
Abstract
Exposures to fine particulate matter (PM2.5) and ozone (O3) ≥US EPA standards are associated with Alzheimer’s disease (AD) risk. The projection of 13.8 million AD cases in the US by the year 2050 obligate us to explore early environmental exposures as contributors to AD risk and pathogenesis. Metropolitan Mexico City children and young adults have lifetime exposures to PM2.5 and O3, and AD starting in the brainstem and olfactory bulb is relentlessly progressing in the first two decades of life. Magnetite combustion and friction-derived nanoparticles reach the brain and are associated with early and progressive damage to the neurovascular unit and to brain cells. In this review: 1) we highlight the interplay environment/genetics in the AD development in young populations; 2) comment upon ApoE ɛ4 and the rapid progression of neurofibrillary tangle stages and higher suicide risk in youth; and 3) discuss the role of combustion-derived nanoparticles and brain damage. A key aspect of this review is to show the reader that air pollution is complex and that profiles change from city to city with common denominators across countries. We explore and compare particulate matter profiles in Mexico City, Paris, and Santiago in Chile and make the point of why we should invest in decreasing PM2.5 to at least our current US EPA standard. Multidisciplinary intervention strategies are critical for prevention or amelioration of cognitive deficits and AD progression and risk of suicide in young individuals. AD pathology evolving from childhood is threating the wellbeing of future generations.
Keywords
INTRODUCTION
The current definition of tauopathies includes morphologically and biochemically heterogeneous neurodegenerative diseases with a wide clinical spectrum, characterized by pathological intracellular deposits of tau [1–3]. Accumulation of pathological tau is a hallmark of Alzheimer’s disease (AD) [3]. Increasing evidence suggests that AD complex pathogenesis includes strong interactions between a harmful environment, systemic, and neural inflammatory, immunological, and metabolic hosts’ responses [4–11]. In the US, we have approximately 5.8 million AD patients and ∼14 million are projected by 2050. In Mexico, the prevalence of AD is 7.3% with an incidence of 27.3 per 1,000 people/year [12]. As of 2015, the Mexican projections [12] calculated 3.5 million people affected by AD by 2050; however, we know today that 99.5% of Metropolitan Mexico City (MMC) young residents (25.36±9.23 years) already have AD hallmarks [4], so that projection will have to change accordingly. In Chile, Patricio Fuentes and Cecilia Albalá [13] stated that the prevalence of AD in people >60 years exceeds 8%, and there is a striking negative association of dementia with years of education: dementia in illiterates reaches 25.2%, while the rate in subjects with >13 years of schooling is only 1.2%. Socioeconomic inequality and the structure and dynamics of Chile’s healthcare system prevent delivery of quality care to the majority of those affected by dementia [14]. Jacqmin-Gadda et al. [15] computed projections of age- and-sex specific prevalence of dementia in France from 2010 to 2030 based on the Paquid cohort, and concluded the dementia prevalence will increase by at least 60% from 2010 to 2030 and France will have to face a dramatic rise of the number of very old demented individuals (+200 % over 20 years in the age range 90–99).
The Apolipoprotein E (ApoE) ɛ4 allele is the most prevalent genetic risk factor for AD [16] and poses a stronger risk in women. In Mexico, Chile, and the US, ɛ3 is the most common allele, while the prevalence of ɛ4 allele in Mexico varies from 8.4% in Guadalajara, to 11.5% in Nayarit and Durango, to 28% among Huichol Indians [17]. In our laboratory, ∼20% of Mexico City residents carry an allele 4; in contrast, about 13% of a predominantly white Caucasian population in the Framingham Heart Study have the allele [18].
Sustained systemic inflammation, metabolic imbalance, and neuroinflammation are recognized factors contributing to neurodegeneration, and the very same effects occur in response to chronic exposure to urban air pollution [19–24]. Based on a 9-year cohort follow-up study of 95,690 individuals, age ≥65 years, Jung et al. [10] estimated that AD risk increases by 211% per 10.91 ppb increase in ozone (O3), and 138% per 4.34 μg/m3 increase in environmental fine particulate matter <2.5 μm (PM2.5). Their findings suggest long-term exposure to concentrations of O3 and PM2.5 that exceed the current United States Environmental Protection Agency (US EPA) standards significantly increase AD risk [10]. Chen et al. have shown an increased in incident dementia in Ontario residents living near major roads with high traffic [11].
In this review, we present published data concerning neuropathological, cognitive, behavioral, and cerebral neuroimaging effects of air pollutant exposure in children, and put forward a hypothesis that several active players in the urban air polluted scenarios—including the concentrations of fine particulate matter (<2.5 μm to >100 nm) and ultrafine particles (<100 nm), their portals of entry, chemistry, and size, set the stage for early progressive neurodegeneration. Combustion and friction-derived, iron-rich, strongly magnetic nanoparticles are strong candidates to produce oxidative stress and protein misfolding and fibrillation with the resultant cellular damage, neuroinflammation, and neurodegeneration [4]. We strongly support that in Mexico City the combination of lifetime exposures (including intrauterine life) to complex mixtures of air pollutants, genetics, lifestyle, and socioeconomic factors cause stresses that drive early systemic inflammation, insulin resistance, hyperleptinemia, structural/metabolic brain changes, neuroinflammation, and neurodegeneration. ApoE ɛ4 is at the centerpiece of this work along with early identification of subjects at risk.
AIR POLLUTANT EXPOSURES AND DEVELOPMENT OF AD
MMC has approximately 5.5 million cars and one of the highest levels of traffic air pollution in the world; residents are being exposed from conception to concentrations of PM2.5 and O3 above US EPA standards. The average MMC resident lives his entire life within less than 300 meters from a street with high volume traffic and is chronically exposed to highly oxidative and ubiquitous combustion and friction-derived nanoparticles (CFDNPs), metals, lipopolysaccharides, polycyclic aromatic hydrocarbons (PAHs), etc. MMC residents spend an average of one hour in traffic every day and travelling in public transportation, i.e., subway, increasing their chance of higher PM2.5 exposures by 6%, including exposures to metals such as Fe, Cu, Ni, Cr, and Mn, resulting from underground sources such as friction, brake systems, and sparking [25]. Minibuses and buses’ emissions expose people to similar concentrations of PM2.5 (∼51 μg/m3), while CO and benzene concentrations are higher in minibuses [26]. MMC has a population density of 9,800 per square kilometer and produces 107,743 tons of solid waste per day. Garbage trucks are seen in every corner selecting the recyclable products and polluting the environment. Around 66% of solid waste are residues with an organic composition that end in landfills and open dumps [27]. People are also exposed to particulate matter from anafre-portable charcoal stoves allowed on street corners and from restaurants, coffee shops, and factories with no control of particulate matter emissions, in addition to 5 million smokers who smoke in public transit, public buildings, schools, workplaces, and streets. Mexico City has the highest prevalence of smokers ages 12–65 in the entire country [28]. MMC residents are exposed to a very complex mixture of air pollutants, with common denominators including fuel combustion from vehicles, industrial and household sources, chemical pollutants in indoor dusts, indoor pollutants such as tobacco, and occupational exposures. The complexity of the exposures and their portals of entry are very important when we try to compare health effects of pollutants in different cities, countries, seasons, etc., or when we read about the pathological effects of controlled exposures in experimental genetically uniform rodents. PM2.5 resulting from gasoline and diesel vehicle emissions and liquefied petroleum gas combustion, produce organic carbonaceous aerosols and black carbon (BC). BC, the result of incomplete combustion of fossil fuels, biofuels, and biomass, is associated with PAHs, semi-volatile species resulting from incomplete combustion of carbonaceous fuels [29, 30]. Trace metals are important components of PM2.5 and include road traffic tracers, i.e., Zn, Cu, Ba, Pb, and Cd, and V and Ni tracers of industrial emissions [30].
The two sites where we first detect hyperphosphorylated tau (P-Tau) in infants are indeed portals of entry of particulate material: the lower medulla and the olfactory bulb (OB) (Fig. 1). The lower medulla sections of Mexico City infants showed P-Tau neurites in the dorsal motor nucleus of the vagus and spinal trigeminal nucleus [4]. Swallowed CFDNPs have easy access to the gastrointestinal epithelium and submucosa, damaging the gastrointestinal barrier integrity and accessing the enteric nervous system [31, 32]. The nasal trigeminal neural pathway is also a potential portal for a direct transport of particles to the brainstem [33]. Intranasal delivery has been shown to non-invasively deliver drugs from the nose to the brain in minutes along the olfactory and trigeminal nerve pathways, bypassing the blood-brain barrier [33].

Representative images of light (immunohistochemistry, IHC) and electron microscopy in children and teens. Key hallmarks defining Alzheimer’s disease: hyperphosphorylated tau and amyloid-β (Aβ) plaques in Mexico City children and teens. A) Medulla section of a 3-year-old boy with ApoE ɛ3/ɛ3 showing a positive P-Tau neurite (upper arrow) alongside a positive P-Tau nucleus (indicative of DNA damage). The lower arrow points to +P-Tau nucleus. IHC AT8, scale bar = 10 μm. B) Mesencephalic section, in the same 3-year-old child as in A, showing P-Tau granular staining in the proximal axons (arrows) of two adjacent neurons. IHC AT8, scale bar = 10 μm. C) Commissure of the superior colliculus in a 3-year-old. Crossing fibers are positive for P-Tau (arrows). The superior colliculus sends small direct projections to the contralateral abducens nucleus and a substantial projection to the reticular formation. It is proposed that the graded density of the cells of origin of this projection is the basic structural mechanism by which the colliculus generates horizontal foveating saccades of different amplitudes [131]. IHC AT8, scale bar = 10 μm. D) Substantia nigra in a 17-year-old male with ApoE ɛ3/ɛ3. Two P-Tau positive neurites are seen (arrows). A pigmented neuron is shown (arrowhead). IHC AT8 counterstained with Hematoxylin, scale bar = 50 μm. E) Same teen as in D to show a substantia nigra neuron with P-Tau+nucleus (arrow). IHC AT8 counterstained with Hematoxylin, scale bar = 20 μm. F) III cranial nerve neurons showing one neuron with P-Tau+ nucleus (arrow) contrasting with one negative (17-year-old). IHC AT8 counterstained with Hematoxylin, scale bar = 10 μm. G) Frontal section IHC for P-Tau from an 18-year-old male with ApoE ɛ3/ɛ4 showing a small tangle (arrowhead) and a positive neurite elsewhere in the neuropil (arrow). IHC AT8 counterstained with Hematoxylin, scale bar = 10 μm. H) Frontal cortex in a 17-year-old male with ApoE ɛ3/ɛ3, double stained with P-Tau and Aβ. A diffuse amyloid plaque (red product, arrowhead) and numerous P-Tau neurites (brown product, arrows). IHC AT8 and Aβ (4G8) counterstained with Hematoxylin, scale bar = 20 μm. I) Frontal cortex in an 11-month-old baby stained for Aβ. A diffuse amyloid plaque is seen (arrowhead). IHC Aβ (4G8) counterstained with Hematoxylin, scale bar = 50 μm. J) Frontal cortex in a 14-year-old female stained for Aβ (4G8), shows increased amounts of amyloid inside neurons (red product, arrows). IHC Aβ (4G8) counterstained with Hematoxylin, scale bar = 50 μm. K) Temporal cortex in a 14-year-old female. Gray matter, stained for Aβ (4G8), shows a blood vessel with abundant amyloid in its wall (arrowhead) and the surrounding positive neuropil (long arrow). IHC Aβ (4G8) red product. Scale bar = 100 μm. L) Electron micrograph frontal section of a 3-year-old boy showing a blood vessel with abundant lipofuscin (Lf). Red blood cells (RBC) are seen in the lumen of the vessel. Scale bar = 500 nm. M) Electron micrograph of a blood vessel in a frontal section of a 3-year-old boy. The endothelial cell is marked EC and abundant lipofuscin is marked Lf. Red blood cells (RBC) in the vessel lumen show clusters of nanoparticles (arrows). A platelet is seen attached to the endothelium (arrowhead). Scale bar = 500 nm. N) Electron micrograph frontal section of a 3-year-old boy showing a cluster of lipofuscin granules (Lf) and nanoparticles (lower portion of the picture). Scale bar = 500 nm. O) Electron micrograph of a frontal section in a 3-year-old boy showing nanoparticles inside mitochondria (M, arrows), clustering in membranes (arrows) and larger nanoparticles elsewhere (arrowhead). Scale bar = 100 nm. P) Two mitochondria (M) in an unmyelinated axon showing abnormal cristae and nanoparticles along the cristae and in the matrix. Notice the accumulation of nanoparticles along the axonal membranes (arrows). Scale bar = 100 nm.
Examination of children and young adults’ brains show early extensive nuclear neuronal P-Tau (Fig. 1), strongly supporting a cerebral stress response previously documented in MMC youth by upregulation of clusters of IL1, NFκB, TNF, IFN, and TLRs and a 15-fold frontal downregulation of the prion-related protein, severely compromising oxidative stress protection [4, 7]. Sultan et al. [34] supported nuclear tau as a key player in early stress responses and stated, “pathological alterations of Tau, e.g., hyperphosphorylation, might impair its ability to shuttle between the cytoplasm and the nucleus and/or affect its affinity for DNA”. The extensive nuclear P-Tau seen in highly exposed MMC children would fail to efficiently protect DNA from oxidative stress damage and likely “contribute to functional failure of neurons” [34] early in life.
The speed of AD progression in MMC residents is given by the presence of one or two ApoE ɛ4 alleles, age, gender, and the lifelong cumulative concentration of fine particulate matter: CPM2.5, related mainly to residency location within the megacity [4]. One ApoE ɛ4 allele increased the odds 23.6 times versus ApoE ɛ3 having similar age and CPM2.5 exposures for developing an advanced stage of AD (neurofibrillary tangle (NFT) stage V) [4]. Age and CPM2.5 independently are also significant for modelling the odds of developing NFT V [4].
The MMC data should be relevant to highly exposed city dwellers, i.e., Santiago, Temuco, Paris, Grenoble, Lyon, Strasbourg, Toulouse, etc. Braak and co-workers have suggested “continual formation of abnormal tau takes place from the beginning until the end-phase of Alzheimer’s disease and is not known to be subject to remission” [35, 36]. Once P-Tau is present, it is likely not a reversible process, particularly in the setting of high pollution. Indeed, when we compared the Braak et al. [35] work with 2,332 autopsies ages 0–100 years (hospitalized German patients from University Hospitals), amyloid-β (Aβ) plaques began to appear between ages 30 and 40, at the time subjects exhibited pretangle stage 1a or 1b and our MMC cohort Fig. 1: subcortical pretangle stage b, and diffuse amyloid plaques are seen at age 11 months, and by the time children/teens reach pretangle stage 1a or 1b they also have Aβ phase 2 [4], it is clear we are witnessing an accelerated AD pathology progression in MMC residents. The intrauterine period and the first two decades of life are key for brain development and dynamic, heterochronus maturation of the cerebral cortex [37], and also for damage associated with environmental pollutant exposures. There is no doubt we have significant variations in AD progression associated with ApoE status, gender, metabolism, nutrition, genetics, occupational history, socioeconomic status, brain, neural, and cognitive reserves, etc. [13, 38].
WILL EVERYBODY RESIDING IN A POLLUTED CITY HAVE DEMENTIA? HOW DO WE CLASSIFY CHILDREN AND YOUNG ADULTS WITH AD MARKERS IN HIGHLY POLLUTED ENVIRONMENTS? IS THE AD NIA-AA 2018 FRAMEWORK APPLICABLE TO YOUNG URBANITES?
The answer to “Will everybody residing in a polluted city will have clinical dementia? is a clear and loud, no. In polluted cities with sustained exposures of PM2.5 above the World Health Organization (WHO) or US EPA standards (PM2.5 10 μg/m3 annual mean and 25 μg/m3 24-hour mean and 12 μg/m3 annual mean and 35 μg/m3 24-hour mean, respectively), subjects will die prematurely of heart attacks, strokes, lung cancer, chronic and acute respiratory diseases, suicide, etc., particularly in low- and middle-income countries, and the greatest number in the WHO South-East Asia and Western Pacific regions. In other words, they will not reach the age to show the full-blown AD clinical picture [39, 40].
The 2018 AD NIA-AA framework [39] establishes: “Although it is possible that β-amyloid plaques and neurofibrillary tau deposits are not causal in AD pathogenesis, it is
We fully agree with these statements, but how we apply them to highly exposed children and young adults? MMC children and young adults have the neuropathological hallmarks of AD and we can stage them [4]. When we studied accidental, homicide or suicide MMC deaths in ‘clinically healthy subjects’, we found cortical tau pre-tangles, NFT Stages I-II by the 2nd decade. Moreover, Aβ1 - 42 concentrations in normal cerebrospinal fluid (CSF) samples are significantly lower in MMC versus clean air controls (p = 0.005) and Non-P-Tau, an early sensitive biomarker of axonal damage, showed a strong increase with age significantly faster among MMC versus controls (p = 0.0055) [41–43]. The low CSF Aβ1 - 42 concentrations (with normal total tau and tau phosphorylated at threonine 181) in MMC 11.2±5.5year old are likely in keeping with predominantly Phase 2 amyloid seen in the first two decades and the choroid plexus epithelium changes related to AD [4, 44].
The question we need an answer for is: How do we apply the NIA-AA research framework to MMC children ages 9.85±2.2 years, with no risk factors for neurological or cognitive deficits who have structural brain changes (white matter hyperintensities) and significant cognitive deficits when compared to matched clean air controls [6, 45]. One decade plus later (age 21.60±5.8 years) young adults have brain atrophy (Drs. Jacqueline Hernández-Luna and Lilian Calderón-Garcidueñas, personal communication, October 30, 2018). If there is a ‘silent clinical stage’, the period is indeed short because by 3rd or 4th grade, children are aware of their memory deficits, parents are concerned their children’s academic performance is not optimal, and teachers’ complain of academic failures. In the first and second decades, we have also described olfactory, auditory, and brain magnetic resonance imaging (MRI)/magnetic resonance spectroscopy alterations in the absence of risk factors, other than their environmental exposures to air pollutants [6, 45–47].
ApoE is a factor that strongly modulates the group effects between the Wechsler Intelligence Scale for Children-Revised, left frontal and parietal white matter, and hippocampus metabolites in children [46]. Preteens have significant olfaction deficits, soap is the predominantly failed odor, and in ApoE ɛ4 carriers, strongly correlated with left hippocampus mI/Cr ratio [20]. Olfaction is compromised in teens and young adults: 62 MMC/25 controls age 21.2±2.7 years were studied with the University of Pennsylvania Smell Identification Test (UPSIT) and olfaction deficits were documented in 35.5% MMC and 12% of controls [20]. ApoE ɛ4 MMC carriers failed 2.4±0.54 items in the 10-item smell identification scale from the UPSIT related to AD, while ApoE ɛ2/ɛ3 and ɛ3/ɛ3 subjects failed 1.36±0.16 items, p = 0.01 [20]. Our odor identification deficits correlate with the findings of P-Tau and Lewy neurites identified in the OBs of MMC children, including toddlers [47]. By the second decade, 84% (48/57) of the bulbs exhibited P-Tau, 68% (39/57) Lewy neurites and vascular amyloid, and 36% (21/57) diffuse amyloid plaques. OB active endothelial phagocytosis of red blood cell fragments containing CFDNPs and the neurovascular unit (NVU) damage, were associated with myelinated and unmyelinated axonal OB damage [47]. OB P-Tau neurites were associated mostly with pretangle stages 1a and 1b in subjects ≤20 years of age, strongly suggesting olfactory deficits could potentially be an early guide of AD pretangle subcortical and cortical P-Tau.
Thus, if the “NIA-AA research framework defines AD biologically, by neuropathologic change or biomarkers, ... ” [39], we could apply the AT(N) biomarker profiles and categories (Table 2 in reference [39]) and take the autopsy data where 99.5% of the MMC 203 autopsy cases ages 1–40 were consistent with AD continuum, then we could add the CSF low Aβ1 - 42 [48], and the structural/volumetric MRI findings (56% of 10.73±2.7 years and 86% of 21.61±6.01 years cohorts) and 55% healthy young subjects with Montreal Cognitive Assessment in the mild cognitive impairment (MCI) and AD score ranges [49] and we have a population younger than 40 years of age with an evolving AD continuum. It is critical to emphasize than in young healthy cohorts ages 21.60±5.8 years, with 13.6±1.28 formal education years, there is a progressive involvement of Visuospatial, Executive, Language, and Memory domains, and the average age for dementia Montreal Cognitive Assessment scores is 22.38±7.7 years [49].
We agree that defining AD by biomarkers indicative of neuropathologic change independent from clinical symptoms represents a profound shift in thinking [39], but we need to go a bit further and explore the AD continuum in the first two decades of life, define the time frame for subjective reports of cognitive decline and the mild neurobehavioral changes coexisting with cognitive decline. This scenario obligates us to find an answer to the question: “What biomarker signature or profile(s) defines the AD continuum in the first two decades of life?” It is clear we need reliable, easy to apply, continuous cognitive instruments, and non-invasive, inexpensive biomarkers that will allow us to document longitudinal changes and much more importantly, the opportunity to intervene. Imaging measures, representing the magnitude of the neuropathologic load or damage accumulated over time [39], are noninvasive and deliver valuable information; the only problem is cost. And sadly, there is no support for early pediatric AD research.
WHY ARE CFDNPs IMPORTANT FOR ANYONE LIVING IN AN URBAN OR RURAL POLLUTED ENVIRONMENT? AND WHY CAN MAGNETIC NANOPARTICLES IN THE BRAIN POSE SPECIFIC RISKS?
Traffic is a major source of nanoparticles and atmospheric nanoparticles are ubiquitous, highly reactive, and mostly anthropogenic [50, 51]. The current definition of nanoparticles adopted by the European Union in 2011 states: “A natural, incidental or manufactured material containing particles, in an unbound state or as an aggregate or as an agglomerate and where, for 50% or more of the particles in the number size distribution, one or more external dimensions is in the size range 1 nm–100 nm” [52]. The nervous system impact of nanoparticles is extensive and includes a number of pathways and targets with many common denominators including oxidative stress [53–60]. CFDNPs are ubiquitous in urban environments [9] and their strong magnetic properties mean they can respond to external magnetic fields and consequently affect the biological outcome of magnetic nanoparticles adding to their neurotoxicity [61, 62]. Gilder et al. [63] explored in 7 human brains (average age 71.57±12.99 years) using two magnetic measurements: natural remanent magnetization and their magnetically saturated state for saturation isothermal remanent magnetization (SIRM); the measurements are essentially consistent with the presence of magnetite [9]. The SIRM values among the several samples obtained of each brain spanned roughly 3 orders of magnitude and yielded a systematic pattern of magnetization intensities [63]. SIRMs from the brainstem, cerebellum, and cortex were the highest, suggesting that concentrated magnetite is not a random process. Their finding of the highest values for the brainstem is very important for the finding of hyperphosphorylated tau precisely in the lower brainstem in Mexico City children [4]. Gilder et al. [63] argue the magnetic carriers could be biologically-engineered, we support external combustion-derived sources [4, 64].
Combustion nanoparticles abundant in MMC brains and present in the abnormal NVUs in the brain are easily transferred from red blood cells to endothelial cells to neural cells and cause extensive damage to mitochondria, endoplasmic reticulum (ER), mitochondria-ER contacts, axons, and dendrites [64]. Indeed, the one critical finding in the evolving AD hallmarks in children in MMC is the extensive and progressive damage to the NVU, the anatomical substrate of neurovascular interactions [65]. Iadecola states that failure of “the coupling between neural activity and blood flow”, will result in neurovascular dysfunction, failure in orchestrating neuronal-astrocytic signaling to local blood vessels and the release of mediators across the cerebrovascular network [65]. A permissive NVU will allow direct exposure of the brain to neurotoxins, pathogens, and harmful chemicals. NVU damage in air pollution-exposed children and young adults is at the core of AD pathology with all the resultant hypoxia and neurovascular impairments during disease progression [24].
The role of oxidative stress in the pathogenesis of AD has been at the center of AD research for several years [66, 67] and certainly reactive oxygen species generation, representing oxidative stress, is key in the mitochondrial effect of nanoparticles, overcoming the antioxidant ability of the cell and inducing mitochondrial dysfunction [67]. In the setting of nanoparticle exposures, there is a striking association with abnormal neuronal mitochondria and the issue of serious damage to an organelle needed for the high energy neuronal requirements [66, 64]. In Xie et al. [68], the size of the magnetic iron oxide (Fe3O4) nanoparticles was key in the type of damage inflicted in human hepatoma cells’ mitochondria: 9 nm magnetic iron oxide particles produced reactive oxygen species generation and the resultant mitochondrial dysfunction and necrosis, while 14 nm particles damaged the plasma membrane and gave rise to massive lactate dehydrogenase leakage. The authors also showed that agglomerated 9 nm nanoparticles resulted in severe oxidative stress and a combination of necrosis/apoptosis. The Xie et al. results are very important in pollution-exposed subjects: nanoparticles caused damage to lysosomal and mitochondrial structures at lower concentrations than concentrations producing disruption of plasma membranes and equally relevant to brain cells: at non-detectable cytotoxic concentrations, the silver nanoparticles persisted inside nucleoli and mitochondria, raising the issue of direct effect to these critical organelles with time [68]. We know mitochondrial damage is prominent in the early stages of AD [66, 67]. Defects on axonal transport of mitochondria interfering with their integrity and their transport could be explained by the presence of nanoparticles with high capacity for oxidative stress. Key to our work is the fact that mitochondria in neurons, glial, and endothelial cells from urban MMC residents are all affected and expectations in terms of mechanisms in mitochondrial dynamics and specific pathology could result in serious physiological consequences [69].
APOE ɛ4, CHILDREN’S BRAIN RESPONSES, RAPID PROGRESSION OF NEUROFIBRILLARY TANGLE STAGES, AND HIGHER SUICIDE RISK IN YOUTH
ApoE ɛ4 allele is the strongest known genetic risk factor for late and early onset AD, and it is strongly associated with oxidative stress, mitochondrial dysfunction, endoplasmic reticulum stress, immune dysfunction, dysregulated lipid and glucose metabolism, and impairment in white matter integrity, relevant to highly exposed air pollution pediatric populations [16, 70–77].
Mechanistically, ApoE ɛ4 influences Aβ clearance and aggregation, and it also impacts AD pathology in Aβ-independent ways [70–72]. ApoE ɛ4 genotype, age, and female sex are established risk factors for AD [73–77]. ApoE ɛ4 is a key modulator of children’s responses to cumulative air pollution exposures, leading to early detrimental effects on cognition [22, 23]. Dean et al. [78] published a brain MRI study of 162 healthy, 2- to 25-month-old infants and toddlers showing ApoE ɛ4 carriers had reduced white matter myelin water fraction and gray matter volume measurements compared with ApoE ɛ4 negative subjects. Altered white matter myelin water fraction measurements were observed in the precuneus, middle and posterior cingulate, lateral temporal, and medial occipitotemporal regions, which are targeted in AD [78]. Chang et al. [79] also demonstrated striking effects in ApoE ɛ4 young children. Those with ɛ2/ɛ4 genotype had the smallest hippocampi, ɛ4/ɛ4 genotype carriers had the lowest measurements of hippocampal fractional anisotropy and age-dependent thinning of the entorhinal cortex, while those with ɛ3/ɛ4 genotype had the largest cortical area measurements in the medial orbitofrontal region. Key to this review paper, the ApoE ɛ4 effects on brain structure were predictive of low scores on executive function and working memory tasks [79].
In Spaniard children [80], ApoE ɛ4 carriers also have significantly higher behavioral problem scores than noncarriers, and interestingly, adverse associations with PAHs and NO2 were stronger or limited to ApoE ɛ4 carriers for behavior problem scores, caudate volume, and inattentiveness trajectories. The finding is very relevant to MMC children because in large cities, the pollutant patterns change with the residency area and thus the behavioral problems are significantly distinct, i.e., North versus South MMC children [22, 23].
We have reported since 2008 [7] that ApoE ɛ4 carriers had greater hyperphosphorylated tau and diffuse Aβ plaques versus ɛ3 carriers (Q = 7.82, p = 0.005), significant neuroinflammation with the upregulation of IL1, NFκB, TNF, IFN, and TLR gene network clusters and the frontal down-regulation of the prion-related protein. Thus, our finding of ApoE ɛ4 carriers having 4.92 times higher suicide odds (p = 0.0006), and 23.6 times higher odds of NFT V (p < 0.0001) versus ApoE ɛ4 non-carriers having similar CPM2.5 exposure and age was not a surprise [4].
But why does suicide among urban ApoE ɛ4 subjects age 25.2±8.4 years matter? A number of factors account for depression and suicide across countries: low socioeconomic status, labor market conditions, persistently low occupational grade, mental disease, addictions, homelessness, traumatic disorders, social exclusion, loneliness, etc. [81–88]. In the US, suicide rates increased >30% in most states during 1999–2016, with nearly 45,000 suicides (15.6/100,000 population [age-adjusted]) among persons aged ≥10 years in 2016. Adults aged 45–64 had the largest absolute rate increase from 13.2 per 100,000 persons [1999] to 19.2 per 100,000 [2016] and 54% did not have any known mental health condition [81]. Thus, ApoE ɛ4 matters in terms of suicide prevention. We have known for some time that older adults with depression are at increased risk of AD [88], but the association between AD neuropathology and suicide in 25-year-old subjects was previously unknown [4]. Gallagher et al. [88] described older adults with depression and MCI had a high rate of progression to AD over a relatively short duration of follow-up, a very important observation because young adults in Mexico City are showing both depression (Lilian Calderón-Garcidueñas, personal communication, November 3, 2018) and MCI [49]. The issue here is to identify young adults with depression and/or MCI whom could be at risk of suicide because they are likely already in the AD continuum at advanced NFT stages (Stage V) [4] if they carry an ApoE ɛ4 allele. ApoE ɛ4 matters.
The clinical history of an urban depressed young patient ought to include the question of whether they exercise on regular basis and if exercise decreases their depressive symptoms, because in a study of Ku et al., 2017 [89], ApoE ɛ4 carriers had no significant difference in depressive symptoms between high active and low active groups. The beneficial effect of exercise on depressive symptoms was restricted to ApoE ɛ4 non-carriers. On the other hand, Krell-Roesch et al. [90], assessing light, moderate, and vigorous physical activities in mid- and late life, showed that light and vigorous physical activity in midlife were associated with lower risk of MCI, and ApoE ɛ4 carriers who did not exercise had a higher risk of incident MCI than non-carriers who reported physical activity. Interestingly, there is an association in older adults between cortical amyloid and loneliness in cognitively intact individuals [91]. Since loneliness and social exclusion [87] are suicide contributors, questions aimed at how the patient feels (UCLA Loneliness scale 92) could be very helpful.
WILL EVERY HIGHLY EXPOSED CHILD DEVELOP AD PATHOLOGICAL CHANGES?
The answer for pediatric Mexico City residents is yes: in 203 autopsies [4], we had 44 children born and raised in Mexico City, and each one of them showed the neuropathological hallmarks of AD. Could we document any cognitive, behavioral, and structural brain changes in these 44 children? We could not, but these people died suddenly, and their relatives stated they were clinically healthy. However, in the studies of clinically healthy Mexico City children, we know they show systemic inflammation, neuroinflammation, and CSF low concentrations of Aβ1 - 42 compared to clean air control children [6, 41–47]. We also know that children and young adults in the 1st–3rd decades of life are showing frontal white matter hyperintensities, metabolic brain changes [22, 46], and cerebral parietal and frontal atrophy. Moreover, in cognition studies using the Montreal Cognitive Assessment test in 517 healthy urbanites, 24.7% and 30.3% scored ≤24 and ≤22 respectively, in the range of MCI ≤24 and AD ≤22 [49]. We all agree children living in highly polluted cities face serious challenges that are multifaceted and dire: socioeconomic (poverty, high urban stress), environmental (severe air pollution, occupational exposures), nutrition and lifestyle-based (poor unbalanced diets, and lack of exercise), maternal-child health based (maternal obesity and diabetes), and social (low safety, poor schools, violence, addictions, adolescent pregnancies, and very low rate of criminal convictions) [93–97]. Over the last two decades, life expectancy in Mexico has stagnated due to the high rates of homicides among men, children, and adolescents [97, 98], type 2 diabetes mellitus and its complications, and childhood and adolescent obesity and metabolic syndrome [99]. Mexican children and young adolescents are affected by obesity, metabolic syndrome, and vitamin deficiencies. Children ages 10.6±2.7 years with exogenous obesity and body mass index numbers greater than 2.0 standard deviations from the mean, showed high prevalence rates (37.5% to 54.5%) of metabolic syndrome [99,100, 99,100]. Parents are afraid of taking their children outdoors for exercise, and there is a lack of exposure to UV light added to the nutrition deficiencies resulting in vitamin D deficiencies among 87% of clinically healthy children residing in Mexico City (serum 25-hydroxyvitamin D below 30 ng/mL) [21]. Chronic exposures to high environmental levels of PM2.5 are associated with basal hyperleptinemia, altered appetite-regulating peptides, and increases in ET-1 (contributing to brain hypoperfusion) in Mexico City children [21]. These findings likely account for the increased rates of hypertension and early development of atherosclerosis among Mexico City adolescents [101] and increased rates of liver injury manifested by elevated serum aminotransferase levels with metabolic syndrome and obesity in adults who grew up and remained living in Mexico City [102].
There are important research findings that physicians have to keep in mind: both systemic and neural inflammation propagate cascades of neurodegeneration by damaging the blood-cerebrospinal fluid barrier (BCSFB) and by driving pathogenic innate and adaptive immune responses in the brain [5, 103–107]. Chronic exposures to PM2.5, nanoparticles and toxic metabolites produce these effects because they readily cross blood-brain and blood-placental-fetal barriers. Kristiansson et al. hypothesized that the effects of poverty and associated air pollution-related stress on impaired cognitive skills are mediated by inflammatory cytokines [108].
SANTIAGO (CHILE) AND PARIS (FRANCE) ARE SELECTED AS EXAMPLES OF HIGH AIR POLLUTION ALONG WITH MEXICO CITY. WHAT SHOULD PEDIATRICIANS DO WITH THE CURRENT INFORMATION?
The metropolitan areas of Santiago and MMC extend on valleys surrounded by high altitude mountains, while the greater urban area of Paris is characterized by uniform and low topography. It is important to be aware of the similarities between Santiago and MMC. While Santiago is located between the mountain ranges of Los Andes, de la Costa, el Manzano, and Angostura with elevations between 3,000 and 6,000 meters above the sea level, MMC is surrounded by the mountain ranges of Monte Alto, Monte Bajo, Las Cruces, Pachuca, and the Sierra Nevada and Chichinautzin with heights in the same range than those for Santiago. The mountain ranges in both urban areas favor the accumulation of air pollutant emissions associated with natural ventilation restrictions. The winds on both regions are driven mainly by differences in thermal heating between the floor of the valleys and the mountains. Surface nocturnal thermal inversions are common, leading to the accumulation of night emissions. As these inversions may persist up to midmorning hours, the emissions that are generated in the morning rush hours add to the trapped nocturnal emissions leading to higher levels of primary air pollutants [109, 110]. Santiago has several regions where the PM2.5 concentrations are several times the recommended WHO standard, and this is critical information for their residents’ health, particularly their children. In turn, the Paris region features a flat topography and restricted ventilation of the local emissions [111].
The three discussed urban areas show significant differences such as their topography, altitude, and populations. Santiago extends over a surface of approximately 1,140 km2 at an altitude of 540 m above mean sea level (AMSL), MMC covers around 2,370 km2 at an altitude of 2,200 m AMSL, and Paris consists of a semicircular region of approximately 1,250 km2 at an average altitude of 35 m AMSL. While the population in Santiago is around 6.5 million inhabitants, the population in MMC exceeds 25 million people, while that for Paris is approximately 12 million people. In spite of the differences in population and extension of the urban areas, levels of PM2.5 are highest in Santiago than in MMC and Paris. Figure 2 shows a comparison between the PM2.5 24 h averages and the annual mean concentrations registered in representative monitoring sites of Santiago, MMC, and Paris during 2016. The PM2.5 short- (24-hour) and long-term (annual) air quality guidelines suggested by the WHO and the US EPA National Ambient Air Quality Standards are included in the figure for comparison.
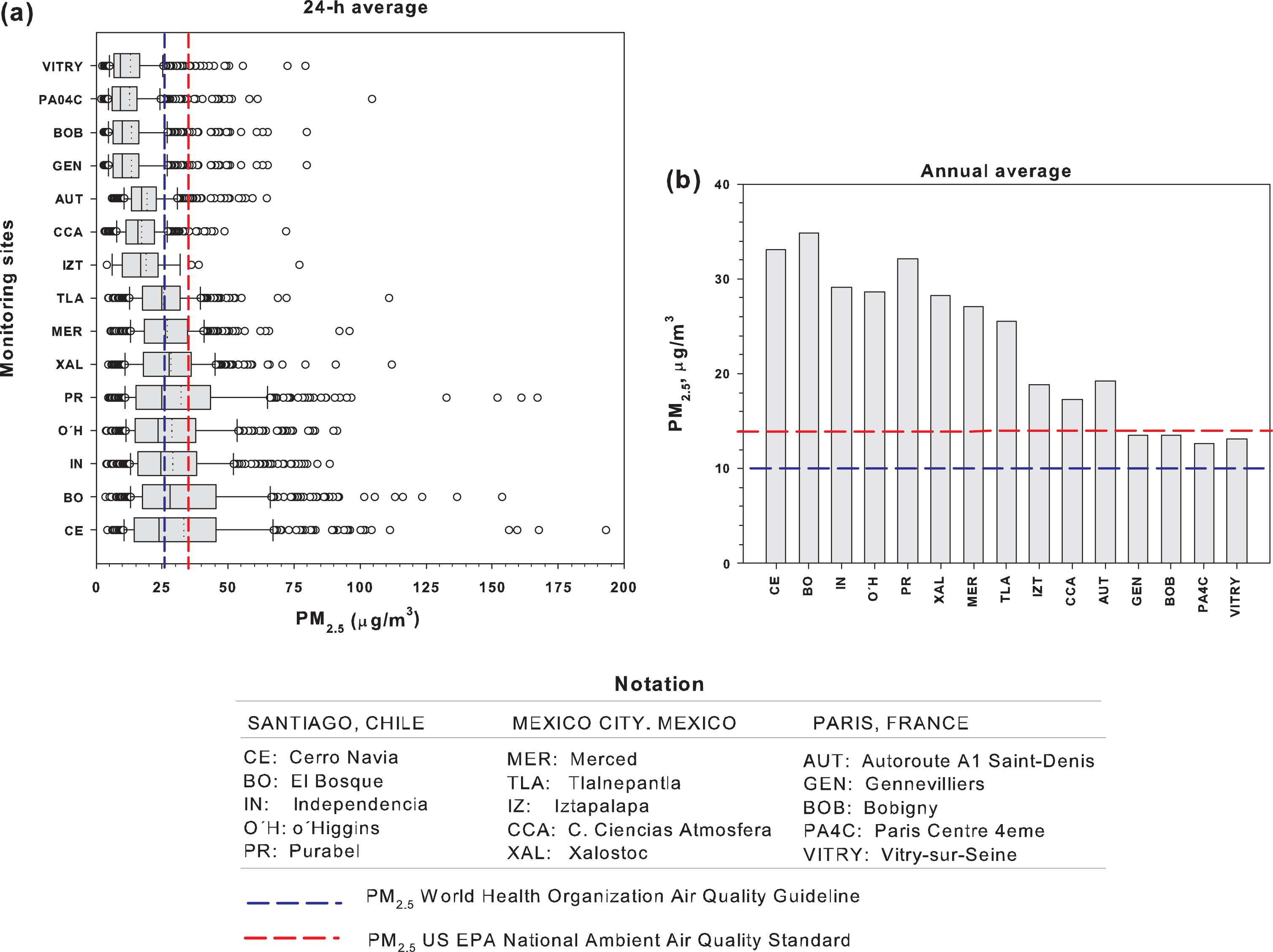
(a) Box and whisker plots of PM2.5 24-hour averages and (b) PM2.5 annual mean concentrations registered in representative air quality monitoring sites of the metropolitan areas of Santiago, Chile, and Paris during 2016. The respective 24-hour and annual mean PM2.5 WHO and US EPA guidelines and air quality standards are included for comparison. Data obtained from: the National Air Quality Information System (Chile) at https://sinca.mma.gob.cl/, the Ministry of Environment of Mexico City at http://www.aire.cdmx.gob.mx/default.php, and the Ministry of Environment to monitor the air quality in Paris and in the Ile de France region at https://www.airparif.asso.fr/en/telechargement/telechargement-station.
The box and whisker plot (Fig. 2) show that in general, higher 24-hour PM2.5 average levels are frequently observed in Santiago, while lower PM2.5 are consistently registered in Paris. The respective WHO guideline is exceeded in practically 50% of the days of the year in all sites of Santiago and three sites of MMC. This guideline is exceeded in < 10% of the days in most sites of Paris. Comparing the levels shown in Fig. 2a with the respective concentration value recommended by the US EPA to prevent effects to short-term exposures, the number of days that the stations exceeded this standard is partially reduced but the trend is kept. On the other hand, when the annual mean PM2.5 levels for the reference year 2016 are compared against the respective WHO guideline and the US EPA standard, all sites in Santiago, MMC, and Paris exceed the WHO standard. Annual PM2.5 means in MMC are strongly dependent on the activity of the urban sector. Three MMC sites located in high population density urban areas with different levels of both vehicular traffic and industrial activity (La Merced, Tlalnepantla, Xalostoc) show levels close to those in Santiago. The annual mean PM2.5 levels in CCA and IZ in MMC and AUT in Paris are representative of the influence of strong vehicular activity.
Using BC as a tracer for the local contribution to PM2.5 for each metropolitan area and assuming BC as a surrogate of elemental carbon, the relationships between BC and PM2.5 concentrations (expressed as μg/m3 BC/μg/m3 PM2.5) in the 3 cities are estimated as: ∼0.12, 0.09 to 0.14, and ∼0.1 for Santiago, MMC, and Paris, respectively [112–114]. BC is continuously used as an indicator in air quality management because it is emitted from the incomplete combustion of carbonaceous fuels from both anthropogenic and natural sources and is always emitted with other particle compounds and gases, such as organic carbon, nitrogen oxides, and sulfur dioxide (SO2) [112]. Carbonaceous particles such as BC have aerodynamic diameters of less than 2.5 microns and typically account for as high as 40% of the PM2.5 mass in urban atmospheres [113]. Taking the PM2.5 annual means from Figure 2b and the BC/PM2.5 ratios mentioned, it is estimated that the annual mean concentration exposure to BC in the study cities are in the ranges of: 2.8–4.2 μg/m3 for Santiago; 1.4–3.6 μg/m3 for MMC; and 1.2–1.9 μg/m3 for Paris.
Based on our Mexican experience, several things come to mind for every physician in an urban or a rural area with PM2.5 sources above WHO standards: Every physician needs to be knowledgeable about the concentrations of the six criteria air pollutants in their city: particulate matter, O3, SO2, NO2, Pb, and CO based on μm/m3 for PM2.5, ppm for O3, etc. If you have extra sources of PM2.5, for example, summer forest fires, the Notre Dame fire, etc., be aware of the changes and the emissions of pollutants out of the ordinary, such as lead in the case of Notre Dame. The clinical history should include specific sources of air pollution for their individual patients (i.e., smelters, cement factories, asphalt facilities, proximity to airports, highways and high transit avenues, etc.). The occupational history is a must, and a careful list of occupations along the patients’ life are needed and of course, the cities where the patient lived, including during the mother’s pregnancy. Also, you should be aware that in many cities, measurements of PM2.5 are only done once a week, and the owners of the pollutant sources are warned of the selected day and thus the cities keep their ‘low PM 2.5’ status (very common, unfortunately). Another common situation is that when PM2.5 levels go well above the standard, somehow all monitoring systems go down, so no recordings are available. Be familiar with the air pollution standards of the US EPA and the WHO applicable for US and European countries, respectively, and compare these standards with your specific country standards (i.e., Chile standards are significantly different) and keep in mind that the standards are meant as a guide and that thousands of people may have symptoms/biological detrimental effects at concentrations below the standards. The Chilean standard for PM2.5, for example,, is twice the WHO standard. Physicians should be knowledgeable about the risk factors for neuroinflammation and neurodegeneration within their own population. What percentage of your children are overweight and obese? Their prevalence of metabolic syndrome? The problem with consumption of sodas and what about diets? ApoE ɛ4 and the prevalence of any high-risk genes in your population? Quality of education in terms of increasing the cognitive reserve? Level of education? Level of exercise? Distribution of socioeconomic status? Multilingual populations? Poor minorities? Recent migrants? Physicians should be aware all biological barriers are potentially damaged by particulate matter. The choroid plexus is the source of CSF production and the BCSFB is critical to the transport of solute transporters, aquaporins, etc.; nanoparticles numbers present in CSF is significantly higher in Mexico City residents versus controls [64], suggesting that the normal function of the choroid plexus could be impaired by nanoparticles and thus, abnormal secretory function and a defective CSF production, along with accumulation of Aβ peptides at the BCSFB may contribute to complications as those seen in AD patients [115]. Penetration of the BCSFB by nanoparticles, bacteria, viruses, etc., could follow different strategies. Transcellular passage or opening of tight junctions, a paracellular mechanism is in place, along with hijacking phagocytic host cells via a ‘Trojan horse’ strategy [116]. Exercise is an important variable. Do children exercise outdoors? Take into account the effects of exercise outdoors versus the benefits, the risk of cardiovascular events in some children, and the fact that exercising outdoors with high concentrations of air pollutants will result in significant concentrations of particulate matter (fine and ultrafine) entering the child’s brain. The authorities ought to report the levels of air pollution in real time and with the real concentrations. Warning should be issued to the population at large and posted in the media, so school authorities and health providers could offer advice to high risk patients and emergency hospital areas are prepared for the increase number of cases arriving within 24-, 48-, and 72-hour gaps. Parents need to be educated about the air pollutants’ health impact, so they can protect their children. They should know about nutrition and cognitive stimulation, the use of smart phones and tablets, and the risk of deafness. Every physician around the world welcomed France’s Education Ministry smartphone ban, which took effect at the beginning of September 2018 and applied to students from first through ninth grades. A major factor impacting cognition is sleep, so we strongly support The American Academy of Pediatrics Statement of Endorsement supporting the American Academy of Sleep Medicine guidelines outlining recommended sleep duration for children from infants to teens [117].
The consensus group recommends the following sleep hours: Infants 4 months to 12 months should sleep 12 to 16 hours per 24 hours (including naps) on a regular basis to promote optimal health. Children 1 to 2 years of age should sleep 11 to 14 hours per 24 hours (including naps) on a regular basis to promote optimal health. Children 3 to 5 years of age should sleep 10 to 13 hours per 24 hours (including naps) on a regular basis to promote optimal health. Children 6 to 12 years of age should sleep 9 to 12 hours per 24 hours on a regular basis to promote optimal health. Teenagers 13 to 18 years of age should sleep 8 to 10 hours per 24 hours on a regular basis to promote optimal health.
CONCLUSIONS AND FUTURE PERSPECTIVES
There is no question that high exposures to ambient air pollutants drive inflammatory cascades that disrupt fetal and brain development and function. Epidemiological human data and experimental animal studies support our thesis that Mexico City young populations with chronic exposures above US EPA and WHO standard concentrations of PM2.5 and O3, together with other significant insulin-metabolic derangements including obesity, type 2 diabetes mellitus, and metabolic syndrome, female sex, and ApoE ɛ4 carrier status, are at high risk for developing neurodegeneration, particularly AD. Factors such as high exposures to endotoxins, metals, PAHs, and the massive presence of nanocluster aerosols in the range of 1.3–3.0 nm [118] and high-temperature, combustion and friction derived iron-rich magnetite [9] capable of entering the brain of young and old MC residents alike ought to be included in this unfortunate equation.
The issue of suicide risk is a serious one: ApoE ɛ4 carriers 25.2±8.4 years in Mexico City have the highest suicide risk, opening the possibility P-Tau and amyloid stages contributed to depression and suicide at this early age. Relevant to suicide risk is the Niculescu et al. work [119, 120] identifying ApoE ɛ4 and IL6 as the top overall biomarkers of interest, both very significant in MMC children. Clearly ApoE ɛ4 status is key as described by Hwang et al. [121]. In the research by Hwang and coworkers, depressed geriatric subjects who were also ApoE ɛ4 carriers showed significantly lower Mini-Mental State Examination scores and an increased suicide attempt history. The relationship between suicide and air pollution has been explored and there are strong correlations with a number of air pollutants and variables such as male sex, the elderly, those with lower education status, white-collar workers, etc. [121–129].
A key challenge for every physician in their own country is to define clinical, laboratory, imaging, and cognitive non-invasive markers for the initial stages of the disease knowing P-Tau is the prime actor (and accumulates even in amyloid negative healthy adults [130]), the disease starts in the brainstem, and ApoE ɛ4 carriers are at highest risk. We strongly recommend that early interventions are integrated in health and educational agendas along with identifying early gender-specific risk trajectories. We are certain air pollution [132] should be included as an early risk factor in the research priorities to reduce global burden of dementia and the concept of preclinical AD ought to be revised. The work of Hochella et al. [133] on nanomaterials, including combustion and friction derived and industrial in origin, emphasized their impact on human health and obligates health workers to keep track of new nanomaterial technologies and their potential detrimental health effects. Finally, there is a need for each country to define their own pediatric and young adult environmental, nutritional, metabolic, and genetic risk factor interactions to prevent a dreadful disease killing millions of people across the world.