
Abstract
Background:
Subclinical cardiac dysfunction is associated with decreased cerebral blood flow, placing the aging brain at risk for Alzheimer’s disease (AD) pathology and neurodegeneration.
Objective:
This study investigates the association between subclinical cardiac dysfunction, measured by left ventricular ejection fraction (LVEF), and cerebrospinal fluid (CSF) biomarkers of AD and neurodegeneration.
Methods:
Vanderbilt Memory & Aging Project participants free of dementia, stroke, and heart failure (n = 152, 72±6 years, 68% male) underwent echocardiogram to quantify LVEF and lumbar puncture to measure CSF levels of amyloid-β42 (Aβ42), phosphorylated tau (p-tau), and total tau (t-tau). Linear regressions related LVEF to CSF biomarkers, adjusting for age, sex, race/ethnicity, education, Framingham Stroke Risk Profile, cognitive diagnosis, and apolipoprotein E ɛ4 status. Secondary models tested an LVEF x cognitive diagnosis interaction and then stratified by diagnosis (normal cognition (NC), mild cognitive impairment (MCI)).
Results:
Higher LVEF related to decreased CSF Aβ42 levels (β= –6.50, p = 0.04) reflecting greater cerebral amyloid accumulation, but this counterintuitive result was attenuated after excluding participants with cardiovascular disease and atrial fibrillation (p = 0.07). We observed an interaction between LVEF and cognitive diagnosis on CSF t-tau (p = 0.004) and p-tau levels (p = 0.002), whereas lower LVEF was associated with increased CSF t-tau (β= –9.74, p = 0.01) and p-tau in the NC (β= –1.41, p = 0.003) but not MCI participants (p-values>0.13).
Conclusions:
Among cognitively normal older adults, subclinically lower LVEF relates to greater molecular evidence of tau phosphorylation and neurodegeneration. Modest age-related changes in cardiovascular function may have implications for pathophysiological changes in the brain later in life.
INTRODUCTION
Heart failure is associated with cognitive impairment [1] and an increased risk of clinical dementia [2]. Emerging evidence suggests even subclinical changes in cardiac function relate to neurodegeneration [3], worse cognitive performance [4], and increased incidence of clinical dementia, including Alzheimer’s disease (AD) [5]. The exact etiology underlying these associations is poorly understood. AD is characterized by the abnormal accumulation of amyloid-β (Aβ) plaques and neurofibrillary tangles comprised of phosphorylated tau. In vivo, these protein abnormalities can be detected in cerebrospinal fluid (CSF) as Aβ42, phosphorylated tau (p-tau), and total tau (t-tau) [6]. It is plausible that associations between cardiac dysfunction and abnormal brain aging may be driven by underlying associations between cardiac dysfunction and increased levels of neuropathology. However, to our knowledge, limited research has studied associations between subclinical cardiac dysfunction and CSF biomarkers of AD and neurodegeneration.
This study aims to elucidate underlying pathological processes that may account for previously reported associations linking subtle reductions in cardiac function to neurodegeneration [3], cognitive impairment [4], and clinical dementia [5]. To achieve this aim, we relate a common standard of clinical care for measuring systolic function, left ventricular ejection fraction (LVEF), to biomarkers of AD and neurodegeneration, including cerebral amyloidosis (Aβ42), tau phosphorylation which relates to neurofibrillary tangle pathology (p-tau), and neurodegeneration (t-tau) [6]. Based on previously reported associations between subclinical cardiac dysfunction and neurodegeneration [3], worse cognitive performances [4], and increased risk of AD [5], we hypothesize that lower (worse) LVEF will relate to lower CSF Aβ42 (reflecting more amyloid sequestered in the brain), higher CSF p-tau (reflecting more tangle formation), and higher CSF t-tau concentrations (reflecting greater neurodegeneration).
METHODS
Study cohort
The Vanderbilt Memory & Aging Project is a longitudinal study investigating vascular health and brain aging, enriched for mild cognitive impairment (MCI) [7]. Inclusion required participants be age≥60 years, speak English, have adequate auditory and visual acuity, and have a reliable study partner. At eligibility, participants underwent medical record review, medical history, clinical interview (including functional assessment and Clinical Dementia Rating [8] with a reliable informant), and neuropsychological assessment. Participants were excluded for a cognitive diagnosis other than normal cognition (NC), early MCI [9], or MCI based on the National Institute on Aging/Alzheimer’s Association Workgroup clinical criteria [10]; MRI contraindication; history of major psychiatric illness, neurological disease (e.g., stroke), head injury with loss of consciousness > 5 min, heart failure, and systemic or terminal illness that would affect follow-up participation. At enrollment participants completed an evaluation, including (but not limited to) fasting blood draw, clinical interview with medication review, physical examination, neuropsychological assessment, echocardiogram, cardiac MRI, and optional lumbar puncture for CSF acquisition. Participants were excluded from the current study for missing echocardiogram, covariate, or CSF data (see Fig. 1 for inclusion and exclusion details).
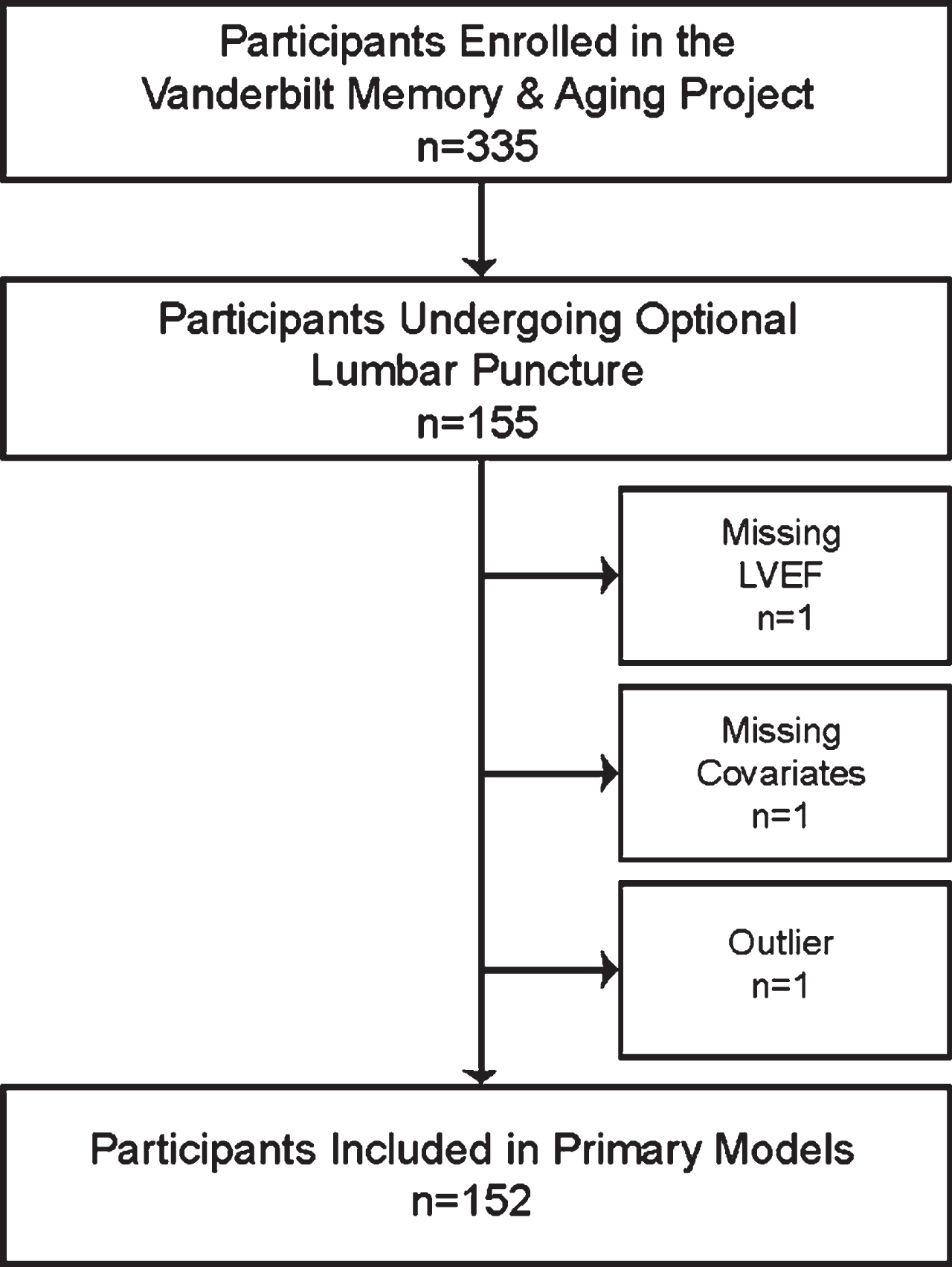
Participant Inclusion and Exclusion Details. Missing data categories are mutually exclusive. Outlier was defined as 7 standard deviations outside the group mean. In secondary models, sensitivity analyses excluded 8 participants with cardiovascular disease or atrial fibrillation. LVEF, left ventricular ejection fraction.

Left Ventricular Ejection Fraction and CSF Biomarkers. Solid and dashed lines reflect unadjusted values of CSF biomarker outcomes (Y axis) corresponding to left ventricular ejection fraction (X axis) for each diagnostic group (solid black line = normal cognition (NC), dashed red line = mild cognitive impairment (MCI)). Shading reflects 95% confidence interval. Aβ42, amyloid-β42, CSF, cerebrospinal fluid; P-tau, phosphorylated tau; T-tau, total tau.
Standard protocol approvals, registrations, and participant consent
The protocol was approved by the Vanderbilt University Medical Center Institutional Review Board. Written informed consent was obtained prior to data collection. Due to participant consent restrictions in data sharing, a subset of data is available to others for purposes of reproducing the results or replicating procedures. These data, analytical methods, and study materials can be obtained by contacting the corresponding author.
Echocardiogram
Standard 2D, M-mode, and Doppler transthoracic echocardiography were completed by a single research sonographer at the Vanderbilt University Medical Center Clinical Research Center on a Philips IE33 cardiac ultrasound machine (Philips Medical, Andover, MD). One of two board certified cardiologists (DKG, LAM) blinded to clinical information confirmed the quantitative measures of cardiac structure and function using commercially available software (HeartLab, AGFA Healthcare, Greenville, SC).
Image acquisition and quantification were completed according to American Society of Echocardiography guidelines. LVEF was calculated by the biplane Simpson’s method from the apical 4 and 2 chamber views as (end diastolic volume – end systolic volume)/end diastolic volume * 100. Final measurements were from a single cardiac cycle for participants in normal sinus rhythm or an average of three cardiac cycles for participants in atrial fibrillation.
Lumbar puncture and biochemical analyses
CSF was collected with polypropylene syringes using a Sprotte 25-gauge spinal needle in an intervertebral lumbar space. Samples were aliquoted in polypropylene tubes, stored at -80°C, and analyzed using commercially available enzyme-linked immunosorbent assays (ELISA; Fujirebio, Ghent, Belgium) to determine Aβ42 (INNOTEST® β-AMYLOID (1 -42) ), p-tau (INNOTEST® PHOSPHO-TAU (181P) ), and t-tau levels (INNOTEST® hTAU). Intra-assay coefficients of variation were < 10%. For all samples, the concentrations of each biomarker were measured in one round of experiments using a single batch of reagents by board-certified laboratory technicians who were blinded to clinical data.
Analytical plan
Analytical covariates were defined as follows: systolic blood pressure was the mean of two measurements. Diabetes mellitus was defined as fasting blood glucose≥126 mg/dL, hemoglobin A1C≥6.5%, or oral hypoglycemic or insulin medication usage. Medication review determined anti-hypertensive medication use. Left ventricular hypertrophy (LVH) was defined on echocardiogram (LV mass index > 115 g/m2 in men, >95 g/m2 in women). Self-report atrial fibrillation was corroborated by any one of the following sources: echocardiogram, cardiac magnetic resonance, documented prior procedure/ablation for atrial fibrillation, or medication usage for atrial fibrillation. Current cigarette smoking (yes/no within the year prior to baseline) was ascertained by self-report. Self-report prevalent cardiovascular disease (CVD) with supporting medical record evidence included coronary heart disease, angina, or myocardial infarction (note, heart failure was a parent study exclusion). Framingham Stroke Risk Profile (FSRP) score was calculated by applying points by sex for age, systolic blood pressure, anti-hypertensive medication usage, diabetes mellitus, current cigarette smoking, LVH, CVD, and atrial fibrillation. Race/ethnicity was treated as a categorical variable with two levels: non-Hispanic White and other (i.e., Hispanic, African American, Native American, Asian). Apolipoprotein E (APOE) genotyping was performed on whole blood samples. APOE ɛ4 carrier (APOE ɛ4) status was defined as positive (ɛ2/ɛ4, ɛ3/ɛ4, ɛ4/ɛ4) or negative (ɛ2/ɛ2, ɛ2/ɛ3, ɛ3/ɛ3). Between-group differences (i.e., NC, eMCI, MCI) were tested using Kruskal-Wallis test for continuous variables and Pearson chi-square test for categorical variables with all cell counts > 5 and Fisher’s exact test for categorical variables with any cell count≤5.
Prior to analyses, data were determined to be normally distributed based on visual inspection and the Shapiro-Wilk’s test. Loess curves on scatterplots of unadjusted data were also inspected and did not reveal any evidence of a nonlinear association between LVEF and CSF biomarkers. For hypothesis testing, linear regression models with ordinary least square estimates related LVEF to CSF biomarkers (one biomarker per model). All models were adjusted for age, sex, race/ethnicity, education, FSRP (excluding points assigned for age), cognitive diagnosis (NC, early MCI, MCI), and APOE ɛ4 status. To determine if effects were driven by participants with cognitive impairment, secondary models restricting the sample to participants with NC or MCI tested an LVEF x diagnosis interaction and then stratified by diagnosis (NC, MCI). These models excluded early MCI participants due to the small sample size. To determine if effects were influenced by biological sex (as AD pathology is more common in women than men) [11], secondary models tested an LVEF x sex interaction and then stratified by sex. Sensitivity analyses were performed for all models by excluding participants with atrial fibrillation and prevalent CVD. Significance was set a priori at p < 0.05. Analyses were conducted using R 3.4.3 (http://www.r-project.org).
RESULTS
Participant characteristics
Participants included 152 adults age 60-90 years (72±6 years), 68% were men, and 93% self-identified as non-Hispanic White. LVEF ranged 51% to 82%. CSF Aβ42 levels ranged 289 pg/mL to 1195 pg/mL. CSF t-tau levels ranged 77 pg/mL to 1542 pg/mL. CSF p-tau levels ranged 13 pg/mL to 157 pg/mL. See Table 1 for total sample characteristics and by cognitive diagnosis. As compared to participants excluded from analyses, participants included were more likely to be non-Hispanic White (p = 0.002) and male (p = 0.003) with a lower FSRP (p = 0.001) but comparable for LVEF (p = 0.65), age (p = 0.38), education (p = 0.07), cognitive diagnosis (p = 0.50), and APOE4 frequency (p = 0.62).
Participant characteristics
Between-group differences were tested using Kruskal-Wallis test for continuous variables and Pearson chi-square test for categorical variables with all cell counts > 5 and Fisher’s exact test for categorical variables with any cell count≤5. Values denoted as mean±standard deviation or frequency. *a modified score was included in statistical models, which excluded points assigned to age (Total = 5.96±2.59; NC = 5.51±2.56; eMCI = 6.73±2.15; MCI = 6.39±2.65). †amyloid positive = Aβ42 ≤ 530 pg/mL [25]. ‡p-tau positive = p-tau≥80 pg/mL [26]. §t-tau positive = t-tau≥400 pg/mL [26]. p-values are presented for the main-effect comparisons across diagnostic groups; statistically significant between group-differences denoted with the following distinctions: ||NC versus eMCI, #eMCI versus MCI, **NC versus MCI; Aβ42, amyloid-β42; APOE, Apolipoprotein E; CVD, cardiovascular disease; CSF, cerebrospinal fluid; eMCI, early mild cognitive impairment; MCI, mild cognitive impairment; NC, normal cognition; p-tau, phosphorylated tau; t-tau, total tau.
LVEF and CSF Aβ42
LVEF was inversely related to CSF Aβ42 levels (β= –6.50; 95% confidence interval (CI) = –12.7, –0.3; p = 0.04), but the result was counterintuitive suggesting better LVEF corresponded to lower Aβ42 levels (i.e., greater cerebral amyloid aggregation). In sensitivity analyses excluding participants with prevalent CVD or atrial fibrillation the association between LVEF and CSF Aβ42 was attenuated (β= –6.0; 95% CI = –12.5, 0.4; p = 0.07; Table 2).
Left Ventricular Ejection Fraction and CSF Biomarkers
*Sensitivity analyses excluded 8 participants with cardiovascular disease or atrial fibrillation. Models were adjusted for age, race/ethnicity, education, Framingham Stroke Risk Profile (FSRP) minus age, and cognitive diagnosis. CI, confidence interval; CSF, cerebrospinal fluid; CVD, cardiovascular disease; MCI, mild cognitive impairment; NC, normal cognition.
The LVEF x cognitive diagnosis interaction term did not relate to CSF Aβ42 levels (β= –12.1; 95% CI = -24.8, 0.6; p = 0.06). In stratified models, better LVEF was counterintuitively associated with lower CSF Aβ42 levels (i.e., greater cerebral amyloid accumulation) among MCI (β= –10.8; 95% CI = –19.4, –2.2; p = 0.01) but not NC participants (β=0.3; 95% CI = –9.1, 9.8; p = 0.95).
To better understand the counterintuitive finding between LVEF and CSF Aβ42 levels in MCI participants, several post-hoc analyses were performed. First, we repeated the main effect model in MCI participants without adjustments and results were similar (β= –14.0; 95% CI = –24.5, –3.5; p = 0.01). Next, we explored potential LVEF interactions with covariates on CSF Aβ42 levels among MCI participants. Most LVEF and covariate interaction terms were unrelated to CSF Aβ42 levels, including LVEF x age (β= –5.4; 95% CI = -24.2, 13.3; p = 0.56), LVEF x sex (β= –4.2; 95% CI = –22.1, 13.6; p = 0.64), LVEF x education (β= –0.5; 95% CI = –20.8, 19.7; p = 0.96), LVEF x race/ethnicity (β= –8.6; 95% CI = –34.3, 17.1; p = 0.50), LVEF x APOE-ɛ4 (β=7.2; 95% CI = -12.3, 26.8; p = 0.46), and LVEF x FSRP (β= –4.9; 95% CI = –25.1, 15.2; p = 0.62). When examining interactions between LVEF and the individual components of the FSRP, a majority of the comparisons were again null, including LVEF x systolic blood pressure (β=5.0; 95% CI = -14.5, 24.6; p = 0.61), LVEF x anti-hypertensive medication usage (β= –0.4; 95% CI = –21.2, 20.5; p = 0.97), and LVEF x LVH (β=10.6; 95% CI = -27.3, 48.5; p = 0.58). The exception was an association between LVEF x diabetes and CSF Aβ42 levels (β= –26.7; 95% CI = –48.8, –4.6; p = 0.02). When results were stratified, LVEF was related to CSF Aβ42 levels in non-diabetic MCI participants (β= –16.1; 95% CI = –26.2, –5.9; p = 0.003).
The LVEF x sex interaction term related to CSF Aβ42 levels (β= –13.0; 95% CI = -25.7, -0.3; p = 0.04), whereby better LVEF counterintuitively related to lower CSF Aβ42 levels (i.e., greater cerebral amyloid accumulation) in female (β= –12.1; 95% CI = –22.6, –1.7; p = 0.02) but not male participants (β= –1.5; 95% CI = –9.6, 6.7; p = 0.72).
To better understand the counterintuitive finding between LVEF and CSF Aβ42 levels in female participants, we performed several post-hoc analyses. First, we repeated the main effect model restricted to female participants without adjustments and results were similar (β= –13.5; 95% CI = –24.4, –2.6; p = 0.02). Next, we explored potential LVEF interactions with covariates on CSF Aβ42 levels just in the female participants. All LVEF and covariate interaction terms were unrelated to CSF Aβ42, including LVEF x age (β= 5.5; 95% CI = –15.1, 26.1; p = 0.59), LVEF x education (β= –9.8; 95% CI = -32.4, 12.8; p = 0.38), LVEF x race/ethnicity (β= –20.2; 95% CI = –50.2, 9.8; p = 0.18), LVEF x APOE ɛ4 (β= 9.2; 95% CI = –15.6, 33.9; p = 0.46), LVEF x diagnosis (β= –9.2; 95% CI = –30.4, 11.9; p = 0.38), and LVEF x FSRP (β= –0.6; 95% CI = –25.5, 24.2; p = 0.96). When examining interactions between LVEF and the individual FSRP components, comparisons were again null (p-values≥0.13).
LVEF and CSF T-tau
LVEF was unrelated to CSF t-tau levels (β= –0.9; 95% CI = –7.6, 5.8; p = 0.79; Table 2). In sensitivity analyses excluding participants with prevalent CVD or atrial fibrillation non-significant results persisted (β= –0.7; 95% CI = –7.6, 6.2; p = 0.85). The LVEF x cognitive diagnosis interaction term related to CSF t-tau levels (β= 20.9; 95% CI = 6.9, 34.9; p = 0.004), such that lower LVEF was associated with higher CSF t-tau levels in the NC cohort (β= –9.7; 95% CI = –17.1, –2.4; p = 0.01). The LVEF x sex interaction term did not relate to CSF t-tau levels (β= –3.0; 95% CI = –16.9, 11.0; p = 0.67).
LVEF and CSF P-tau
LVEF was unrelated to CSF p-tau levels (β= –0.3; 95% CI = –1.1, 0.5; p = 0.47; Table 2). In sensitivity analyses excluding participants with prevalent CVD or atrial fibrillation non-significant results persisted (β= –0.2; 95% CI = –1.0, 0.5; p = 0.53). The LVEF x cognitive diagnosis interaction term related to CSF p-tau levels (β= 2.6; 95% CI = 1.0, 4.2; p = 0.002), such that lower LVEF was associated with higher CSF p-tau levels in the NC cohort (β= –1.4; 95% CI = -2.3, –0.5; p = 0.003). The LVEF x sex interaction term did not relate to CSF p-tau levels (β= –0.5; 95% CI = –2.1, 1.1; p = 0.55).
DISCUSSION
The aim of this study was to elucidate underlying pathways that may provide biological insights into previously reported associations linking subtle cardiac function reductions to worse cognitive outcomes among aging adults [4, 5]. While main effect models were mostly null, we found a diagnostic interaction between LVEF and CSF biomarkers emerged in which lower LVEF related to greater molecular evidence of neurodegeneration (t-tau) and neurofibrillary tangle pathology (p-tau) in participants with normal cognition. All statistically significant associations remained when adjusting for stroke volume (data not shown) and excluding participants with prevalent CVD and atrial fibrillation, suggesting results cannot be explained by comorbid vascular disease.
Previous work, including our own, has reported that subclinical reductions in cardiac function relate to worse cognition in aging adults [4], including incident clinical dementia [5]. These associations may be due to decreased blood flow delivery to the brain, an observation supported by a recent report that subclinical reductions in cardiac output relate to modestly decreased cerebral blood flow (CBF) in aging adults [12]. Our current findings build on these prior observations by suggesting subtle cardiac dysfunction relates to in vivo molecular biomarker evidence of phosphorylation of tau and neurodegeneration. Thus, if age-related cardiac changes impact blood flow delivery to the brain (even subtly), decreased CBF may induce the earliest biochemical changes underlying neurofibrillary tangle formation and contribute to neuronal death prior to the onset of memory loss or other cognitive symptoms [13].
There are multiple pathways by which subclinical cardiac dysfunction and subsequent reductions in CBF may affect phosphorylation of tau and neurodegeneration. Previous research suggests that tau may be normally modified via the attachment of a monosaccharide to prevent phosphorylation [14]. This modification, however, has been shown to be downregulated when CBF is reduced, resulting in increased phosphorylation of tau [15]. Research in transgenic AD mouse models has also shown that acute CBF reductions inhibit enzymes that function to dephosphorylate tau [16]. Additionally, chronic subclinical reductions in CBF could also result in blood-brain barrier (BBB) breakdown due to a loss of shear stress required to maintain vascular endothelial cells [17]. Such breakdown creates vulnerability in which blood-derived proteins (e.g., thrombin, plasminogen, fibrinogen) can enter the brain [18], resulting in neuronal toxicity and death [19, 20]. This vulnerability can be exacerbated in older adults who have been shown to have increased age-related BBB breakdown in the hippocampus [21], which anatomically corresponds to where neurofibrillary tangles first evolve in AD [22]. Therefore, subclinical reductions in LVEF may interfere with CBF delivery, creating or exacerbating a vulnerable environment for BBB breakdown, and inducing phosphorylation of tau, neuronal toxicity, and neuronal death. Alternatively, while a direct pathway between age-related subclinical cardiovascular dysfunction and molecular biomarkers of tau phosphorylation and neurodegeneration is plausible, we cannot rule out the possibility of a shared upstream mechanism or epiphenomenon explaining our results. Additional studies are needed to better understand mechanisms underlying this association.
It is noteworthy that associations between lower LVEF and higher CSF t-tau and p-tau levels were observed in NC but not MCI participants. The lack of a similar association in MCI participants may be due to these participants having higher levels of neuropathological burden from concomitant disease processes (e.g., amyloidosis, cerebral small vessel disease) that drive their cognitive symptom manifestation. That is, other competing factors likely explain more variance in p-tau and t-tau levels among MCI participants, reducing the relative contribution of subclinical cardiac changes on these biomarker outcomes. The observation that subclinical cardiac changes relate to molecular biomarkers of neurofibrillary tangle pathology and neurodegeneration prior to onset of cognitive symptoms suggests subtle cardiovascular dysfunction may initiate or contribute to early tangle pathology or cell death rather than accelerate it. Indeed, a comprehensive data driven model in older adults recently showed that vascular dysfunction precipitates downstream changes in brain aging, including neurodegeneration and cognitive impairment [23].
We observed a counterintuitive finding whereby better LVEF related to decreased CSF Aβ42 concentrations (reflecting greater cerebral amyloid aggregation) particularly among MCI participants and among our female participants. These results were present in adjusted and unadjusted models suggesting overfitting cannot explain the unexpected results. In post-hoc analyses, we failed to identify an interaction between LVEF and most covariates (e.g., age, education, race/ethnicity, FSRP, APOE ɛ4) on CSF Aβ42 levels within the MCI or female participant subgroups. Among MCI participants, we did identify an interaction between LVEF and diabetes on CSF Aβ42 levels. Stratified models aligned with the initial counterintuitive finding, such that higher LVEF was associated with lower CSF Aβ42 levels (greater cerebral amyloid aggregation) in non-diabetic MCI participants. It is worth noting that although results did not meet our a priori threshold for significance, we observed a similar counterintuitive effect in our MCI cohort between LVEF and CSF levels of both t-tau and p-tau. It is plausible that in the presence of abnormal protein accumulation, cardiac function increases in a compensatory manner to support clearance. Alternatively, and more likely, this counterintuitive finding may be due to participant selection bias (e.g., recruitment into a memory-focused study in which participants were excluded for clinical heart failure) or due to a more complex, multivariable (e.g., 3-way) interaction or confound not captured by our methods. Replication is needed in larger cohorts to fully elucidate associations between LVEF and CSF biomarkers and determine if these effects are truly counterintuitive among individuals with prodromal dementia.
Our study has several strengths, including using a well-validated and commonly used measure of cardiac function, a core laboratory for processing both cardiac and CSF measurements where technicians were blinded to participant information, comprehensive ascertainment of potential confounders, and stringent quality control procedures. However, several limitations must be considered. Due to the observational design of our study we cannot draw any conclusions regarding causality. Additionally, while LVEF is a common standard of clinical care for measuring cardiac function, it may be less sensitive than other cardiac measures at detecting early changes to systolic function (e.g., cardiac strain). Lastly, participants were well-educated, predominantly White, and have less cardiovascular burden compared to the general population. Generalizability to other races, ethnicities, ages, or adults with poorer health is unknown. In a cohort with increased vascular risk or disease, we might expect stronger associations between cardiovascular dysfunction and in vivo molecular biomarkers of neuropathology than observed here. Replication is needed, especially to better understand the counterintuitive findings among MCI participants and to rule out the possibility of a false positive finding.
In summary, subclinical cardiac changes, captured by lower LVEF, relate to in vivo molecular biomarker evidence of neurofibrillary tangle pathology and neurodegeneration among cognitively normal older adults without clinical heart failure. Modest age-related changes in cardiovascular function may have implications for abnormal biochemical changes in the brain in late life, presumably through subtle reductions in blood flow delivery to the brain. Future research is needed to determine the exact mechanism and direction of effect underlying these associations.
Footnotes
ACKNOWLEDGMENTS
The authors would like to thank the dedicated Vanderbilt Memory & Aging Project participants, their loved ones, and our devoted staff and trainees who contributed to recruitment, screening, and enrollment of the cohort. We also thank our skilled laboratory technicians at the Clinical Neurochemistry Laboratory in Mölndal, Sweden. This research was supported by Alzheimer’s Association IIRG-08-88733 (ALJ), R01-AG034962 (ALJ), R01-AG056534 (ALJ), R01-NS100980 (ALJ), K24-AG046373 (ALJ), Paul B. Beeson Career Development Award in Aging K23-AG045966 (KAG), Paul B. Beeson Career Development Award in Aging K23-AG048347 (SPB), K01-AG049164 (TJH), K12-HL109019 (DKG), T32-GM007347 (EEM), F30-AG064847 (EEM), F32-AG058395 (KEO), UL1-TR000445 (Vanderbilt Clinical Translational Science Award), S10-OD023680 (Vanderbilt’s High-Performance Computer Cluster for Biomedical Research), the Vanderbilt Memory & Alzheimer’s Center, the Torsten Söderberg Foundation, Stockholm, Sweden, the Swedish Research Council (#2017-00915), the Swedish Alzheimer Foundation (#AF-742881), Hjärnfonden, Sweden (#FO2017-0243), and a grant (#ALFGBG-715986) from the Swedish state under the agreement between the Swedish government and the County Councils, the ALF-agreement.