
Abstract
A major impediment in early diagnosis of Alzheimer’s disease (AD) is the lack of robust non-invasive biomarkers of early brain dysfunction. Metropolitan Mexico City (MMC) children and young adults show hyperphosphorylated tau, amyloid-β, and α-synuclein within auditory and vestibular nuclei and marked dysmorphology in the ventral cochlear nucleus and superior olivary complex. Based on early involvement of auditory brainstem centers, we believe brainstem auditory evoked potentials can provide early AD biomarkers in MMC young residents. We measured brainstem auditory evoked potentials in MMC clinically healthy children (8.52±3.3 years) and adults (21.08±3.0 years, 42.48±8.5 years, and 71.2±6.4 years) compared to clean air controls (6.5±0.7 years) and used multivariate analysis adjusting for age, gender, and residency. MMC children had decreased latency to wave I, delays in waves III and V, and longer latencies for interwave intervals, consistent with delayed central conduction time of brainstem neural transmission. In sharp contrast, young adults have significantly shortened interwave intervals I–III and I–V. By the 5th decade, wave V and interval I–V were significantly shorter, while the elderly cohort had significant delay in mean latencies and interwave intervals. Compensatory plasticity, increased auditory gain, cochlear synaptopathy, neuroinflammation, and AD continuum likely play a role in the evolving distinct auditory pathology in megacity urbanites. Understanding auditory central and peripheral dysfunction in the AD continuum evolving and progressing in pediatric and young adult populations may shed light on the complex mechanisms of AD development and help identify strong noninvasive biomarkers. AD evolving from childhood in air pollution environments ought to be preventable.
Keywords
INTRODUCTION
In the scenario of severe exposures to air pollutants including combustion and friction-derived nanoparticles, starting in utero, Metropolitan Mexico City (MMC) residents are developing early Alzheimer’s disease (AD) brainstem pathology as infants, with the pathological changes relentlessly progressing in the first four decades of life [1–6].
Hyperphosphorylated tau is first detected in the lower medulla in subjects as young as 11 months, and cochlear and vestibular nuclei are involved by the second decade [1, 8]. MMC 6-year-olds already have significant central delays in brainstem auditory evoked potentials (BAEPs) [7], and the auditory pathology and evoked auditory potential abnormalities are not restricted to humans, but also significantly affect young ≤5-year-old dogs [8].
The literature provides clear evidence for both auditory pathology and dysfunction in central auditory processing in AD [9–21]. Testing of central auditory processing-enabling comprehension of auditory stimuli amidst a distracting background has established a strong correlation with cerebrospinal fluid (CSF) tau levels, entorhinal and hippocampal cortex volumes and cognition deficits [18]. The strong association between sensory and motor changes may precede the cognitive symptoms of AD by several years [17], and the AD definition as a biological construct [22] enables an accurate characterization and understanding of the sequence of events that lead to cognitive impairment and certainly obligates us to explore children and young adults, who have been highly exposed to air pollution, for auditory system dysfunction [1, 6].
Given the progression of the AD continuum [22] in the first four decades of life [1] and the central brainstem delay observed in 6-year-old MMC children [7, 23], our primary objective was to measure BAEPs in healthy elementary school children and study three cohorts of clinically healthy adults in the 3rd, 5th, and 8th decades of life who reside in highly polluted MMC. Accordingly, non-invasive physiological assessment of auditory brainstem pathways may serve as a screening tool of brainstem and supratentorial AD pathology (Fig. 1) [1, 13–17]. Recording of BAEPs is non-invasive and will guide clinicians regarding the risk for individuals. Understanding auditory dysfunction in early AD in pediatric and adult populations may shed light on the mechanisms of disease progression and characterization of temporally complex auditory dysfunction in a fatal heterogeneous disease [1, 23]. Air pollution is a risk factor for the development of AD and as the number of patients with this condition around the world continues to grow, it is imperative that research efforts focus on preventable, early causes and appropriate interventions.

The auditory pathway and brainstem auditory evoked potentials (BAEPs). A) Schematic of the human auditory pathway. Neurons in the spiral ganglion project to the cochlear nuclei (CN) via the auditory nerve (AN). The CN projects bilaterally to the superior olivary complex (SOC) and the contralateral inferior colliculus (IC). Projections from the CN, SOC, and nuclei of the lateral lemniscus project to the IC via the lateral lemniscus (LL). The BAEPs include five waves (I through V; shown schematically in B). Waves I and II correspond to activity in the AN, wave III corresponds to activity in the CN and SOC, wave IV corresponds to activity in the LL and wave V corresponds to activity in the IC. C-F) Representative BAEPs from Metropolitan Mexico City subjects: a 10-year-old female (C), a 20-year-old male (D), a 41-year-old male (E), and 64-year-old female (F).
MATERIALS AND METHODS
Study design
This prospective protocol was approved by the review boards and ethics committees at the Universidad del Valle de México and the University of Montana. Written consent was obtained from all subjects; parents signed a written consent for the children and all minors gave their assent for the study. The geographic areas selected for this study were MMC and Polotitlán, in the Mexico State. Mexico City residents are exposed year-round to complex mixtures of air pollutants [24–29] including fine particulate matter (PM2 ·5) and ozone (O3) concentrations above the United States National Air Ambient Quality Standards (NAAQS). Both the PM2 ·5 annual standard 12μg/m3 averaged in three years, and the 24-h 35μg/m3 standard have been historically exceeded across the metropolitan area for the last 20 years [27–29]. Polotitlán is located 114 km NW of MC at 2300 m above sea level, and its selection as a control city was based on four key factors: 1) concentrations for all major air pollutants below the current USEPA standards, 2) access to a healthy population of children and adults, and 3) altitude above sea level similar to that of Mexico City, and relative proximity to Mexico City to facilitate the follow-up of the control cohorts.
Study population
This work includes data from 123 clinically healthy children and adults, including 69 children (control, n = 17; mean age 6.53±.072 years and MMC, n = 52; mean age 8.52±3.3 years) and three adult cohorts: ages 18–30 years (n = 24; mean 21.09±3.03 years), 31–65 years (n = 23; mean 42.48±8.55 years), and older than 66 years (n = 7; mean 71.29±6.42 years) (Table 1). All participants, except two, were right handed. Clinical inclusion criteria were: negative smoking history and environmental tobacco exposure, lifelong residency in MMC or in the control city, residency within 5 miles of the city monitoring stations, full term birth, unremarkable clinical histories, including no hearing impairments, negative history of hospitalizations for respiratory illnesses, negative histories of repeated upper respiratory, ear infections, and iron deficient anemia, personal and familial histories of deafness, febrile episodes or vaccinations in the previous 3 months. Cohorts were matched by socioeconomic status and parent educational level (in the children’s cohorts). Participants were from middle class families living in single-family homes with no indoor pets, used natural gas for cooking, and kitchens were separated from the living and sleeping areas. Participants slept in bedrooms with no carpeting and had open windows for ventilation. MMC and control subjects underwent physical exams and a standard otoneurological examination and BAEPs.
Comparison of mean latencies and inter-peak differences of the replicable waveform components of the BAEPs in Metropolitan Mexico City and control group. The normal wave latencies in italics correspond to normal healthy female and male subjects from Patel et al. [31]
*p-values adjusted for age, gender and residency.
Audiometry examination
All participants had a complete otorhinolaryngological exam and audiometry was carried out using Interacoustics Diagnostic Audiometer AD229 with a peltor H7A headphone in a sound proofed room. The subject-controlled Highson-Westlake procedure was used in accordance with ISO 8253-1. A threshold was defined as 2/3 correct responses in a procedure with 5 dB increases and 10 dB decreases. Pure-tone air-conduction hearing thresholds were measured at 125, 250, 500, 700, 1000, 1500, 2000, 3000, 4000, 6000, and 8000 Hz. Subjects admitted into the study had normal results in the practiced studies.
Brainstem auditory evoked potentials
The BAEPs were recorded using an adapted version of the standard clicks procedure [30] with an Eosate Biomedica System. Disc electrodes were attached to the mastoid processes (M1 - 2, reference electrode), vertex (Cz active electrode), and midline forehead (Fpz, ground). Impedance was kept below 5 K Ohms. Stimulation was delivered by model ER-3A tube earphone. The contralateral ear was masked by white noise of 40 dB during stimulation. The auditory stimuli were 100μs monoaural unfiltered rarefaction clicks at a rate of 11.4/s. The clicks were delivered at intensities of 80 dB above hearing threshold, 5 blocks of 2000 repetitions were recorded for each ear. Responses were amplified and averaged 1,000 times for each ear, respectively (thus, 2,000 times in total). Bandpass filters were set at 300 Hz (Low) and 3000 Hz (High). Absolute latency for waves I, III, V and interpeak latency I–III, III–V, and I–V were recorded (Fig. 1). The latency obtained from left and right ears were evaluated separately and then averaged to represent each subject by one value.
Statistical analyses
We first calculate the sample means and sample standard deviations of I, III, and V waves, and their differences within each cohort of Control children, MMC children, MMC 18–30 years old, MMC 31–65 years old, and MMC 66 years and older. Then, we calculate the p-values, after adjusting age and gender, for testing the differences between Control children and various age groups of MMC. We accomplish this by fitting appropriate linear regression models including age, gender, and residency as explanatory variables. Table 1 shows the results of Control children versus MMC children and the MMC adult results in the models adjusting age, gender, and residency. We also included the normal latency values of waves I, III, and V according to the work of Patel et al. [31]. The analyses were performed using Microsoft Excel, SAS version 9.4 (SAS Institute, Cary, NC), and R statistical software version 3.4.1 (R-project.org) with 2-sided significance set at α= 0.05 type I error rate.
RESULTS
Air pollution levels
Mexico City residents in this study have been exposed to concentrations of ozone (O3), and particulate matter (PM) above the USEPA standard their entire life [25, 27–29]. Control subjects were residents in a clean air city (Polotitlán) with levels of all criteria pollutants below USEPA standards.
BAEPs
The absolute latencies of Waves I, III, and V (Fig. 1) and the mean interpeak differences for the children and adult groups are shown in Table 1 and in selected samples in Fig. 1. Inspection of data on Table 1 finds that the latencies of waves I, III, and V in the control children are within normal limits at 1.7, 3.7, and 5.5 ms. On the other hand, the MMC children have clinically significant increases in waves III and V that occur at 3.95 and 5.91 ms while wave I is shorter at 1.6 ms. MMC children had significantly longer latencies than control children for interwave intervals I–III and III–V, but a normal latency for interweaves I–V, thereby indicating delayed central conduction (Fig. 1C). Table 1 shows significant differences in MMC in the adult ages from age 18 to older subjects ages 71.2±6.4 years (Fig. 1D-F). Specifically, the shortening of wave I at age 8 persist in the 3rd and 4th decades but certainly becomes prolonged in the older individuals examined (p < 0.0001, Fig. 1F).
By the second decade, the interpeak I–III is shortened and significant. By the 5th decade, wave V is shorter and the interval I–V becomes significantly shorten compared to previous decades (Fig. 1E). By the 8th decade, waves I, III, and V mean absolute latencies are prolonged and interpeaks I–III and III–V are significantly different from previous decades. Wave I is definitely prolonged in the elderly population.
DISCUSSION
Clinically healthy children residing in MMC showed a strong delay in both mean absolute wave latencies III and V and interpeak latency intervals I–III and III–V indicative of a central conduction delay. The central delay is seen in waves III and V generated rostral to the auditory nerve and the prolongation of the I–III and III–V interpeak latency, with a normal I–V interval. Thus, these delays as described by Legatt [32] are likely associated with bilateral lesions in the auditory brainstem extending from the cochlear nuclei, superior olivary complex, nuclei of the lateral lemniscus, and central nucleus of the inferior colliculus [32]. These regions have been shown to have inflammatory perivascular cells and accumulation of both hyperphosphorylated tau and α-synuclein in autopsy materials from MMC children of the same age as these clinical cases (Fig. 2) [1, 5–7]. Strikingly, MMC children demonstrate shortening of the earliest BAEP component, wave I, related to activity in the auditory nerve [32]. We believe the decreased latency in wave I and the auditory nerve might result from a number of different changes. First, the increased gain in the spiral ganglion, auditory nerve, and cochlear nucleus as a consequence of excitatory/inhibitory imbalance. Second, the possibility of an abnormal pattern of input from the trigeminal system, posterior columns, and reticular formation should also be entertained (Fig. 2). Third, it is possible that the brainstem neurodegenerative changes that we have observed have a specific impact on smaller diameter axons in the auditory nerve resulting in faster conductions times from the spiral ganglion to the cochlear nucleus. Finally, our previous work has revealed dysmorphology and Aβ1-42 in the superior olivary complex (SOC). The SOC includes olivocochlear neurons that project to the cochlear nucleus and organ of Corti. These projections normally function to suppress and modulate activity in the auditory nerve. The loss of these neurons in the SOC might lead to faster conduction in the auditory nerve. Such a finding would be consistent with changes observed after stimulation of olivocochlear axons in the work of Guinan and Gifford [33].
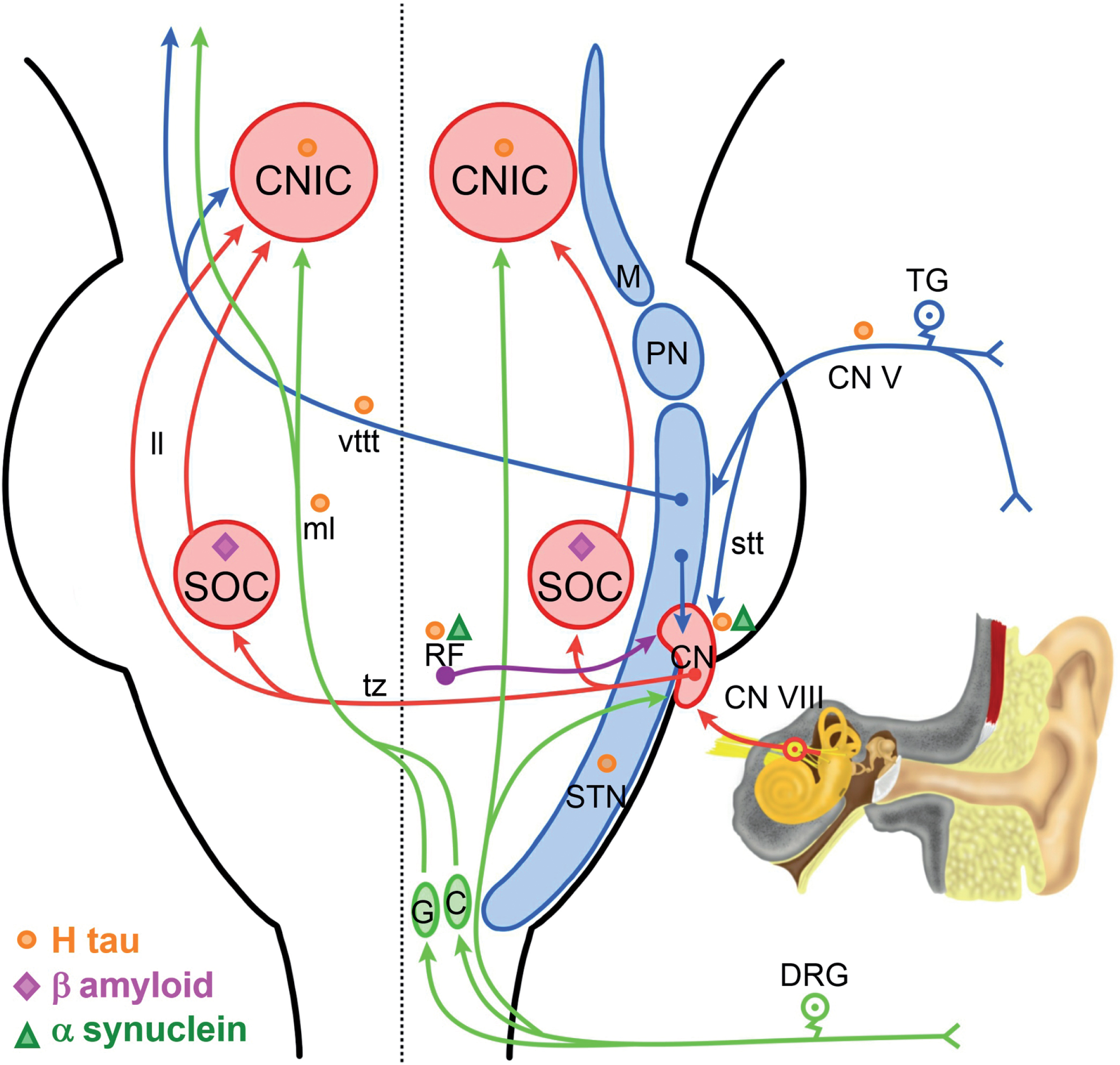
Shown on the right side is a schematic of auditory brainstem circuits (red arrows) and other circuits that we believe are involved in increased gain. Somatosensory inputs from the posterior column/medial lemniscus pathway are shown in green. Somatosensory input from the trigeminal system are shown in blue. Inputs from the reticular formation are shown in purple. There are direct and indirect projections from these somatosensory systems to the auditory system. Previous neuropathological studies have revealed degenerative changes in many of these structures [1, 7]. Sites of H tau are indicated by orange circles, sites with amyloid-β are indicated by magenta triangles, and sites of α-synuclein with green triangles. C, cuneate nucleus; CN V, trigeminal nerve; DRG, dorsal root ganglion; G, gracile nucleus; ll, lateral lemniscus; M, mesencephalic nucleus; ml, medial lemniscus; PN, principle nucleus; RF, reticular formation; STN, spinotrigeminal nucleus; stt, spinotrigeminal tract; TG, trigeminal ganglion; tz, trapezoid body; vttt, ventral trigeminalthalamic tract.
The remarkable findings in this work (Figs. 1 and 2) are progressive changes associated with shortening of absolute latencies and interpeak differences as subjects go into adulthood and involvement of wave I (cochlear hair cell involvement and spiral ganglion pathology) [9] and significant central and peripheral delays as people reach the 7th decade (Fig. 1F).
Given our studies in children and young adult [1, 7] where AD neuropathology progressively evolves from subcortical (brainstem) pretangle stages in babies, cortical tau pre-tangles, neurofibrillary tangles (NFT) Stages I-II by the 2nd decade, and NFT stages III–V in the 3rd and 4th decades, we proposed that the BAEPs progressively evolving changes are a consequence of early brainstem inflammation, the extensive medullary, pontine, and mesencephalic accumulation of hyperphosphorylated tau and α-synuclein involving the dorsal cochlear and vestibular nuclei, the inferior colliculus, the central nucleus of the inferior colliculus, the dysfunction of the olivocochlear bundle, and the late significant involvement of the cochlea and spiral ganglion (Fig. 2) [23]. The SOC is an early target nucleus in both MMC children and dogs [7, 8] showing statistically striking differences in cell body area, perimeter, major axis, and circularity versus clean air controls [7]. SOC immunolabeling for amyloid-β is present in highly exposed subjects (Fig. 2) [7].
Further supporting the connection between central auditory processing as described in AD [9–21], we have published the results of the Montreal Cognitive Assessment (MoCA) in 517 clinically healthy Mexican urbanites, ages 21.60±5.88 years, with 13.69±1.28 years of formal education [34]. The average MoCA scores are 23.92±2.82 and relevant to the BAEPs current findings: 24.7% and 30.3% of the clinically healthy cohort scored ≤24 at the level of mild cognitive impairment and ≤22 at the level of AD, respectively [34]. Strikingly, average age forAD MoCA scores across PM2.5 polluted cities in Mexico was 22.38±7.7 years [34].
The progressive changes observed in clinically healthy children and their transition to adulthood cohorts exhibit a temporal progression pattern likely the result of compensatory plasticity, a phenomenon certainly described in neurodegenerative diseases and also associated with acute cerebral events [35–43]. Neuronal plasticity understood as an ability of the brain to reorganize and rebuild resulting from either brain progressive pathology and/or changed environmental conditions takes place concomitantly with expression of particular genes at work, as well as the individual brain functional reserve [44–50].
Age is a major factor in the auditory deficits [51] as exemplified by the cochlear synaptopathy and the compensatory downstream plasticity in the form of altered gain. In our young cohorts, it is clear that the damage is initially central at the level of the SOC, followed by gradual compensatory changes including the altered gain phenomenon described by Parthasarathy et al. [51] ending in the fully irreversible central auditory delay and the involvement of the cochlea (Fig. 2). The concept of “modulation frequency selective increase in the representation of envelope cues at the level of the auditory midbrain and cortex” discussed by Parthasarathy et al. [51] is the expected trajectory of regional auditory degeneration in still young urbanites [7]. So, although age determines the ultimate irreversible changes, the process is a long one and goes hand in hand with the involvement of supratentorial regions as part of the neurodegenerative process in both AD and Parkinson’s disease [1, 52].
Indeed, the cascade of central gain changes and compensation leads to permanent changes late in the process, i.e., in our cohorts at the 8th decade (71.2±6.42 years). The issue is very critical because these progressive abnormalities could be considered potential electrophysiological biomarkers for an initial brainstem and cerebellar dysfunction, followed by supratentorial involvement [39]. In this regard, Sheppard et al. [39] puts forward a factor that ought to be taken in consideration in modern daily life: prolonged exposures to low-level sounds. The model of continuous exposure to low-level noise produces a cochlear-mediated central gain and the neural output of the cochlea is significantly reduced [39]. The cochlea could certainly respond in a polluted environment with an increase in the antioxidant defenses as described by Fetoni et al. [40] or components of air pollution could have detrimental effects on mitochondria and oxidative stress as described by Tan et al. [41].
The expectation along the compensatory plasticity is certainly transcriptional changes in receptors associated with normal auditory functions, including GABA, glutamatergic AMPA, and NMDA receptor distribution in the brainstem, inferior colliculus, and the auditory cortex [38, 54]. Caspary et al. [53] described changes in pre- and post-synaptic GABAergic and glycinergic inhibitory neurotransmission in relation to holeoastatic plasticity. In the work of Balaram et al. [38], the authors stated that in the auditory system, homeostatic mechanisms could support rebalanced excitation and inhibition in the inferior colliculus and auditory cortex after either developmental, trauma and we will add neurodegenerative changes. Central gains are variable depending on the neuronal type and this variability results in either suppression or excitability [55–57].
Tinnitus, defined as a phantom sound and present in 15% of the adult population, is a good example of inhibitory neurotransmitter dysfunction in response to noise-induced peripheral deafferentation [44]. Caspary and Llano [44] emphasized the role of the auditory thalamus-medial geniculate body, as an obligate auditory brain center in a unique position to gate the percept of sound as it projects to auditory cortex and to limbic structures. The issues discussed for tinnitus apply in the general sense to the maladaptive plasticity/Gain Control Theory of tinnitus pathology as discussed by Auerbach et al. [42] and Richardson et al. [46]. Essentially the theory states [42] that the reduced inhibition associated with increased spontaneous and abnormal neuronal activity, including bursting and increased synchrony takes place throughout the central auditory pathway, including the inferior colliculus, ventral cochlear nucleus, and dorsal cochlear nucleus, and critical to our findings in highly exposed air pollution individuals, the central gain levels are expected to be a dynamic process with changes in the types of cells involved over time and variations in the balance between excitatory and inhibitory projections (Fig. 2). Auerbach et al. [42] have three mechanisms potentially associated with gain: 1) a decrease in inhibitory synaptic responses, 2) an increase in excitatory synaptic responses, and 3) changes intrinsic to neuronal excitability. All of these mechanisms could be at work in the setting of a progressive neurodegenerative process. In addition, Noreña et al. [58] discussed the role of the trigeminal nerve and the trigeminal complex in the generation of gains triggered by acoustic shock. Strikingly, the involvement of the trigeminal nuclei (TGN) is early in MMC children and teens [1], and since the TGN innervates the cochlea and the vestibular labyrinth [59, 60] has projections to the central auditory system (Fig. 2) [43, 61] and significantly, TGN electrical stimulation impacts the firing activity of dorsal cochlear nucleus neurons [43], neurodegenerative changes in the TGN certainly will translate in excitatory impact on the dorsal cochlear nucleus and the stria vascularis of the cochlea (Fig. 2) [58].
Auditory-somatosensory interactions likely also play a role, including the dorsal root ganglia and the TGN and their projections to the trigeminal sensory complex [48]. The somatosensory information is fed into the auditory system via the cochlear nucleus [61]. Cochlear nucleus projection neurons target the cuneate and gracilis nuclei and aggregate in the ventral edge of the dorsal column nuclei [48]. It is very interesting to emphasize that both gracilis and cuneatus are involved in an inflammatory process in MMC children [7] (Fig. 3B, C) as evident by trafficking of cells immunoreactive for CD163, a marker of perivascular microglia—marking the transition from M1 to M2 phenotype in macrophages—likely activated by vascular compromise [62].
Since the limbic system is involved in processing aversive auditory stimuli [63], it is expected that the amygdala, involved in the AD process and related direct and indirect influences on brain reserve in regions that are most vulnerable to the evolving and progressive AD continuum, will also play a key role in the processing of auditory stimuli [47].
We argue that the evolving BAEPs changes across 8 decades of life in clinically healthy MMC residents are complex responses to continuous exposure to air pollution components (i.e., iron-rich, magnetic, nanoparticles associated to combustion and friction), neurodegenerative changes, including AD continuum [1–3, 32], compensatory plasticity, and environmental (including low and high-level noise exposures) damage to peripheral and central auditory centers ending in a maladaptive auditory responses [44, 64–68]. We illustrate the different mechanisms potentially at work in Fig. 2. Essentially, children exposed to the severe MMC air pollution versus controls manifest systemic inflammation, immunodysregulation, endothelial dysfunction [69, 70], neuroinflammation [5, 6], auditory brainstem dysmorphology [7, 8], and AD continuum [1]. Strikingly, MMC children exhibited significant decreases in the numbers of natural killer cells and increased numbers of mCD14+ monocytes and CD8+ cells along lower concentrations of interferon gamma and granulocyte-macrophage colony-stimulating factor and a state of endotoxin tolerance [69, 70]. In the first decade of life, upregulation of cyclooxygenase-2, interleukin-1β, and CD14 in olfactory bulb, frontal cortex, substantia nigra and vagus nerves are documented together with disruption of the blood-brain barrier, endothelial activation, oxidative stress, and inflammatory cell trafficking [5, 6]. CSF changes are also seen in the first decade [71–73] and reflect alterations in key neuroimmune mediators (i.e., macrophage inhibitory factor) and prion cellular protein with important antioxidant activity, as well strong markers of AD including low CSF concentrations of amyloid-β1-42 [72] and high concentrations of non-phosphorylated tau [73].
The widespread infra and supratentorial inflammation, along with the accumulation of hyperphosphorylated tau, amyloid-β, and α-synuclein in key auditory regions, i.e., cochlear and vestibular nuclei, the inferior colliculus, the central nucleus of the inferior colliculus, and involvement of the SOC and trigeminal nuclei likely play a role in the progressive BAEPs changes (Fig. 2) [7, 23]. It is noteworthy to comment that the interpeak I–V representing to the entire activity of the auditory fibers and reflecting activity including caudal to the inferior colliculus and the rostral portion of the lateral lemniscus [31] does not reach statistical significance in the older group of subjects explored (Table 1). As it is well known, the inferior colliculus is the common target of separate pathways that transmit different types of auditory information and it is likely that their tonotopic characteristics will be translated in different plasticity responses. Swords et al. [19] support the notion that both peripheral and central auditory dysfunction occur in the prodromal stages of AD, we fully agreed on the presence of peripheral and central deficits; however, we support the changes are progressive and at least in the context of air pollution and AD development, the earliest change is indeed central delays in childhood [7]. What Swords et al. [19] and Tuwaig et al. [18] are seeing in AD patients is a terminal stage of auditory dysfunction that took at least 6 decades to evolve.
Our findings are very important because the evolving BAEPs changes reflect cochlear and brainstem progressive changes that might serve as indicators of early neurodegenerative changes as seen in AD and Parkinson’s disease.
It is indeed striking that the late AD changes include dichotic listening impairment, enhanced right ear advantage, loss of top-down modulation of incoming auditory stimuli, and loss of sensory gating ability and although the current AD consensus favors the audiological deficits are not directly related to maladaptive plastic changes [19], our data suggests otherwise.
It will be of extreme importance to confirm our data in larger cohorts and integrate other tests to the study of young individuals exposed to severe air pollution [21, 23].
Pediatric ear, nose & throat specialists in MMC are recording tinnitus in 1 of 3 children older than 9 years attending the hospital for potential audiological deficits; the complaint is significantly higher in adult populations with 2 out of 3 complaining of tinnitus in relation to a routine otoneurological exam or because of audiological deficits.
Given the early development of AD pathology in MMC children, teens, and young adults [1], the otoneurologists are recommending both an audiologic and otoneurologic full clinical examination, including a complete medical and prenatal history, recording of the region within MMC the child has lived (high exposures to fine particulate matter and heavy metals and noise in NW MMC versus endotoxins and high ozone in SW MMC), history of exposure to tobacco and E-cigarettes, an ear examination with an otoscope, tonal audiometry, logoaudiometry, and timpanometry with Jerger scale in the hands of a Board Certified Otoneurologist and Audiologist with significant expertise. Speech tests and the dichotic digits test should be explored [21]. Additionally, BAEPs are recommended as a baseline study. The use of specific filters, i.e., condensation and rarefaction to observe waves I, II and III, and III, IV, and V, respectively, are highly recommended. Evaluation of sensorineural and conduction hearing loss are a must.
There is no question of the heterogeneity and temporal progression of AD and its impact on the auditory system. There is also a need to consider auditory gain in a broader sense as William Sedley has said [68]. The need to diagnose early brainstem dysfunction by non-invasive procedures is critical in a population that essentially has no complaints related to audition and when a major difficulty in the early diagnosis of AD pathology has been the lack of robust non-invasive early biomarkers.
The complexity of AD is accompanied by the clinical task of diagnosing young individuals at early stages and by the fact that in late stages there are no treatments. We have major challenges ahead. Indeed, the evolving auditory changes in MMC subjects can be explored easily in a non-invasive manner and these changes likely go hand in hand with early cognitive impairment.
There is converging evidence in highly exposed air pollution young subjects that the brainstem and the olfactory areas are initial targets, followed in less than two decades of supratentorial involvement with significant cognitive deficits [34]. The opportunity to explore the auditory system is unique at early stages and we should advantage of the system plasticity to find out neuroprotective mechanisms for the millions of people exposed to air pollution across the globe. AD evolving from childhood in particulate matter (including PM2.5 and magnetic combustion and friction-derived nanoparticles) exposed subjects is preventable.