reactions. The sequences of primers were as follows: human EHMT1:5’-GGACGGAATTGACCCCAACT, 5’-AATATTAGCGCC-CGCCTGAA; human EHMT2:5’-TCCAGCATTTCCGCATGAGTGA, 5’-CAACTGTTCAG-CTAGAGCTTCG; human GAPDH: 5’-GACAACAGCCTCAAGATCATCAG, 5’-ATGGCATGGACTGTGGTCATGAG; rat EHMT1:5’-CATAGCAAAAGCAGACACGA, 5’-ACTTTCCAAGGTTTCCTTTC; rat EHMT2:5’-TCCAGCATTTCCGCATGAGTGA, 5’-CAACTGTTCAGCTAGAGCTTCG; rat GAPDH: 5’-GACAACTCCCTCAAGATTGTCAG, 5’-ATGGCATGGACTGTGGTCATGAG.
Electrophysiology in human cortical cultures
Human ES-derived cortical neurons (DIV110-140) were used to measure ionic currents (voltage-dependent sodium, potassium and calcium currents), synaptic currents (AMPAR- and NMDAR-mediated currents, spontaneous (sEPSC) and miniature excitatory postsynaptic currents (mEPSC)), action potentials and network activity using whole-cell voltage-clamp or current-clamp techniques as we previously described [29–32]. The long-term culture was used because only at this stage, all the electrophysiological properties of mature cortical neurons could be reliably detected in most of the recorded neurons. Recordings were obtained with an Axon Instruments 200B amplifier that was controlled and monitored by an IBM PC running pClamp 10.0 with a DigiData 1320 series interface (Axon instruments). Tight seals (2–10 GΩ) from visualized neurons were obtained by applying negative pressure. With additional suction, the membrane was disrupted into the whole-cell configuration following series resistance (4–10 MΩ) compensation (70–90%).
For the recording of voltage-dependent sodium, potassium and calcium currents, cells (held at –70 mV) were depolarized by brief pulses of voltage steps (–90 mV to +50 mV) or a ramp protocol. For the recording of APs, membrane potentials were kept at –70 mV, and a series of hyperpolarizing and depolarizing step currents were injected. Spontaneous APs were recorded with cells (held at –55 mV) without current injection. AMPAR and NMDAR-mediated currents were evoked by application of AMPA (100μM) or NMDA (100μM) for 2 s every 30 s in neurons held at –70 mV. Drugs were delivered with a “sewer pipe” system. The array of drug capillaries (∼150μm inner diameter) was positioned a few hundred microns from the cell under recording. Solution changes were controlled by the SF-77B fast-step solution stimulus delivery device (Warner Instruments). Neurons were perfused with ACSF (in mM): 130 NaCl, 26 NaHCO3, 3 KCl, 1 CaCl2, 5 MgCl2, 1.25 NaH2PO4, 10 Glucose, pH 7.4, 300 mOsm, bubbled with 95% O2 and 5% CO2.
For the recording of voltage-dependent Na+, K+, Ca2 + currents, and APs, internal solution contained (in mM): 60 K2SO4, 60 N-methyl-D-glucamine, 20 HEPES, 1 MgCl2, 0.5 EGTA, 12 phosphocreatine, 2 Na2ATP, 0.2 Na2GTP and 0.1 leupetin, pH 7.2-7.3, 265–270 mOsm. External solution was ACSF. A modified ACSF (3 mM CaCl2, 20 mM CsCl, 0.5μM TTX) was used for Ca2 + current recordings.
For the recording of AMPAR- or NMDAR-mediated ionic currents, internal solution contained (in mM): 180 N-methyl-D-glucamine, 4 MgCl2, 40 HEPES, 0.5 BAPTA, 12 phosphocreatine, 3 Na2ATP, and 0.5 Na2GTP, pH 7.2-7.3, 265–270 mOsm. External solution for AMPAR current recording contained (in mM): 127 NaCl, 20 CsCl, 1 MgCl2, 10 HEPES, 5 BaCl2, 12 glucose, 0.001 TTX, pH 7.3-7.4,300–305 mOsm. External solution for NMDAR current recording was the same except for the removal of MgCl2 and the addition of 1 mM CaCl2 and 20μM glycine.
For sEPSC and network activity measurements, cells were held at –70 mV. Internal solution contained (in mM): 130 cesium methanesulfonate, 10 CsCl, 4 NaCl, 1 MgCl2, 10 HEPES, 5 EGTA, 2.2 lidocaine, 12 phosphocreatine, 5 MgATP, 0.5 Na2GTP, 0.1 leupeptin, pH 7.2-7.3, 265–270 mOsm. External solution contained (in mM): 127 NaCl, 5 KCl, 2 MgCl2, 2 CaCl2, 12 glucose, 10 HEPES, pH 7.3-7.4, 300–305 mOsm. Tetrodotoxin (TTX, 0.5μM) was added for mEPSC recordings.
Data analyses were performed with Clampfit (Axon instruments), Mini Analysis Progam (Syanptosoft, Leonia, NJ) and Kaleidagraph (Albeck Software).
Electrophysiology in rat cortical cultures
Cultured rat cortical neurons (DIV 16–21) were used for whole-cell recordings of AMPAR-mediated ionic and synaptic currents with standard voltage-clamp techniques as we previously described [28]. Neurons were perfused with background solution containing (in mM): 140 NaCl, 2 KCl, 2 MgCl2, 1 CACl2, 15 HEPES, 23 glucose, pH 7.3-7.4, 300–305 mOsm. Internal and external solutions for recording AMPAR-mediated ionic currents and mEPSC were the same as those used in human neurons.
Statistics
All data are expressed as mean±s.e.m. Statistical analysis for experiments with two groups was performed using Student’s t-test. Experiments with more than two groups were subjected to one-way or two-way ANOVA, followed by Bonferroni’s post hoc tests using Prism 5.0 (GraphPad Software).
RESULTS
Generation and characterization of human cortical neurons differentiated from human embryonic stem cells
hES cells were cultured and differentiated using an embryoid body (EB)-based system [8, 25]. The hES cells (Fig. 1A) were first differentiated to EBs (Fig. 1B). After the initial differentiation, EBs were plated on matrigel-coated plates to enable the formation of neuroepithelial rosettes that express neuroectodermal differentiation marker Pax6 (paired box protein 6) and neuronal stem cell marker Nestin (Fig. 1C, E). Cells from these rosettes were continually cultured and further differentiated into forebrain cortical neurons (Fig. 1D). After 8–10 weeks of in vitro cultivation, matured neurons were immunocytochemically stained for the neuronal marker Tuj1 (neuron-specific β-III Tubulin), dendritic marker of matured neurons MAP2, and glutamatergic neuronal marker vGluT1 (vesicular glutamate transporter 1) (Fig. 1F, G). The positive staining of these markers indicated that we successfully differentiated hES into matured glutamatergic neurons.

Generation of human cortical neurons from human embryonic stem (hES) cells. Images of distinct stages of in vitro differentiation of hES cells, including human ES cells (A), embryoid bodies (EBs, B), neuroepithelial cells (C), and neurons (DIV40, D). E) Images of neuroepithelial cells (DIV 20) immunostained for neuroectodermal differentiation marker PAX6 (green), neural stem cell marker Nestin (red) and DAPI (blue). F) Images of hES cell-derived neurons (DIV 120) immunostained for neuronal marker Tuj1 (green), dendritic marker MAP2 (red), and DAPI (blue). G) Images of hES cell-derived neurons (DIV 120) immunostained for Tuj1 (green), glutamatergic neuronal marker vGluT1 (red) and DAPI (blue). Scale bar: 20μm.
Next, electrophysiological recordings were performed to test whether these human neurons (∼DIV110) were functionally active. We found that most of the neurons exhibited voltage-dependent Na+ and K+ currents (90% of recorded cells, n = 27, Fig. 2A), voltage-dependent Ca2 + current (Fig. 2B), and action potentials elicited by depolarizing current pulses (75% of recorded cells, n = 23, Fig. 2C), indicating that the intrinsic membrane properties of these human neurons are normal. AMPAR-mediated and NMDAR-mediate currents were also observed (Fig. 2D), indicating the presence of ligand-gated receptor channels in the human neurons. Moreover, sEPSC and mEPSC (Fig. 2E) were found in 75% of recorded cells (n = 22). The cortical neuron-specific rhythmic network activity (Fig. 2F) and the synaptic-driven spontaneous action potentials (sAP) (Fig. 2G) were also detected (50% of recorded cells, n = 16), indicating the formation of glutamatergic synaptic connections and transmission in these human neurons. Taken together, the data confirm that all the major features of mature glutamatergic neurons are present in the generated human neurons, which makes them a valid in vitro system.

Electrophysiological characterization of human ES cells-derived mature cortical neurons (DIV110). A) Representative traces of voltage-dependent Na+ and K+ currents in response to depolarization steps. B) Representative traces of voltage-dependent Ca2 + currents in response to depolarization protocols. C) Action potentials in response to current injections. D) Representative traces of whole-cell AMPA- and NMDA-elicited ionic currents. E) Representative traces of spontaneous and miniature excitatory postsynaptic currents. F) Representative traces of large rhythmic network activity. G) Representative traces of spontaneous action potentials.
Alteration of histone methyltransferases by Aβ treatment in human and rodent neurons

The histone methyltransferase EHMT1/2 was elevated in human cortical neurons and rat neuronal cultures. qPCR data on the mRNA level of EHMT1 and EHMT2 in human ES cells-derived mature cortical neurons (A) and rat cortical cultures (B) without or with the treatment of Aβ oligomers (1μM, 3 days). *p < 0.05, t-test.
To study the pathophysiological basis and treatment strategy of AD using the generated human neurons, we exposed them to Aβ, since Aβ oligomers have been found to induce synaptic dysfunction and cognitive impairments in rodent models [3–5]. Postmortem studies of AD human brains have found elevated H3K9me2, a repressive histone mark, in the cortex [33, 34], so we examined the mRNA level of EHMT1/2, the H3K9me2-catalyzing enzyme, in mature hES-derived cortical neurons exposed to Aβ (1μM) for 3 days [28]. The qPCR assays indicated that EHMT1 mRNA was significantly increased in Aβ-treated human neurons (Fig. 3A, 1.42±0.13 of control, n = 5, p < 0.05, t-test). Similarly, Aβ treatment significantly elevated the mRNA level of EHMT1 and EHMT2 in rat cultures (Fig. 3B, EHMT1:1.42±0.18 of control, EHMT2:1.29±0.11 of control, n = 6, p < 0.05, t-test).
Loss of AMPAR currents by Aβ treatment and its rescue by an EHMT inhibitor in human and rodent neurons
Next, we examined the Aβ-induced alteration of whole-cell AMPAR current and its normalization by EHMT1/2 inhibitor in human neurons. As shown in Fig. 4A and 4B, AMPAR-mediated ionic current density (pA/pF) was significantly decreased in Aβ-exposed human neurons (Con: 15.68±1.62, n = 22; Aβ: 6.60±0.70, n = 20, p < 0.001, ANOVA). Treatment with the specific EHMT1/2 inhibitor BIX01294 (0.5μM, 12 h) [27] significantly increased AMPAR current density (Aβ+BIX01294:12.42±1.95, n = 22; Con+BIX01294:17.04±1.95, n = 17), bringing it close to the control level. Consistently, in rat cortical cultures, Aβ significantly decreased AMPAR current density, which was rescued by subsequent BIX01294 treatment (Fig. 4 C and D, Con: 28.09±1.78, n = 26; Aβ: 16.86±1.77, n = 24; Aβ+BIX01294:23.94±1.75, n = 23; Con+BIX01294:26.53±2.43, n = 18, p < 0.001, ANOVA).
To find out the impact of Aβ and BIX01294 on glutamatergic synaptic transmission in human cortical neurons, we examined AMPAR-mediated EPSC. As shown in Fig. 5A and B, Aβ-treated human cortical neurons exhibited significantly decreased sEPSC frequency and amplitude (Frequency, Con: 3.31±0.99 Hz, n = 8; Aβ: 0.48±0.10 Hz, n = 8, p < 0.05, ANOVA; Amplitude, Con: 17.14±0.58 pA, n = 8; Aβ: 11.52±0.53 pA, n = 8, p < 0.001, ANOVA). Subsequent treatment with BIX01294 induced a complete recovery of sEPSC frequency and a slight increase of sEPSC amplitude (Frequency, Aβ+BIX01294:3.07±0.70 Hz, n = 9; Amplitude, Aβ+BIX01294:13.00±1.29 pA, n = 9). mEPSC, a synaptic response resulting from quantal release of single glutamate vesicles, showed a marked reduction in its frequency by Aβ treatment, which was dramatically restored by BIX01294 treatment in human neurons (Fig. 5C and D, Con: 3.15±0.70 Hz, n = 9; Aβ: 0.85±0.14 Hz, n = 8; Aβ+BIX01294:2.79±0.43 Hz, n = 8, p < 0.01, ANOVA). The amplitude of mEPSC was not significantly altered by Aβ and BIX01294 in human neurons (Con: 10.61±0.38 pA, n = 9; Aβ: 10.49±0.39 pA, n = 8; Aβ+BIX01294:10.39±0.39 pA, n = 8). Similarly, in Aβ-treated rat cortical cultures, BIX01294 successfully rescued mEPSC frequency and amplitude (Fig. 5E and F, Con: 5.72±1.05 Hz, 28.95±1.66 pA, n = 16; Aβ: 0.78±0.21 Hz, 22.68±1.54 pA, n = 11; Aβ+BIX01294:5.55±1.54 Hz, 25.01±1.06 pA, n = 11; Con+BIX01294:4.61±0.98 Hz, 26.03±1.44 pA, n = 17, p < 0.05, ANOVA).
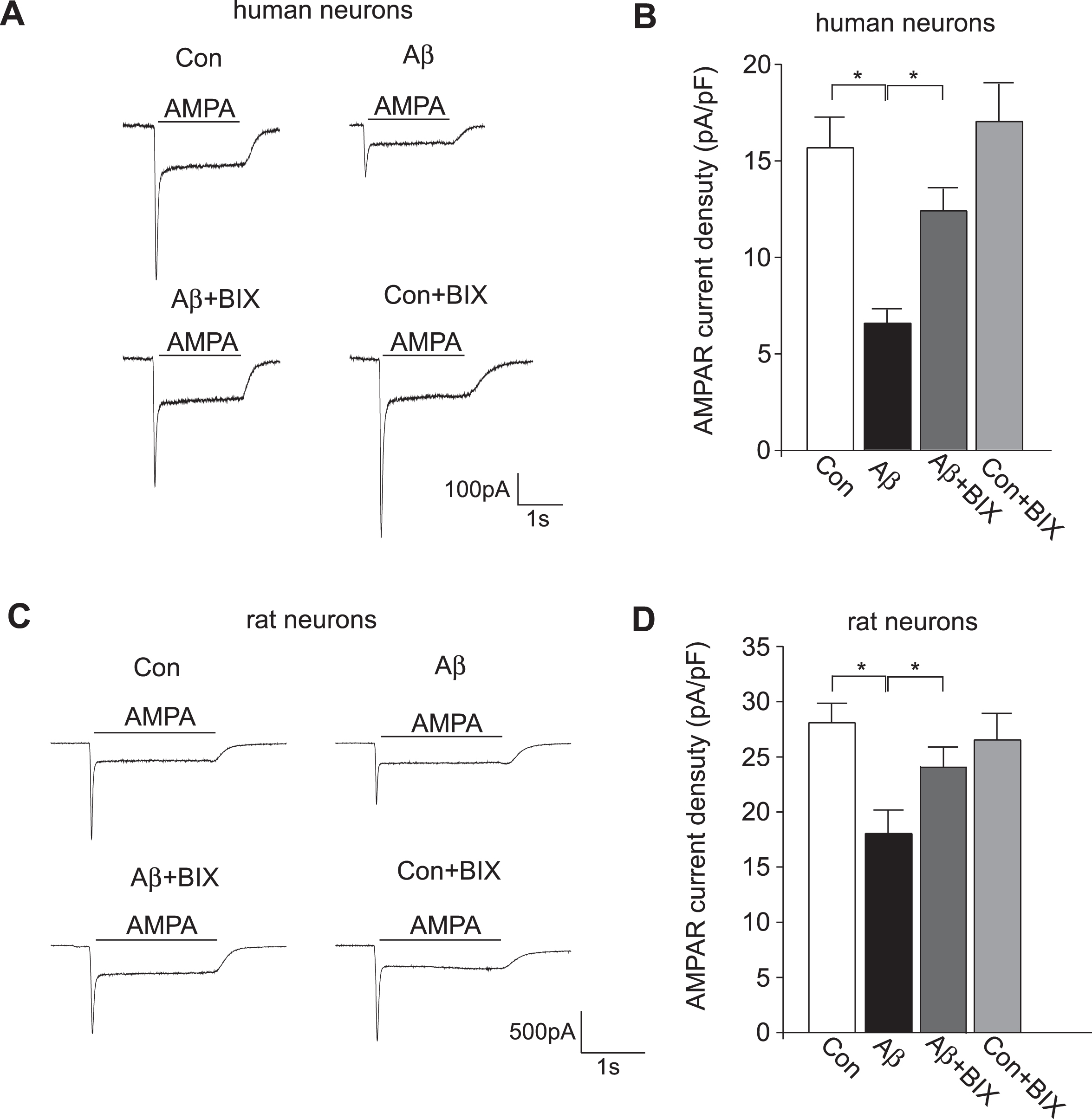
Aβ treatment decreased AMPAR current density in human cortical neurons and rat neuronal cultures, which was rescued by the EHMT inhibitor BIX01294. Representative traces of AMPAR-mediated ionic currents in human cortical neurons (A) or rat cultures (C) without or with the Aβ exposure (1μM, 3 days) and the subsequent BIX01294 treatment (0.5μM, 12 h). Bar graph (mean±SEM) of AMPAR current density in different groups of human cortical neurons (B) or rat cultures (D). *p < 0.05, ANOVA.

Aβ treatment decreased AMPAR-mediated synaptic transmission in human cortical neurons and rat neuronal cultures, which was rescued by the EHMT inhibitor BIX01294. Representative traces of sEPSC (A) and mEPSC (C, E) in human cortical neurons (A, C) or rat cultures (E) without or with the Aβ exposure (1μM, 3 days) and the subsequent BIX01294 treatment (0.5μM, 12 h). Bar graph (mean±SEM) of sEPSC frequency & amplitude (B) and mEPSC frequency & amplitude (D, F) in different groups of human cortical neurons (B, D) or rat cultures (F). *p < 0.05, **p < 0.01, ANOVA.
DISCUSSION
Most AD research is based on animal models, which has provided insights into the pathogenic mechanisms and drug targets for this neurodegenerative disease. However, human conditions cannot be entirely recapitulated in rodents. The translational value of animal models could be enhanced when combined with in vitro models derived from patient-specific iPSCs and isogenic controls generated using CRISPR/Cas9 genome editing [17, 35]. Induced neurons from FAD patients carrying risk factors, such as mutant presenilins or APOE ɛ3/4 allele, recapitulate pathological signatures of AD [12, 36]. Human PSC-derived cortical neuronal precursors transplanted into the brain of a murine AD model display human-specific pathological features, including abnormal tau and neurodegeneration [37]. By using a frontotemporal dementia patient-derived iPSC carrying the Tau P301 L mutation and the isogenic lines in which p35 is replaced with a noncleavable mutant, researchers have confirmed the role of p25/Cdk5 in tauopathy [38]. Thus, human stem cell-derived neurons offer a new platform for investigating molecular mechanisms of AD and identifying therapeutic agents [39, 40]. However, the heterogeneity of the human population, the extended temporal course of the disease, and the accurate modeling of common non-familial “sporadic” forms of AD remain challenges for human reprogramming-based cell models [17].
Genetic and epigenetic variation associated with the initiation and progression of AD can also be investigated using iPSCs [41]. Epigenetic mechanisms have been proposed to play a key role in the deregulation of vulnerability genes underlying AD pathogenesis [20, 42]. Animal studies suggest that histone post-translational modifications, particularly histone acetylation, are prospective drug targets for AD [43–46]. Histone methylation marks and enzymes are implicated in senescence and cognitive functioning [47], but their links with AD await to be firmly identified [20, 45]. Human postmortem studies have found aberrant histone methylations at frontal cortex of AD patients [33, 48], suggesting that targeting histone methylation is a new avenue to treat AD-related abnormalities.
In the current study, we have used Aβ exposure of human cortical neurons differentiated from hES cells as an in vitro model for sporadic AD. Choosing this system instead of human iPSCs is based on several considerations. First, H9 hES cells are the gold standard in stem cell research widely used in a variety of studies. Data generated on H9 hES cells can be readily compared to data generated on the same cells by other investigators. Second, the diverse genetic background of idiopathic AD patients makes it hard to use patient-specific iPSCs to compare with those from normal control subjects. Third, as idiopathic AD has complex and unclear genetic contributions, it is not possible to generate isogenic control lines. Fourth, even if iPSCs from AD patients with monogenic mutations are used, it is unclear whether the results are generally applicable to idiopathic AD. In this study, we have demonstrated that EHMT1 mRNA is significantly increased in Aβ-treated human neurons, and the Aβ-induced impairment of AMPAR-mediated ionic currents and synaptic transmission is rescued by in vitro treatment with an EHMT1/2 inhibitor. The potential relevance of these findings to AD treatment is confirmed by our recent study in a familial AD mouse model carrying APP mutations, which shows that in vivo administration of EHMT1/2 inhibitors rescues synaptic and cognitive deficits by restoring glutamate receptor expression [34]. Our current results have further validated the potential of using human stem cell-derived neurons for disease modeling and drug discovery.
In rodent neurons, Aβ is found to impair glutamatergic signaling by affecting presynaptic vesicle cycling [49, 50] and disrupting postsynaptic AMPAR trafficking [28, 51]. In human stem cell-derived neurons, the Aβ-induced substantial reduction of sEPSC and mEPSC frequency suggests a presynaptic and a possible postsynaptic component, while the Aβ-induced reduction of AMPAR ionic current suggests a postsynaptic component. The restoration of glutamatergic transmission in human neurons by EHMT1/2 inhibitor is likely achieved by the collective upregulation of the transcription of multiple presynaptic and postsynaptic genes. Human studies have revealed the causal role of EHMT1 in intellectual disability [52, 53]. Our current study suggests that targeting EHMT1/2-associated chromatin-modification module might also be able to restore transcriptional homeostasis and synaptic function in neurodegenerative disorders.
Footnotes
ACKNOWLEDGMENTS
We would like to thank Xiaoqing Chen for her excellent technical support. This work was supported by National Institutes of Health (NIH) grant (AG056060) to ZY, NIH grant (NS102148) to JF, and Department of Veterans Affairs Merit Awards (BX002452, BX003831) to JF.