
Abstract
Exaggerated Ca2+ signaling might be one of primary causes of neural dysfunction in Alzheimer’s disease (AD). And the intracellular Ca2+ overload has been closely associated with amyloid-β (Aβ)-induced endoplasmic reticulum (ER) stress and memory impairments in AD. Here we showed for the first time the neuroprotective effects of Xestospongin C (XeC), a reversible IP3 receptor antagonist, on the cognitive behaviors and pathology of APP/PS1 AD mice. Male APP/PS1-AD mice (n = 20) were injected intracerebroventricularly with XeC (3μmol) via Alzet osmotic pumps for four weeks, followed by cognition tests, Aβ plaque examination, and ER stress-related protein measurement. The results showed that XeC pretreatment significantly improved the cognitive behavior of APP/PS1-AD mice, raising the spontaneous alteration accuracy in Y maze, decreasing the escape latency and increasing the target quadrant swimming time in Morris water maze; XeC pretreatment also reduced the number of Aβ plaques and the overexpression of ER stress proteins 78 kDa glucose-regulated protein (GRP-78), caspase-12, and CAAT/enhancer-binding protein (C/EBP) homologous protein (CHOP) in the hippocampus of APP/PS1 mice. In addition, in vitro experiments showed that XeC effectively ameliorated Aβ1 - 42-induced early neuronal apoptosis and intracellular Ca2+ overload in the primary hippocampal neurons. Taken together, IP3R-mediated Ca2+ disorder plays a key role in the cognitive deficits and pathological damages in AD mice. By targeting the IP3 R, XeC might be considered as a novel therapeutic strategy in AD.
Keywords
INTRODUCTION
Alzheimer’s disease (AD), an irreversible neurodegenerative disease, mainly occurs in the elderly and imposes a heavy burden to the AD families and society. The typical pathological features include senile plaques composed of amyloid-β (Aβ), neurofibrillary tangles, and neuronal death. Although a mass of therapeutic strategies based on eliminating Aβ have been proposed, nearly no chemical directly against Aβ can prevent or reverse the progress of AD currently [1]. Since the calcium hypothesis of AD was proposed [2], a lot of experiments indicated that abnormal Ca2+ signaling is a robust and proximal factor associated with the pathological features of AD such as Aβ aggregation and tau hyperphosphorylation in cultured cells and AD brain [3–6]. Intracellular Ca2+ overload was derived from exaggerated Ca2+ entry and intracellular Ca2+ release. The transmembrane Ca2+ entry is mediated by plasma membrane-embedded channels including voltage-gated Ca2+ channel (VGCC), non-specific ionotropic glutamate receptor (iGluR), and store-operated calcium entry (SOCE), while the intracellular Ca2+ release pathway is composed mainly of inositol trisphosphate receptors (IP3Rs) and ryanodine receptors (RyRs) on the membrane of endoplasmic reticulum (ER) [7].
ER, as an important organelle, plays key roles in protein synthesis and fold, Ca2+ signaling, and cellular stress. The disorder of ER function has been found in presenilin (PS)-associated familial Alzheimer’s disease (FAD) [8], and even the ER-regulated Ca2+ signaling has been viewed as an effective way to cure AD [4, 9]. For example, exaggerated Ca2+ signaling through IP3 R were found in PS mutant FAD cells [10]; IP3 R antagonists 2-APB [10] and heparin [11] protected cells from Aβ-induced cytotoxicity; Activation of RyR promoted the Aβ deposition in HEK293 cells and postmortem AD patients’ brains [12], while inhibition of RyR with dantrolene decreased Aβ formation and cognitive deficits in Tg2576 mice [13] and 3×Tg-AD mice [14]. Further studies show that IP3Rs makes larger contribution than RyRs in the Ca2+ signaling disorder of AD mice. For instance, Lopez et al. reported that elevated intracellular Ca2+ concentration in neurons from 3xTg-AD mouse could be reduced by inhibiting IP3Rs but not by blocking RyRs, suggesting RyR channels do not contribute to the elevation of intracellular Ca2+ concentration [15]. Shilling et al. reported that genetic reduction of the IP3R1 by 50% not only normalized exaggerated Ca2+ signaling in hippocampal neurons but also restored normal RyR and rescued memory in 3xTg mice [16].
Xestospongin C (XeC), a compound isolated from the Xestospongia species, is a selective, specific, and membrane-permeable inhibitor of IP3 R [17]. It is reported that XeC suppressed Aβ-induced calcium overload in neurons and reduced subsequent cell death [18]. In addition, XeC also reversed neuropathy caused by hyperanesthesia [19, 20], improved the working memory in rats [21], affected the antidepressant efficacy of a sigma1 receptor agonist [22], and was even used to treat HIV as a part of drug combination [23]. However, it is still an open question whether XeC could alleviate the cognitive and pathological impairments in AD mice. In the present study, we investigated for the first time the in vivo neuroprotective effects of XeC by examining multiple cognitive behaviors, Aβ accumulation, and ER stress in the hippocampus of APP/PS1 mice. The effects of XeC on Aβ1 - 42-induced neuronal damage and intracellular Ca2+ disorder were also investigated in primary cultured hippocampal neurons.
MATERIALS AND METHODS
Animals and grouping
Male APPswe/PS1dE9 (APP/PS1) transgenic mice (n = 40) and wild type (WT) control mice (n = 40) were ordered from Institute of Laboratory Animal Sciences (Beijing, China). As a classical AD animal model, the APP/PS1 mice well demonstrate the phenotypes of AD, showing Aβ plaque deposition by 4–6 month of age [24, 25] and cognitive deficits by 8–12 month of age [26, 27]. All animals were fed in a temperature-controlled room (20–25°C) with sufficient food and water supply. At 7 months old, all mice were divided into four groups (WT+Phosphate buffer solution (PBS), WT+XeC, APP/PS1 + PBS, and APP/PS1 + XeC) and were continuously injected with 3μmol XeC or PBS (with the same amount of dimethylsulfoxide (DMSO) for dissolving XeC) by using Alzet osmotic pumps at a constant rate 0.15μl/h for 4 weeks [28]. All animal experiments were carried out in accordance with the guidelines of Ethical Committee of Shanxi Medical University in Taiyuan, China.
Drugs and reagents
XeC was purchased from Abcam (UK), dissolved in Dimethy1 sulfoxide (10 mM) and stored in –20°C, then diluted in PBS before experiments. 10μM XeC was used in cell culture experiments and 3μM XeC (200μl) was applied by an injection pump in APP/PS1 mice. Aβ1 - 42 was purchased from China Peptides (Shanghai, China). Preparation of Aβ oligomers was as described [29]. Briefly, 1 mg Aβ powder was dissolved in 1 ml hexafluoroisopropanol (HFIP), then packed into 10 small tubes (100μl/tube), flushed with nitrogen, and stored at –80°C. Before experiments, 6μl DMSO and 34μl PBS were added to each tube and aged at 37°C for 36 h.
Surgical placement of injection pumps
As previously described [30], mice were anesthetized with 5% chloral hydrate (0.007 ml/g), and a 42-day Alzet osmotic pump (model 2006) loaded with XeC or PBS was implanted in a subcutaneous pocket on the back of the mice. A catheter connected to the pump was inserted into the lateral ventricle at the following coordinates based on bregma: 0.6 mm posterior, 1.1 mm lateral, and 2 mm depth. The Alzet pumps deliver drugs or PBS at a rate of 0.15μl/h and a total 200μl over 4 weeks. This chronic injection by osmotic pump avoids the fluctuation of drug concentration and decreased the damage induced by repeated intracerebral injection. The drug injection was kept during behavioral tests.
Open field test (OFT)
Mice were first subjected to the OFT to examine the locomotor activity and anxiety-like behavior [31, 32]. The apparatus of OFT was a box of 40 cm×40 cm×40 cm, and the central square of 20 cm×20 cm was defined as central area. After 30 min adaptation in the testing room, mice were placed at the center of the apparatus and allowed them to explore freely for 5 min. The total distance and the percentage of dwelling time in the central area were recorded by Smart 3.0 software system. After each trail, the apparatus was cleaned with 75% ethanol.
Y-maze test
To evaluate the working memory ability of mice, a spontaneous alternation test in Y-maze was performed [33, 34]. Y-maze consists of three arms (30 cm long, 7 cm high, 15 cm wide), symmetrically disposed at 120° from each other. Each mouse was placed individually in the triangular central region of the maze and allowed to move freely for 8 min. The numbers of total arm entry and correct alternation of mice in the Y-maze were recorded with a camera and Smart 3.0 software system. A correct alternation was defined as the entries different from the former two entries. The percentage of spontaneous alternation was calculated as (number of correct alternations/ (total number of arm entries-2))×100%.
Morris Water maze (MWM) test
MWM was used to test long term spatial learning and memory of mice. The maze was a circular swimming pool (diameter 120 cm; high 50 cm) filled with tap water (22±2°C) and the surrounding was decorated with eye-catching visual cues [35]. The MWM was divided into four quadrants and a 14 cm (diameter) platform was placed 1 cm below the water surface in the center of one quadrant. The quadrant containing the hidden platform refers to the target zone. MWM test lasted for 6 days. In the first 5 days of place navigation tests, each mouse was subjected to 4 trials per day in the maze containing underwater platform as acquisition trials. The time for mice to reach the platform refers to escape latency. On the 6th day, probe test was performed without platform, two trials for each mouse (60 s/trial). The number of platform crossings and the swimming time percentage in the target quadrant was calculated. To avoid the influence of visual and motor ability, a visible platform test was performed, in which a platform was elevated 1 cm above the water surface and the mice were put into other random quadrants two times. Ethovision 3.0 software (Noldus Information Technology, Wageningen, Netherlands) was used for recording swimming traces and acquiring data above.
Immunohistochemistry
After behavioral tests, mice were sacrificed and perfused intracardially with 25 ml of saline. The brains of mice were removed and fixed with paraformaldehyde for 24 h before dehydrating in 30% sucrose. Thereafter, brains were sectioned at a thickness of 20μm with freezing microtome (LEICA, Germany). The sections were transferred into PBS and blocked with normal goat serum (Solarbio, Beijing, China) for 30 min, followed by primary antibody (6E10, 1:1000, Biolegend, USA) incubation at 4°C overnight, second antibody for 4 h, and DAB detection for 15–30 min. All slices were processed in parallel. The Aβ burden was calculated as the ratio of immunopositive region area to the entire hippocampus area.
Western blot
Bilateral hippocampi of mice were removed and homogenized by ultrasonic grinder. After centrifugation and protein concentration measurement, protein samples were separated by 10% sodium dodecylsulphate polyacrylamide gel electrophoresis (SDS-PAGE) and transferred to polyvinylidene fluoride film (PVDF) membrane (Millipore, Temrculla, CA). The membranes were incubated with 3% bovine serum albumin (BSA) at 4°C for 2 h. After that, they were incubated with primary antibodies anti-GRP78 (BiP) (1:2000, ab21685, Abcam), anti-caspase-12 (1:2000, ab62484, Abcam), anti-CHOP (1:500, D46F1, CST), and anti-β-actin (1:4000, Chinese fir, Beijing, China) overnight at 4°C, followed by secondary antibody (1:5000, anti-rabbit IgG) for 2 h at room temperature. Finally, the immunocomplex was visualized by the enhanced chemiluminescence and detected by Azure Biosystems. The density of all target bands was analyzed by using Alpha View software.
Primary cell culture and apoptosis assay
Primary hippocampal neurons were cultured from 1-day SD rats as described previously [36]. Briefly, pups were sterilized in 75% ethanol for several seconds, and their brains were quickly removed and placed into 4°C dissection solution. Bilateral dorsal hippocampi were dissected and cut into 1 mm3 pieces, then dissociated with 0.125% trypsin and DNase I (Sigma) into single-cell suspensions. Neurons were seeded onto poly-D-lysine coated culture plates and grow in Dulbecco’s modified eagle medium (Gibco), fetal bovine serum, and horse serum (Gibco). After 16 h, the culture medium was changed as Neurobasal medium (Gibco) supplemented with B-27 serum-free supplement (Gibco), GlutaMAX, and penicillin–streptomycin (Gibco) at 37°C with 5% CO2. Half of the medium was replaced every third day. Experiments were performed on 10-day-old cultures. Apoptosis assay was performed using flow cytometry (BD Biosciences) with an Annexin V-FITC/PI detection kit (Dojiton, AD10-10, Japan) according to manufactory’s direction. Cells were pre-incubated with 20μM Aβ1 - 42 for 24 h. 10μM XeC was applied 1 h before Aβ.
Intracellular Ca2+ imaging
To observe the changes in intracellular Ca2+ level, Fluo4-NW Calcium Assay kit (Molecular Probes, Invitrogen) was used. Pre-washed primary neurons on 35 mm well were loaded with 1 ml of the dye loading solution containing Fluo4-NW and probenecid, according to the manufacturer’s instructions. After incubation for 30 min at 37°C and 15 min at room temperature in the dark, the Ca2+ imaging of neurons was taken at a rate of 1 picture per 3 s on an Olympus IX70 microscope with a CCD camera controlled by MetaFluor software (Universal Imaging). The changes of Ca2+ fluorescence was expressed as the ratio of current fluorescence density relative to resting fluorescence (ΔF/F0). Cells were pre-incubated with XeC (10μM) for 1 h and then of Aβ1 - 42 (20μM) was acutely applicated for 100 s.
Statistical analysis
Statistical analysis was performed using SPSS 19.0 and SigmaPlot 12.3. The data from MWM acquisition trails was analyzed by using a repeated-measure analysis of variance (ANOVA), and the others were analyzed using two-way ANOVA followed by Tukey’s Post Hoc Test. All graphical data were presented as mean±SEM, significance p < 0.05.
RESULTS
XeC ameliorated disinhibition-like behavior of APP/PS1 mice
Figure 1A showed representative running traces of mice in the open field. As indicated in the histograms (Fig. 1B), the total running distance of mice did not show significant difference among the four groups (p > 0.05), suggesting that genotype and XeC treatment basically did not affect the locomotive activity of mice. Since previous studies have shown that bilateral hippocampal CA1 region injection of Aβ induced a significant behavioral disinhibition [37], we further compared the percentage of time spent in the central area of the open field. Two-way ANOVA showed that APP/PS1 gene mutation and XeC treatment had a significant main effect on the time in central area (APP/PS1: F (1,30) = 42.177, p < 0.001; XeC: F (1,30) = 11.649, p = 0.002) and a significant interaction (F (1,30) = 13.477, p = 0.001). Tukey’s post hoc test showed that the running time of mice in the central area had a significant increase in APP/PS1 + PBS group (10.95±0.97%) compared with WT+PBS group (4.58±0.49%, p < 0.01), whereas this increase was significantly reduced in the APP/PS1 + XeC group (6.5±0.54%) (p < 0.05) (Fig. 1C). These results indicate that 8-month-old APP/PS1 mice displayed disinhibitory behavior in the open field, while XeC could ameliorate the behavioral disinhibition in APP/PS1 mice.

XeC ameliorated behavioral disinhibition and working memory deficit in APP/PS1 mice. A) Representative running traces of mice in open field test (OFT). The green and red circles represent initial position and stop position respectively, and the green pane is the defined central area. B,C) Histograms showing the total running distance of mice within 5 min (B) and the percentage of running time in the central area (C) in the OFT. D,E) Histograms showing the total arm entries of mice (D) and the percentage of correct spontaneous alternation (E) in Y maze test. *p < 0.05, **p < 0.01, ***p < 0.001. All values were shown as mean±SEM (n = 10).
XeC improved short term working memory of APP/PS1 mice
We next analyzed short term working memory of APP/PS1 mice using Y-maze. As shown in Fig. 1D and E, there was no significant difference in the number of total arm entries among groups (Fig. 1D) (p > 0.05), but the spontaneous alternation behavior displayed a significant difference (Fig. 1E). Two-way ANOVA showed that the genotype and treatment had significant main effects (genotype: F (1,36) = 14.682, p < 0.001; treatment: F (1,36) = 14.979, p < 0.001) and an interaction effect (F (1,36) = 12.215, p = 0.001). Tukey’s post hoc test showed that the correct alternation percentage had a significant decrease, from 67.41±1.85% in WT+PBS group to 52.4±2.27% in APP/PS1 + PBS group (p < 0.001); while XeC treatment restored it to 67.49±1.77% (p < 0.001), nearly equal to the level of normal control mice. This result reveals that XeC could effectively improve the working memory of APP/PS1 mice in Y maze.
XeC ameliorated the long term spatial memory deficits of APP/PS1 mice
MWM was used to test the spatial learning and memory ability of APP/PS1 mice. In the first five days of hidden platform tests (Fig. 2A), repeated measures ANOVA showed that the average escape latency of mice to find the hidden platform gradually decreased, with a significant main effect (F (4,128) = 105.64, p < 0.001). Meanwhile, genotype and treatment had significant interact effect with days on the escape latency (day×genotype: F (4,128) = 6.713, p < 0.001; day×treatment: F (4,128) = 2.75, p < 0.05). Tukey’s post hoc test showed that from the second training day, the average escape latency in APP/PS1+ PBS mice (days 2–5:56.56±1.6, 55.82±2.31, 47.22±2.34, 45.51±2.5) was significantly longer than that in WT+PBS mice (days 2:46.56±1.88, p < 0.01; day3–5:36.11±3.83, 29.6±1.8, 25.66±2.52; p < 0.001 for each). However, treatment with XeC (APP/PS1 + XeC) significantly decreased the escape latencies (day 3:45.53±1.07, p < 0.01 day 4:34.55±1.28, p < 0.001; day 5:31.42±1.12, p < 0.001), indicating that XeC ameliorated the deficits of spatial learning ability in APP/PS1 mice.

XeC improved spatial learning and memory of APP/PS1 mice in MWM. A) A tendency chart showing the changes of average escape latency of mice in the hidden platform test. **p < 0.01, ***p < 0.001 versus C57 + PBS; # #p < 0.01, # # #p < 0.001 versus APP/PS1 + PBS. B) Representative swimming traces on the fourth training day. C,D) Histograms showing the number of platform crossing (C) and percentage of swimming time in the target quadrant (D) in the probe trail. ***p < 0.001. E) Representative swimming traces of mice in the probe trail. F,G) Histograms showing the average escape latency in visible platform test (F) and the average swimming speed of mice (G). All values were shown as mean±SEM (n = 8–9).
On the last training day, a probe test without platform was performed to examine the spatial memory of mice. Two-way ANOVA showed that genotype and treatment had main effects and an interaction on the crossing times (genotype: F (1,32) = 19.042, p < 0.001; treatment: F (1,32) = 108.282, p < 0.001; genotype×treatment: F (1,32) = 22.085, p < 0.001) and the swimming times (genotype: F (1,32) = 5.611, p < 0.05; treatment: F (1,32) = 8.478, p = 0.006; genotype×treatment: F (1,32) = 24.779, p < 0.001). Tukey’s post hoc test showed that the mice in APP/PS1 + PBS group had fewer times of crossing platform (2.11±0.16, p < 0.001) and percentage of swimming time in target quadrant (21.2±1.64%, p < 0.001) than in WT+PBS group (4.61±0.15 and 34.68±1.36%). After treatment with XeC (APP/PS1 + XeC), both the times of crossing platform (3.61±0.17) and swimming time percentage (33.75±2.46%) had significant recovery. These data indicate that XeC improved the long term reference memory of APP/PS1 mice. Figure 2F and G showed average escape latency and swimming speed of mice in visible platform test. No difference was found between the four groups (p > 0.05), suggesting that the differences appeared in the acquisition phase and probe trail did not result from the change in the motor ability and visual acuity of mice.
XeC treatment reduced Aβ plaques in the hippocampus of APP/PS1 mice
We further examined the effects of XeC treatment on the brain pathology by detecting Aβ plaques in the hippocampus of mice. As shown in Fig. 3, there were few Aβ immunopositive plaques in the hippocampal slices in WT+PBS (0.08±0.03%) and WT+XeC (0.06±0.01%) groups. As expected, larger number of Aβ plaques were observed in the hippocampal slices of APP/PS1 + PBS mice (3.37±0.72%, p < 0.001 versus WT+PBS). Importantly, the area of Aβ plaques were significantly reduced by XeC treatment in APP/PS1 + XeC group (1.19±0.23%) (p < 0.01 versus APP/PS1 + PBS). Two-way ANOVA manifested that the genotype and treatment had significant main effects and an interaction effect on the Aβ plaque area (genotype: F (1,12) = 34.185, p < 0.001; treatment: F (1,12) = 8.47, p = 0.013; genotype×treatment: F (1,12) = 8.196, p = 0.014). This suggests that XeC treatment reduced the Aβ deposition in the brain of APP/PS1 mice.
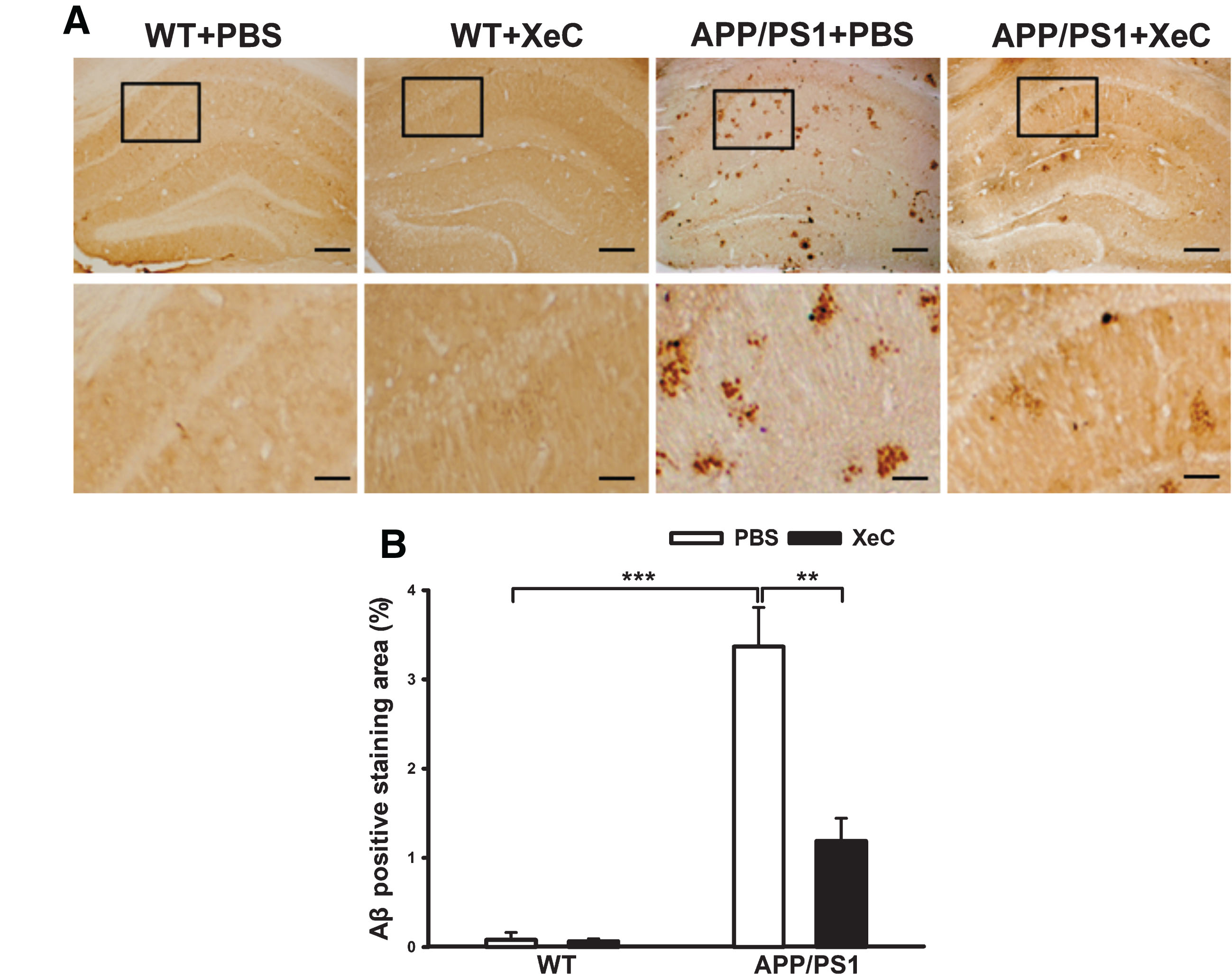
XeC reduced Aβ plaques in the hippocampus of APP/PS1 mice. A) Immunohistochemistry photographs showing the Aβ-immunopositive plaques stained by 6E10 in hippocampal slices. Scale bar = 200μm (top) and 50μm (bottom). B) Histograms indicating the quantitative analysis of Aβ plaques in the hippocampus by comparing Aβ-immunopositive staining area (%) (n = 4).
XeC downregulated ER stress-associated proteins in the hippocampus of APP/PS1 mice
To explore the specific molecular mechanism underlying the neuroprotection of XeC, we next analyzed the expression levels of ER stress-related signaling molecules including 78 kDa glucose-regulated protein (GRP-78), caspase-12, and CAAT/enhancer-binding protein (C/EBP) homologous protein (CHOP) using western blotting (Fig. 4). Two-way ANOVA showed that genotype and treatment had significant main effects and an interaction on the expression level of GRP-78 (genotype: F (1,20) = 6.804, p < 0.05; treatment: F (1,20) = 5.291, p < 0.05; genotype×treatment: F (1,20) = 2.166, p < 0.05). Post hoc test indicated a remarkable increase in GRP-78 (Fig. 4B) in the mice of APP/PS1 + PBS (2.199±0.25) compared to that in the control mice (WT+PBS, p < 0.01), while XeC restored the level of GRP-78 in the mice of APP/PS1 + XeC group (1.07±0.21, p < 0.05). Similarly, two-way ANOVA also showed great main effects and an interactional effect on the caspase-12 (Fig. 4C) and CHOP (Fig. 4D). The expressing levels of caspase-12 (2.32±0.19) and its downstream target-CHOP (2.67±0.21) in APP/PS1 + PBS mice were significantly elevated compared with that in control mice (p < 0.01 for each), while the XeC pretreatment effectively antagonized their increases, declining to 0.80±0.15 (p < 0.001) and 0.83±0.095 (p < 0.01), respectively. These data suggest that XeC ameliorated ER stress by down-regulating the expression levels of GRP-78, CHOP, and caspase-12, which might be associated with the neuroprotection of XeC in the alleviating behavioral and pathological impairments in APP/PS1 mice.
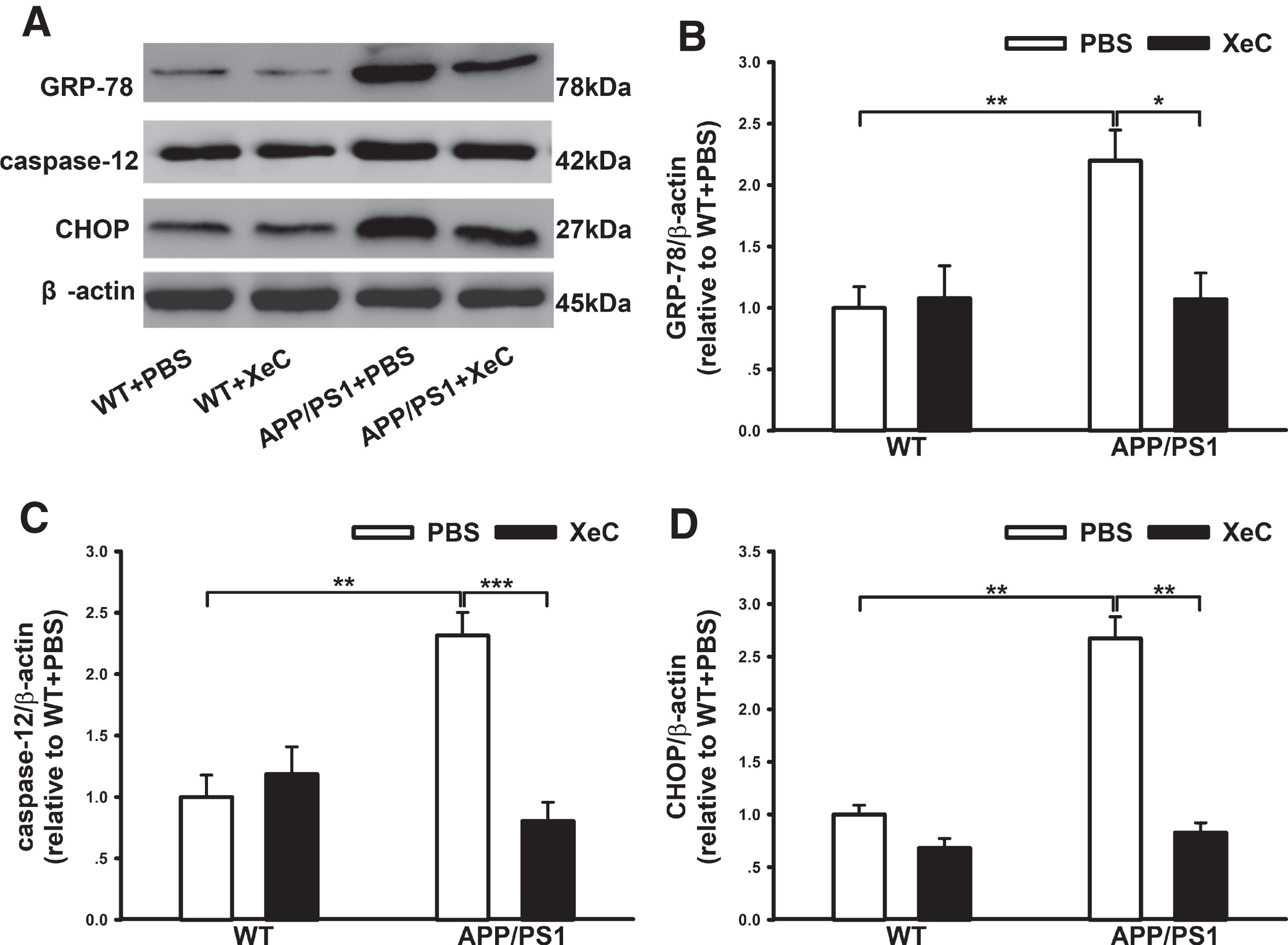
XeC down-regulated ER-stress proteins in the hippocampus of APP/PS1 mice. A) Representative immunopositive bands showing the expression levels of GRP-78, caspase-12 and CHOP in the hippocampus. β-actin was used as loading control. B-D) Histograms showing the quantitative analysis of GRP-78 (B), caspase-12 (C), and CHOP (D), respectively. *p < 0.05, **p < 0.01, ***p < 0.001. All values were shown as mean±SEM (n = 6).
XeC pretreatment ameliorated Aβ1 - 42-induced early apoptosis in primary cultured hippocampal neurons
Prolonged ER stress could result in cell apoptosis [38, 39]. Thus we used flow cytometry to test the effects of XeC on Aβ1 - 42-induced cell apoptosis in cultured hippocampal neurons. Figure 5A displayed representative hippocampal cells activated by Annexin V-FITC/PI in the four groups. The quantified data in the histogram Fig. 5B showed the early apoptotic rate of cells. Notably, the percentage of early apoptosis in Aβ-treated cell group (18.38±1.15%) was apparently more than that in the control group (4.9±0.4%, p < 0.001), while XeC pretreatment before Aβ application displayed a significant decrease in the early apoptotic rate (9.87±0.93%, p < 0.001) compared to the Aβ alone treated groups (Aβ1 - 42: F (1,20) = 94.759, p < 0.001; XeC: F (1,20) = 14.736, p = 0.001; Aβ1 - 42×XeC: F (1,20) = 39.157, p < 0.001). Therefore, we concluded that XeC attenuated Aβ-induced early cell apoptosis.
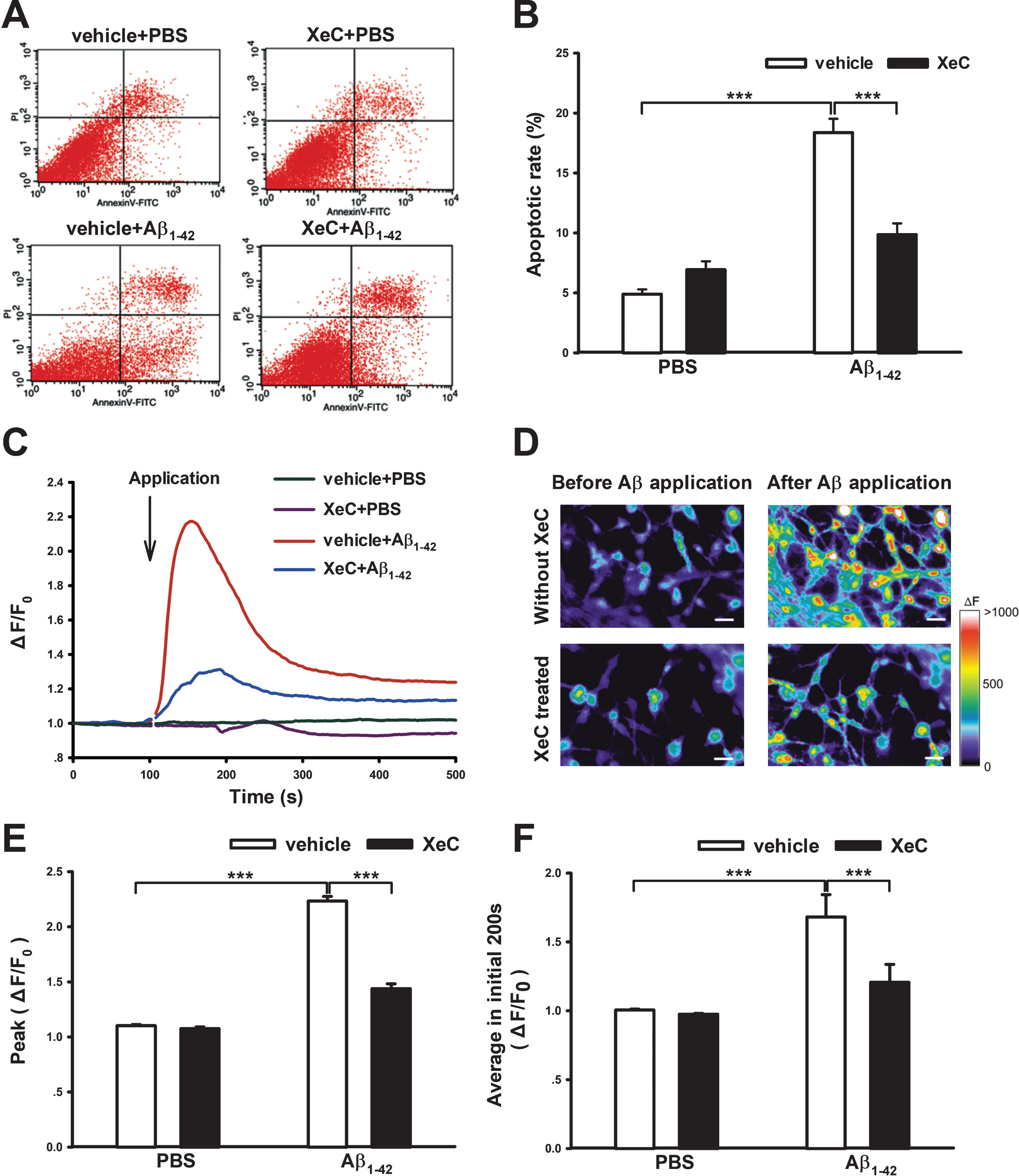
XeC pretreatment ameliorated Aβ1 - 42-induced early cellular apoptosis and [Ca2 +]i elevation in primary cultured hippocampal neurons. A) Representative fluorescence-activated cells detected by Annexin V-FITC/PI and flow cytometry. In each plot, the lower right quadrant distributes early apoptotic cells and the lower left quadrant indicates viable cells. B) Quantity of early apoptotic rate in each groups. ***p < 0.001. All values were shown as mean±SEM (n = 6). C) Averaged [Ca2 +]i change traces from 25 neurons, showing acute application of Aβ1 - 42 (20μmol/L) induced increase of [Ca2 +]i in the presence or absence of XeC. D) Representative intracellular Ca2+ images in primary cultured hippocampal neurons. The increased fluorescence intensity by Aβ (upper row) was obviously suppressed by XeC pretreatment (lower row). Scale bar = 20μm. E,F) Histograms showing the peak (E) and average (F) fluorescence intensities.
XeC suppressed Aβ1 - 42-induced intracellular Ca2+ overload in primary cultured hippocampal neurons
Intracellular Ca2+ overload can lead to ER stress and cell damage like cell apoptosis [40]. To investigate the exact mechanism of the protective effect of XeC, intracellular Ca2+ imaging experiments were performed in primary cultured hippocampal neurons. After loading with Ca2+ indicator Fluo4-NW and obtaining stable baseline recording, the hippocampal neurons were acutely exposed to Aβ1 - 42. Figure 5C showed the intracellular Ca2+ concentration ([Ca2 +]i) changing traces averaged from 25 neurons in different groups. Figure 5D showed representative Ca2+ images before and after application of Aβ1 - 42 in the absence or presence of XeC. Two-way ANOVA showed that Aβ1 - 42 and XeC treatment had significant main effects on the peak value and average values (200-s post Aβ1 - 42 application) of fluorescence intensity changes (ΔF/F0), as well as a significant interaction. Tukey’s post hoc test indicated that, compared with control (Vehicle + PBS), acute application of Aβ significantly increased the ratio of ΔF/F0, being 2.23±0.042 (peak value) and 1.68±0.163 (average value in initial 200 s) (p < 0.001 for each). Interestingly, Aβ-induced increases in the fluorescence intensity were greatly suppressed by the XeC pretreatment (XeC+Aβ1 - 42), becoming 1.44±0.044 and 1.21±0.131, respectively (Fig. 5E, F, p < 0.001 for each). All of these results suggest that XeC significantly reduced Aβ-induced intracellular Ca2+ elevation, which might be associated with its neuroprotection against Aβ-induced cell death.
DISCUSSION
XeC, a kind of macrocyclic bis-1-oxaquinoli- zidines originally isolated from Australia sponge, has more specific effect in inhibiting IP3R-mediated calcium signaling than other antagonists [17]. It is reported that XeC showed great efficiency in improving isoflurane-induced neurodegeneration [19] and forced-swimming induced depression in mice [20]. In the present study, we demonstrated for the first time the neuroprotective roles of XeC in APP/PS1 AD mice. In Y-maze test, XeC did not affect the number of total arm entries, but significantly increased correct alternation percentage in APP/PS1 mice, nearly back to the normal level of control mice. This result suggests that XeC can improve the short-term working memory of AD mice. In MWM tests, XeC treatment significantly decreased the escape latencies of APP/PS1 mice in place navigating tests and increased the swimming time percentage in probe best, implying the long-term spatial learning and memory in AD mice has been partly recovered by XeC. In addition, we noticed that the APP/PS1 mice spent more time in the central area of open field than WT mice. This behavioral disinhibition may reflect a mental disorder in the AD mice. It is reported that hippocampal injury leads to behavioral disinhibition [41, 42]; bilateral hippocampal injection of Aβ1–42 also evoked an obvious behavioral disinhibition in elevated plus-maze test, with an increased residence time in the open arm [37]. Generally, mice prefer the marginal area of a new environment. Therefore, the increased dwell time in the central area of the open field reflected poor cognition of AD mice [43, 44]. Interestingly, XeC ameliorated this behavioral disinhibition of APP/PS1 mice. Taken together, these data indicate that chronic treatment with XeC improves the cognitive and mental behaviors in APP/PS1 mice.
The cognitive deficits in AD are mostly associated with the accumulation and neurotoxicity of Aβ in the AD brain [45]. By examining the distribution of Aβ plaques in the brain of mice using immunohistochemistry, we found that more Aβ aggregated in the hippocampus of APP/PS1 mice, which is consistent with the spatial cognitive impairments in the mice. Importantly, XeC treatment not only alleviates the cognitive impairments but also reduces Aβ burden in the hippocampus. The area percentage of Aβ immunopositive plaques in the hippocampus had a significant reduction, from 3.37% to 1.19% after XeC treatment. It is reported that the pathological changes were also found in the cortical area [46, 47]. In view of the close association of neurotoxic Aβ with ER stress in AD [48], we examined the expression levels of ER stress-related proteins including GRP-78 (an ER resident chaperone and stress response protein), CHOP (a pro-apoptosis protein), and caspase-12 (an apoptotic initiator) in APP/PS1 mice [49, 50], and Aβ-induced apoptosis in cultured hippocampal neurons. Compelling evidence indicate that ER stress could be an early event in AD, which in turn accelerates Aβ production and neurotoxicity [51]. In the present study, we noticed that ER stress had an obvious activation in the AD mice, with high expressing levels of GRP-78 and its downstream target-CHOP and caspase-12. The activation of ER stress in AD mice is supported by the results of Chafekar et al. by using oligomeric Aβ1 - 42 [52]. As mentioned above, ER stress and Aβ deposition promote mutually, probably forming a positive feedback and finally leading to neurotoxicity, such as the cell apoptosis and behavioral disorder in the study. On the contrary, alleviation of ER stress reduces Aβ, improves cognitive function, enhances learning and memory ability, and also eases inflammation in AD models [53, 54]. The present study found that XeC pretreatment effectively downregulated the expression levels of GRP-78, CHOP, and caspase-12 in the hippocampus of AD mice. This result probably partly accounts for why XeC could reduce Aβ deposition in the hippocampus, alleviate cultured hippocampal neuron apoptosis, and improve cognitive behaviors of the APP/PS1 mice. We noticed that the rescue of memory loss of rats [55] and cell apoptosis [56] by liraglutide from Aβ25 - 35 was also through restoring ER stress.
The accumulation of Aβ in AD leads to intracellular Ca2+ dysregulation and thus synaptic dysfunctions, spine loss, and memory deficits, while the calcium mishandling in turn enhances the accumulation and deposition of Aβ [5, 57]. Therefore, the maintenance of intracellular Ca2+ homeostasis is particularly essential for reducing the pathological Aβ and improving memory deficits in AD. As a ubiquitous second message, Ca2+ regulates numerous cellular processes and many neuronal functions such as neurotransmitter release, synaptic plasticity, gene expression, and even cell destiny [58]. ER is the largest Ca2+ pool. Recently, it has been shown that handling ER Ca2+ signaling has therapeutic potential for the treatment of AD [9, 59]. In particular, IP3R-mediate Ca2+ signal disorder plays a critical role in Aβ-induced calcium overload, because suppression of IP3 R reduces more resting calcium levels in neurons from 3×Tg-AD mice than suppression of RyR [15]. We also found that blocking IP3 R with XeC and blocking IP3 R and RyR with XeC plus dantrolene produced nearly the same inhibitory effects on Aβ1 - 42-induced Ca2+ elevation (data not shown). In addition, the connection between ER and mitochondrial influences the function of mitochondrial and its related fundamental cellular processes especially the synaptic activities [60, 61]. The exaggerated Ca2+ signaling through an IP3R-PS interaction is a robust proximal gain-of-function molecular mechanism in FAD [10, 62], which increased the open probability of mitochondrial permeability transition pore and induced cell death [28]. Hedskog et al. found Aβ oligomers also upregulated the mitochondria-associated ER membrane function in postmortem AD brains and APPswe/Lon mice which is associated with IP3 R and VGCC [63]. Therefore, targeting IP3 R may also restore impaired mitochondrial function in AD. As the downstream signal molecules, Ca2 + /CaM kinase β, CaM kinase IV (CaMKIV) and transcription factor cAMP response element binding protein (CREB) have been found to have a great activation in PS-associated FAD which strongly linked to IP3 R [64]. Indeed, in vivo research demonstrated that reducing IP3 R expression rescued the deficits of hippocampal LTP and memory loss, and decreased the deposition of Aβ and tau hyperphosphorylation in 3×Tg-AD mice [16].
In the present study, our Ca2+ imaging experiments showed a significant stimulation effect of Aβ1 - 42 on the [Ca2 +]i, which is consistent with the neuronal hyperactivity in AD mouse [48]. Gilson et al. also found that Aβ oligomer rapidly and dose-dependently (0.1-1μM) increased [Ca2 +]i in PC12 cells and cortical neurons [65]. Recent publications also provided more solid evidence that soluble Aβ1 - 42 could induce a robust increase in [Ca2 +]i in cultured hippocampal neurons [66, 67]. Ca2+ fluxes across the plasma membrane and release from intracellular stores have both been reported to underlie the Aβ-induced intracellular Ca2+ overload. A number of mechanisms by which Aβ disrupts intracellular Ca2+ homeostasis have been put forward such as forming cation-conducting pores [68–70], activating plasma membrane receptors such as NMDAR [71–73] and altering neuronal excitability [74]. Importantly, it has been reported that extracellular Aβ oligomers increased IP3 production by activation of mGluR5 [75, 76]; Aβ-induced Ca2+ release from ER mostly depended on activating PLC and subsequent IP3 generation [77]; intracellular Aβ evokes Ca2+ liberation through InsP3Rs in an IP3-dependent manner [78]. Therefore, it is not strange that Aβ-induced increases in [Ca2 +]i were greatly suppressed by the XeC pretreatment in the present study. These results indicate that IP3R-mediated intracellular Ca2+ release is involved in the Aβ-induced Ca2+ overload, and blocking the over activation of IP3 R with XeC would be helpful to the maintenance of Ca2+ homeostasis and improvement of memory in AD.
In conclusion, the present study demonstrated for the first time that IP3 R antagonist XeC could ameliorate the cognitive impairments and pathological damage in APP/PS1 mice. The neuroprotection of XeC might be attributed to its blocking effects on Aβ-induced Ca2+ disruption, ER stress, and early cell apoptosis. Our results also indicate that IP3R-mediated Ca2+ disorder plays a key role in the pathogenesis of AD, and by targeting the IP3 R, XeC might be considered as a novel therapeutic strategy in AD.
DATA AVAILABILITY STATEMENT
The data that support the findings of this study are available from the corresponding author upon reasonable request.
Footnotes
ACKNOWLEDGMENTS
This research was supported by the following programs: 1) National Natural Science Foundation of China (No. 31471080 and No. 31600865); 2) Fund Program for “Sanjin Scholars” of Shanxi Province; 3) Shanxi Province Science Foundation for Excellent Young Scholars, Grant Number: 201801D211005; 4) Shanxi Province Innovation Foundation for Postgraduate (No. 2018SY051); 5) Fund for Shanxi “1331 Project” Key Subjects Construction (1331KSC); 6) Fund for Shanxi Key Subjects Construction (FSKSC).